|
|
|
- Kristin Francis
- 5 years ago
- Views:
Transcription
1
2
3
4
5
6
7
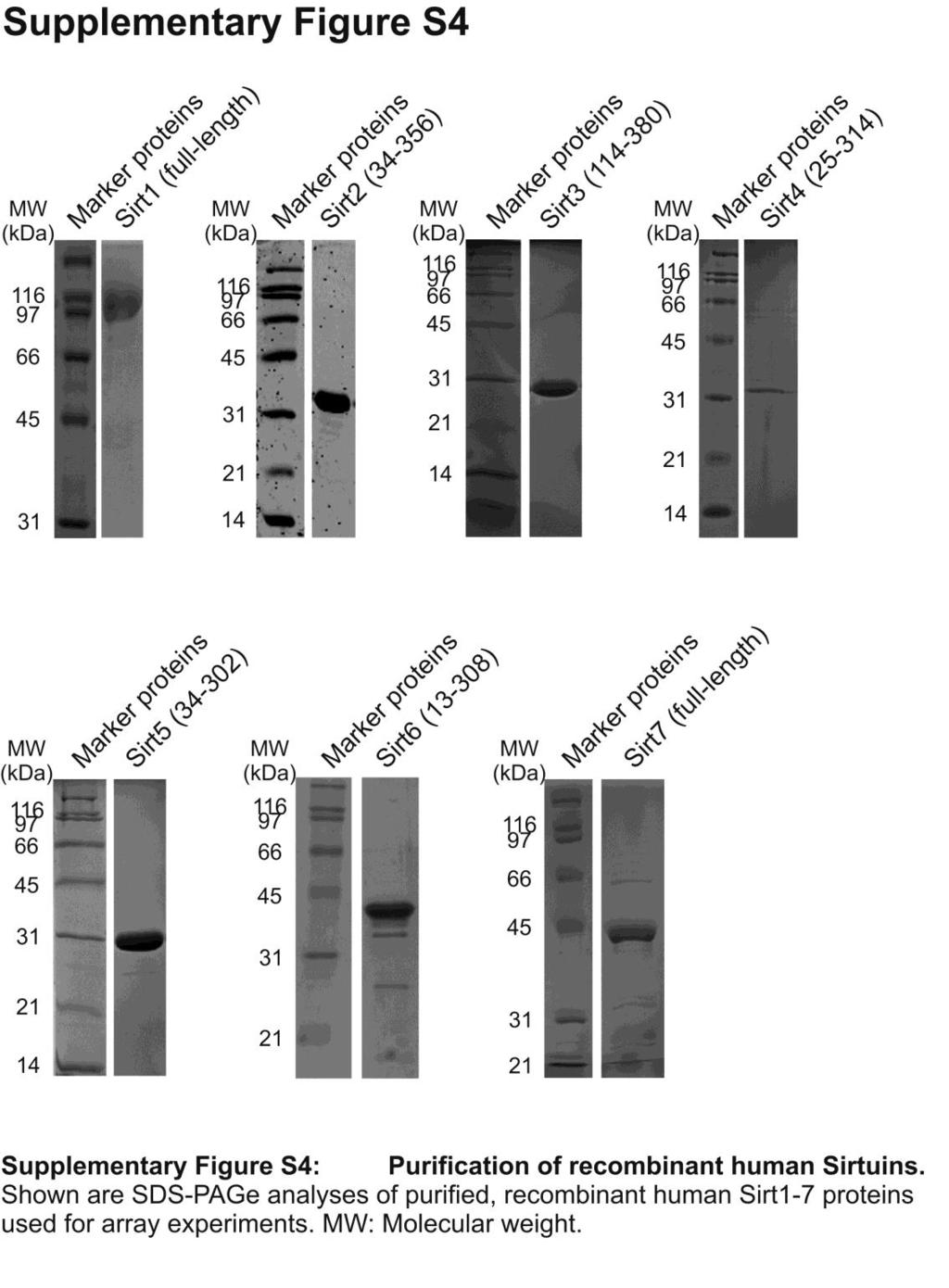
8 SUPPLEMENTARY METHODS Peptides HPLC purified peptides from GL Biochem (Shanghai, China): mthsp75-k332, RYTLHY(acetyl-K)TDAPLN; PDH-K81, KADQLY(acetyl-K)QKFIRG; NADH-DH-K243, IENAYK(acetyl-K)TFLPE; ANPtransloc2-K268, ARDEGG(acetyl-K)AFFKGA; AATase- K159, VFLP(acetyl-K)PTWG; HMGB-K12, KKPRG(acetyl-K)MSSY; TFIID3-K625, DREKGK(acetyl-K)DKDKRE; p53-k382, STSRHK(acetyl-K)LMFKTE; Nnt-K331, APVLFN(acetyl-K)EMIESM; CPS1-K527, FKRGVL(acetyl-K)EYGVKV. C-terminal Prx1 peptides CKQK and C(acetylK)QK (residues 196 to 199 with S196C substitution) were prepared using Fmoc-based solid phase synthesis and O-benzotriazole- N,N,N,N-tetramethyluroniumhexafluorophosphate (HBTU) activation. Fmoc-protected amino acids and HBTU were from Novabiochem (Hohenbrunn, Germany), and Fmoc-Lys(Boc)- Wang resin from Iris Biotech (Marktredwitz, Germany). Peptides were deprotected and cleaved from resin using 92.5% TFA, 5% TIS, and 2.5% water. Peptides were HPLC purified, analyzed by ESI-MS on an LCQ Advantage (Thermo Scientific, Dreieich, Germany), and pooled accordingly. The masses obtained for C(acetylK)QK and CKQK were Da and Da, in good agreement with calculated masses (548.7 Da and Da, respectively). Expression and purification of human Sirtuins Human Sirt1 residues and , Sirt , Sirt and , Sirt , and Sirt and were expressed in E. coli with N-terminal His-tag by using available constructs, and the proteins were purified through affinity and size exclusion chromatography 29, 44, 45. Sirt and Sirt , Sirt and Sirt were cloned into pet15b (Sirt4; Merck Biosciences, Darmstadt, Germany) and popin S (Sirt7; OPPF, University of Oxford, UK), respectively, resulting in constructs with N-terminal His-
9 tag (Sirt4) or N-terminal His- and His-SUMO-tag (Sirt7). Sirt4 was produced and purified similar to Sirt1. Shortly, cells were lysed in 25 mm Hepes, ph 7.0, 150 mm NaCl, and 10 mm imidazole, and protein bound to Talon resin (Clontech, Mountain View, USA) and eluted with the same buffer with imidazol increased to 200 mm. The protein was run on a Superdex200 size exclusion column (GE Healthcare, Waukesha, USA) in 20 mm Hepes ph 7.0, 150 mm NaCl and concentrated in amicon units (Millipore, Billerica, USA). Sirt7 fulllength was expressed in BL21-CodonPlus-(RIL) cells using auto-induction at 25 C (Overnight Express Instant TB media, Novagen). After lysis in 50 mm Hepes ph 8.0, 300 mm NaCl, 1 mm TCEP, 20 mm imidazole, cleared lysate was run over a HisTrap FF crude column (GE Healthcare). After washing with the same buffer containing 30 mm imidazole, protein was digested on-column with SUMO protease and eluted with 5 column volumes lysis buffer. The protein was subjected to size-exclusion chromatography on a Superdex75 column (GE Healthcare) in 25 mm Hepes ph 7.5, 40 mm NaCl, 2% (v/v) glycerol and 1 mm TCEP. Sirt was produced as full-length Sirt7, except that protein was eluted during affinity