Approaches to gene manipulation
|
|
|
- Malcolm Morton
- 5 years ago
- Views:
Transcription
1 Experimental Genetics 1
2 Approaches to gene manipulation 1) Gain of Function mutation (hyperactive, overexpression): In cells in culture, the usual way to determine what a gene does is to overexpress the gene and protein and see what happens. Another way is to alter the gene such that the protein is consitutively expressed in cells. This means that it is ALWAYS expressed, which is NOT NORMAL for most proteins. Usually gene transcription (leading to protein production) is carefully regulated so the protein is active only at certain times (i.e. like only during cell division)
3 Gain of Function: Constitutively Active One way to gain function is to make a gene constitutively active (means the gene is ALWAYS on)! Most genes are regulated; they are not always being transcribed (which means protein is not always made). 1) A deletion mutation can make a gene constitutively active (CA): Ex: A protein has a part, A, that, by folding, inactivates the protein (i.e. by hiding the domain that is an effecter = makes the protein active). This is autoinhibition. Now, if the gene is mutated, and no part A is made (you make a truncated (shortened) protein), then the effecter domain is always exposed and always makes protein. 2) A point mutation can make a gene CA: One common regulatory scheme is that a protein needs to be phosphorylated to make it active, and this works because the PO4 group is negative. Phophorylation occurs primarily on three aas: tyrosine, serine, threonine. These are usually infrequent amino acids, occurring only where they will affect regulation of the protein. So if you make a point mutation and change a tyrosine to a glutamate, which has a negative group that mimics the phosphorylated group. Then this protein will be active all the time. It will not need to be phosphorylated now to be active. And dephosphorylating enzymes cannot inactivate it.
4
5
6
7 Loss of function strategies 1) Get rid of the whole gene at the DNA level, 2) degrade the mrna 3) inactivate i the protein. OR: make a dominant negative mutant
8
9 Loss of function strategies 4) Make a dominant negative mutation. This is a numbers game. You swamp the cell with bait that binds but doesn t work: You engineer a gene that makes a protein that a) binds to the correct partner protein, but b) is not able to perform the function (i.e. has no enzyme action) (This assumes that function requires A binding to B and then A activating protein B) B.) Now you get the cell/animal to make LOTS of this mutated protein. Soon, the cell is packed with the bad protein. This binds to all the partner proteins, but then the pair are NOT ACTIVE! So no function occurs. The few numbers of normal protein A are now unable to find partners to activate.
10 Gene Cloning
11
12 Gene cloning: Isolate the gene of interest: MAKE A cdna Library is the first step! 1) Collect all mrna from the tissue or cells of interest. This will be a snapshot of what proteins are being made at that one time. So if you want to know what genes are involved in how the uterus functions during late pregnancy, you would wait until late pregnancy and isolate the uterus tissue. You grind it up and use fancy techniques to get just RNA. THEN, to isolate mrna, you make use of the fact that mrna, but not other types of RNA, have polya tails. Use cellulose beads complexed with poly dt in a column. The poly A tails will hybridize (anneal) with the poly dt and stay in the column. Rinse and then elute your mrna out with high salt.
13
14
15 Gene cloning: Isolate the gene of interest: 2) Reverse transcribe the mrna into cdna: Add oligo dt RNA primers to start the reaction. Use reverse transcriptase to make DNA that is complementary to the mrna. This single stranded DNAwill be hybridized (annealed) to the single stranded mrna.
16
17 Gene cloning: Isolate the gene of interest: 3) Use a restriction endonuclease to cut the mrna into fragments and remove it, leaving single stranded cdna. (RNase chews up nicked RNA) 4) Make double stranded cdna (ds cdna) by adding DNA polymerase, DNA ligase and primers. The two strands will hybridize (anneal). THIS ds cdna is a DNA library, containing i DNA encoding for a lot of different genes, from the tissue and time of interest.
18
19 Gene cloning: Isolate the gene of interest: 5) Go to the library and pull out the cdna encoding your particular gene of interest. Do this by PCR. (PCR is a method to highly amplify a gene or part of the gene you are interested in). The trick is in finding the suitable primers for the DNA. Designing primers is one of the hardest areas of cloning, requiring the most innovation.
20
21 Gene cloning: Make lots of your gene with plasmids 6) Now, you need LOTS of the cdna: So you will propogate it (grow it up) in E coli bacteria. Do this by making use of bacterial PLASMIDS. = Plasmid based propagation.
