IDENTIFYING A DISEASE CAUSING MUTATION
|
|
|
- Brittany Gibbs
- 5 years ago
- Views:
Transcription
1 IDENTIFYING A DISEASE CAUSING MUTATION Targeted resequencing MARCELA DAVILA 3/MZO/2016

2 Core Facilities at Sahlgrenska Academy












3 Statistics Software

4 Increasing statistical and bioinformatics knowledge Personalized training (software/programming) Courses Genomics and Advanced NGS data analysis Perl for life science researchers Unix with applications to NGS data Seminars and workshops
5 Supporting local bioinformaticians


6 Sanger sequencing Dye-labeled terminator Capillar electrophoresis C h r o m a t o DNA template Laser beam g r a m


 Sequencing reaction")
7 Next Generation Sequencing Roche 454 Solexa Illumina SOLiD Life Technologies Ion Torrent Life Technologies Qiagen DNA library preparation Amplification (epcr, bridge PCR) Sequencing reaction Imaging Decoding





8 Third Generation Sequencing Single molecule- real time No optics Increased sequencing speed Nanopore Ion Torrent SMRT

9 Sequencing Costs 2nd NGS Sanger HGP 3rd NGS HiseqX






10 Illumina workflow Library prep Cluster generation Sequencing, imaging and base calling Fastq files
11 Fastq format :instrument:run:flowcell:lane:tile:x:y pair:fail:control:index 2) sequence 3) marker 4) quality 1:N:0:TACAGC 2) GCATTTTAGTAGAACCAGNCATTTCCCCCNACNTCNNTNCGNNANNNNTAA 3)

12 Phred quality score Probability that the base has been erroneously called Phred score P(called wrong) 10 1 in 10 90% 20 1 in % 30 1 in ,9% Accuracy base call 40 1 in ,99% 50 1 in ,999% Phred = 10 LIMS Phred = 50


13 Sequencing run Quality QScore Distribution A succesful run should have 80% >= Q30 Illumina Sequencing Analysis Viewer


14 Sequencing run Quality Data by Cycle Illumina Sequencing Analysis Viewer


15 Sequencing run Quality Demultiplexing Illumina Sequencing Analysis Viewer - CASAVA
16 Different recepies Single end (SE) Paired-end (PE) R1 R bp Mate-pair (MP) R1 R2 2-5 kb
17 Data handling workflow Exome-seq Quality Check Quality Filter Mapping to ref genome De novo assembly Methyl-seq SNV detection CpG patterns Metagenomics ChIP-Seq RNA-seq Taxa summary Peak detection Transcript estimation



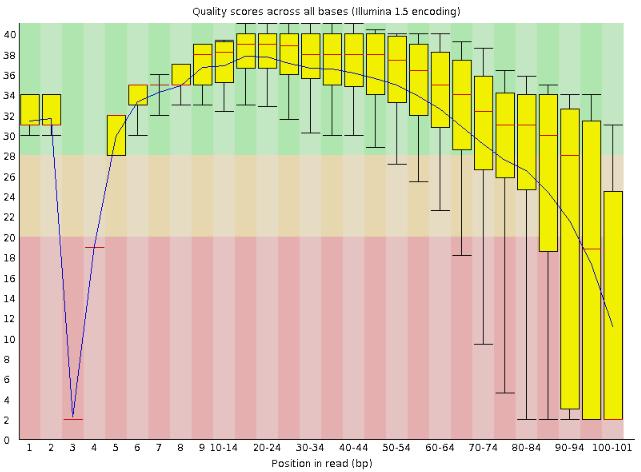
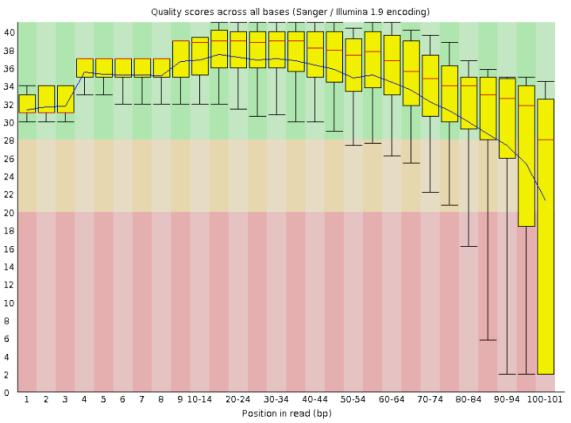
18 Quality check with FastQC