chromatography with 5 column volumes 300 mm imidazole instead of using a proteolytic digest. Acetyl-HMG-B1 and specifically acetylated Prx1 preparation Prx1 residues were cloned with N-terminal 6x His-tag into ptxb3 (New England Biolabs, Ipswich, USA) resulting in a C-terminal Mxe intein/chitin binding domain (CBD) fusion. Prx1-intein fusion was expressed in E. coli BL21(DE3)Rosetta2. Cells were grown at 37 C to an OD 600 of 0.6, expression induced with 0.5 mm isopropyl- -Dthiogalactopyranosid, and culturing continued at 20 C overnight. Harvested cells were resuspended in buffer A (20 mm Tris/HCl ph 7.8, 150 mm NaCl) plus 0.3 mm phenylmethyl sulfonylfluorid and protease inhibitor mix (Serva, Heidelberg, Germany) and disrupted using a french press. Lysate was cleared by centrifugation, supplemented with 10 mm imidazole
10 and subjected to affinity chromatography using Talon resin. After washing beads with 10 volumes 20 mm Tris/HCl ph 7.8, 500 mm NaCl and 10 volumes buffer A plus 20 mm imidazole, Prx1-intein fusion was eluted in buffer A plus 150 mm imidazole and the buffer was exchanged against buffer A using a Nap25 column (GE Healthcare). Prx1-intein fusion (20 µm) was cleaved in Prx1-buffer (20 mm Tris-HCl, ph7.8, 150 mm NaCl) by adding 300 mm sodium 2-mercaptoethanesulfonate (MESNa) 53 overnight at 4 C. The protein was incubated with chitin beads for 2 h at 4 C. Beads were washed (3x) with Prx1-buffer to obtain protein-thioester in the supernatant (yield of 75%). The Prx1-thioester was verified by MALDI-MS analysis (calculated mass: 22,749.0 Da; observed mass: 22,729.1 Da; A mass difference of approximately -20 Da was found for all MALDI-MS measurements of Prx1 variants and is within the error margin of the MALDI-MS system). Ligation reactions were performed in 3 ml Prx1-buffer containing 300 mm MESNa at ph , with 50 μm protein-thioester and 1 mm CKQK and C(acetylK)QK, respectively (Prx1 C-terminus with S196C substitution). After 72 hours at 4 C, ligations were complete according to MS analysis. Excess peptide was removed by dialysis against Prx1-buffer and ligation products were isolated with an average total yield of 60%. The products Prx1-CKQK and Prx1- C(acetylK)QK were verified by MALDI-MS (Prx1-CKQK: calculated mass: 23,113.4 Da; observed mass: 23,092.0 Da; Prx1-C(acetylK)QK: calculated mass: 23,155.5 Da; observed mass: 23,137.7 Da) and analyzed by blue native PAGE. DNA encoding human HMG-B1 amino acids 1-84 was cloned in a modified pet15b resulting in an N-terminal His- and C-terminal Strep2-tag, and expressed in E. coli BL21(DE3). The protein was purified through NiNTA affinity chromatography, SP sepharose ion exchange chromatography, and gel filtration on a Superdex-75 column (GE Healthcare) in 20 mm HEPES, ph7.5, 150 mm NaCl, 10 % (v/v) glycerol, and 1 mm DTT. HMG-B was chemically acetylated with acetic anhydride according to 61.
11 SUPPLEMENTARY REFERENCE 61. Means GE, Feeney RE. Chemical modification of proteins. Holden-Day: San Francisco, 1971.