22
23 Gene cloning: Make lots of your gene with Plasmid based propagation A. Get a plasmid: now they have commercially available plasmids that basically have all of the cloning sites (sites for restriction enzymes) in a a multiple cloning site = polylinker region. This plasmid is called a cloning vector. These plasmids all have an ORI (origin of replication) and an antibiotic resistance gene.
24
25

26
27 Gene cloning: Plasmid based propagation B. Insert your cdna of interest into the MCS site of bacteria. Cut the plasmid with RESTRICTION ENZYMES. Add your PCR made cdna (which was engineered tobe cut by the same restriction enzymes) to the cut plasmid. Use DNA LIGASE to join all the cut ends together, resulting in a plasmid containing your cdna.
28
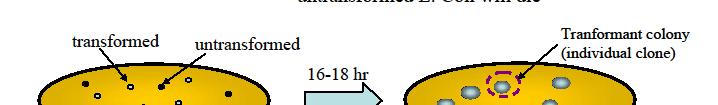
29 Gene cloning: Plasmid based propagation C. Take your new cloned gene and transform it into E Coli using heat shock and CaCl2 (open cell wall and membrane). This is where A miracle occurs. Not all bacteria take in plasmids. So you have to figure out what htyou have. Plate E Coli in antibiotic medium. E. Coli WITHOUT PLASMIDwill diebecausetheydonothavetheplasmid. do have Wait until colonies of bacteria grow up that are resistant to the antibiotic. Cut out the small colony and grow those bacteria up in a big flask. Then extract the cdna using restriction enzymes. YOU will have lots of specific cdna.



30

31
32 Gene cloning: Now have the gene cloned: Now what do you do with it? Alter it! Now, we have the DNA in a vector that we can work with (the plasmid) and we have made lots of it, specific for our DNA/RNA/protien. This means that we have cloned our gene. Now we want to do gene manipulation: i.e. gene overexpression or mutations.


33
34 Gene Manipulation This is going to be either: Gain of function OR Loss of functionof function
35 Gene Manipulation: Over expression Gi Gain of function: Overexpression in eukaryotic cells in culture. We want to have eukaryotic cells express lots of our protein ti and see what htthat t does to the cell. How do you express a protein?
36 Gene Manipulation: Over expression Overexpression/Expression: 1) Must ttransfer your plasmid idf from a cloning l vector to an expression vector plasmid BECAUSE bacteria do not have the same promoters (genes) and regulatory proteins as eukaryotic cells. The Expression vector (EV) differs from the cloning vector in that the EV has a strong promoter that will allow expression of the gene (transcription) when the gene is in eukaryotic cells.
37
38 Gene Manipulation: Over expression We are going to get mammalian cells to have our gene and then express our protein and in fact, OVERexpress our protein. How? 1) First: Must transfer the plasmid (with cloned gene) from the cloning vector (plasmid) to an expression vector plasmid BECAUSE bacteria do not have the promoters (genes) to regulate expression of eukaryotic genes. ALSO: have to put this in eukaryotic cells (not bacteria) because ONLY eukaryotic cells have the cellular machinery to fold the protein and finish it off
39 Gene Manipulation: Over expression Transfer your gene from cloning vector to expression vector: Steps: 1) Use PCR to make LOTS of the cdna! PCR needs primers. ***SOME DNA polymerases and primers used in PCR will copy the ENTIRE plasmid. Those are the ones used here. So get lots of copies of the entire cloning vector with ihpcr. 2) Now, cut out the gene from cloning vector with restriction titi enzymes
40 Gene Manipulation: Over expression Now: Put your clone in the expression vector (plasmid) with restriction enzymes to remove the plasmid DNA, and DNA ligase to join in the new DNA. NOW: TRANSFECTION: You must put this expression vector in EUKARYOTIC cells. Can t use heat shock and CaCl2 as you did before with bacteria, because this will kill eukaryotic cells! Must use milder means.
41 Gene Manipulation: Over expression Transfection: Putting a DNA plasmid in cells Many Methods: 1) put your plasmid in a liposome (combine lipids and plasmid together under certain conditions). The liposomes naturally merge with and become part of the cell membrane, releasing the contents into the cell (like fusion of soap bubbles): Pros: very mild. 2) electroporation: ti electrical lpulses open the membrane and force in the slightly charged plasmid. 3) microinjection: use fine tipped pipet pp and poke cell and squirt stuff in. [4) Extra: Gene gun: modified 22 caliper gun actually blows gold coated with your DNA into cells. Used mostly for plants.]