19 Per base sequence content A Exome-seq BS-seq C B bad adapter clipping RNA-seq D

20 Per base sequence content Amplicon seq
21 Human Mouse Rat Ecoli Yeast PhiX Adapters Vectors Wasp No hits Human Mouse Rat Ecoli Yeast PhiX Adapters Vectors Wasp No hits %mapped Contamination check with FastQScreen Good Bad
22 Quality Filter with PRINSeq Ambiguous 1:N:0: GCATTTTAGTAGAACCAGNCATTTCCCCCNACNTCNNTNCGNNANNNNTAA X nts Low quality TRIM-galore
23 Data handling workflow Quality Check Quality Filter Exome-seq Mapping to ref genome De novo assembly Methyl-seq SNV detection CpG patterns Metagenomics ChIP-Seq RNA-seq Taxa summary Peak detection Transcript estimation
24 Mapping CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF TCGATCGACG CACGTGCTGG ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA GGAACGC CTACT CGACGCA CTACT TGGAACGC READS WHERE to place the reads? a) Unique reads b) Everywhere possible c) Choose one randomly d) Use pair-end data mean DNA fragment size: 40 Bfast, BioScope, Bowtie, BWA, CLC bio, CloudBurst, Eland/Eland2, GenomeMapper, GnuMap, Karma, MAQ, MOM, Mosaik, MrFAST/MrsFAST, NovoAlign, PASS, PerM, RazerS, RMAP, SSAHA2, Segemehl, BAM files
25 Mapping HOW to place the reads? Ungapped, Gapped RNA-seq Exome

26 IGV Integrative Genome Viewer coverage reads My BAM gene

27 UCSC browser gene variants My BAM My VCF Variation track





28 Data handling workflow Quality Check Quality Filter Exome-seq Mapping to ref genome De novo assembly Methyl-seq SNV detection CpG patterns Metagenomics ChIP-Seq RNA-seq Taxa summary Peak detection Transcript estimation
29 Monogenic diseases Modifications of a single gene over 10,000 of human diseases (½ have a gene associated) DISEASE GENE MUTATION Thalassaemia HBB frameshift Sickle cell anemia HBB G6V Cystic Fibrosis CFTR G542X Fragile X syndrome FMR1 CGG expansion Huntington s HTT CAG +36 repeats UTRs Coding regions Splice sites/branch site

30 Enrichment kits


31 Enrichment kits mirna UTR s



32 Realignment and recalibration Correct alignments due to the presence of indels Differenciate between polymorphisms and sequencing errors GATK

33 Variant calling CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF TCGATCGACG CACGTGCTGG CTACT CGACGCAGATACT ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA READS Is it a variant? What is the most likely genotype? SOAP2, samtools, GATK, Beagle, CRISP, Dindel, FreeBayes, SeqEM, VarScan, Mutect VCF files

34 Amplicon, quite noisy Variant calling

35 BODY HEADER VCF format Variant call format
36 Variant annotation CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF TCGATCGACG CACGTGCTGG CTACT CGACGCAGATACT ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA In which gene is it located? Name, Description, OMIM, Pathway, GO, Expression profiles... Is there any AA change? GAA -> GAG = E->E GTT -> CTT = V->L TGG -> TGA = W->X TGA -> CGA = X->R Where in the gene is it located? Intron, exon, UTR, intergenic region, splice site What impact does the AA change have? Damaging, benign Is it a known SNP? CSV files READS Annovar, SIFT, PP2, dbsnp, GO, KEGG, OMIM 1000G
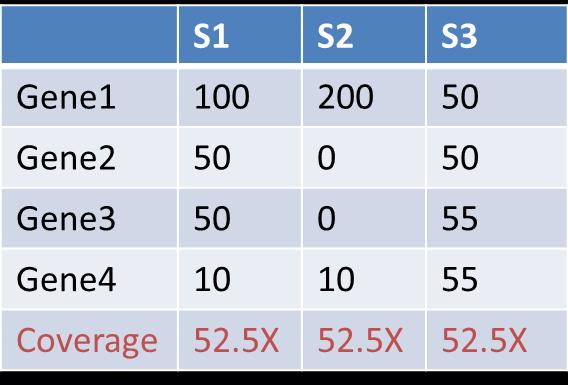
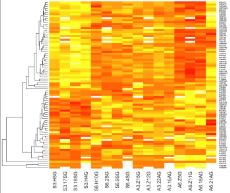
37 Coverage
")
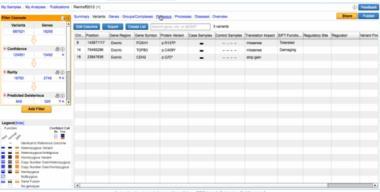
38 Variant Analysis Ingenutity Variant Analysis (IVA)


39 Identification of disease causing mutation

40 Alpers syndrome: progressive neurodegenerative dissorder POLG1 Alpers Huttenlocher FARS2 encoding enzyme to charge mt trna with Phe 19 patients: 6 had POLG1 mutations For this study:
 and NARS2")
41 Exome sequencing Mutations in PARS2 (Pro) and NARS2 (Asn)

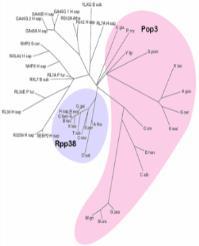
42 NARS NARS2 P. horokoshii monomer-monomer interaction

43 PARS PARS2