42
43 Transfection: Overexpression Transient Transfection: If you put plasmid DNA in a eukaryotic cell, it will last for ONLY up to 1 2 weeks before being degraded. Stable Transfection: A miracle occurs In a small percentage of the cells, the plasmid somehow gets incorporated into the chromosomal DNA (don t know how). This means that fromthat point on, with each division, the genes in the plasmid will be then copied faithfully into daughter cells and forever carried on.
44
45 Overexpression: Stable Transfection Stable Transfection: You just plate out your cells in antibiotic containing medium and wait. After 2 4 weeks, you hopefully will have some surviving colonies of cells. Each colony is a clone of a stably transfected cell, which is hopefully expressing your gene (making the protein) and also passing the gene on with each division. [You then rejoice and sell your cell line to any company that will take it!]
46 Over expression [Note: Bothtransienttransient and stable transfections are used FREQUENTLY in molecular biology to see what a gene or a mutated gene does in its natural environment, the cell. However, sometimes overexpression of a protein can add complications because it can kill the cell!]
47 Gene Mutations (point or deletion) Here is where the real cleverness comes in doing gene work. There is a lot of skill and knowledge needed to figure out how to make a specific mutation appropriately. Then, there is skill needed also in interpreting the results once you put the mutated gene back into either cells or the animal.
48 Gene Mutations (point or deletion) A really good, proficient molecular biologist will sit down with their gene and figure out the appropriate cuts and primers to use to make the desired mutation. A less proficient person will go to a website from a company! See next slide!
49 DNA work TO make the primers that you need for either a point mutation, deletion or insertion: Go to Stratagene Website: QuikChange Then to the Help button: htm Example: Load sequence by pasting text or typing in the provided text area or by locating a file containing the sequence using the Browse button. Notes: 1) All non "a", "c", "g", or "t" characters in the sequence will be ignored. Line breaks within the sequence are also removed during uploading. 2) The sequence can be either in FASTA format or plain ASCII text. Press the Upload Now button. The program will display a series of checkboxes, each corresponding to a single "sense" nucleotide from the DNA sequence. Check the Delete a nucleotide or a region radio button above the checkboxes. Using the checkboxes, select two nucleotides immediately flanking the deleted region as shown below: Sequence:...acgtgctagctgcacgtacgtagctacgtagcccgatcgtagc... Region to delete cacgta Check these bases:...acgtgctagctgcacgtacgtagctacgtagcccgatcgtagc... Note 1) The two checked nucleotides will not be deleted 2) If you are deleting a single nucleotide you must check the two flanking bases. Press Design Primers button. The program will open two tables with all primer characteristics, sequences, and primer template duplex schemes.
50 Gene Mutations (point or deletion) Why do we do this? The heart of a large segment of Mol. Biol. research is to determine which parts of the gene (and protein) are responsible for function of the protein or regulation of the gene (why, when and how is the gene turned on). To do that we need to alter one specific part of the gene or its regulatory DNA and then see what happens. Can t predict without doingthis!
51 Point mutations Dr. Roy outlined one way to make a point mutation. That is by having two overlapping primers. Then you change one base pair (in each primer). Then, when the primers direct transcription, BECAUSE they are ovelapping, they encode for amplifying the ENTIRE plasmid. In doing this, they direct the new DNA to have the change in the base pair.
52 Point mutations NOTE: IN the PCR outlined by Dr. Roy, you use two overlapping primers and a DNA polymerase that transcribes the ENTIRE loop of plasmid DNA. Because the primers overlap, you end up copying the entire plasmid (and if you use the appropriate polymerase). So you end up with two new copies of the plasmid DNA, with the new base pairs in them. The parent plasmid has methylated bases, but the PCR created DNA does NOT (you are in a test tube not a bacteria!). SO newly made DNA is NOT METHYLATED! Use restriction enzymes that cut DNA ONLY at methylated bases (highly unusual restriction enzyme, mostwill NOT cut methylated bases) ONLY the parent DNA is methylated, so this results in destruction of the parent, methylated DNA. Then your PCR amplifies only the mutated DNA.
53
54
55 Point mutations Once you have your new plasmid with the change in the single base, you transfect that into eukryotic cells, overexpress it and see what the cells do. (You Ask: what does that protein do to the cells?)
56 Deletion Mutations To understand Dr. Roy s Deletion mutations you have to realize the following: In this example of PCR, you have primers that DO NOT OVERLAP. Therefore, during the amazing amplification of PCR (see movies or your class notes) ONLY the region BETWEEN the primers ends up being amplified and is your product at the end.
57 Deletion Mutations So: TO make a deletion mutation in which you want to copy only A and B exons out of a gene with A, B and C exons, you choose primers that start on one side of the DNA at A. On the otherside side, thesecond primer starts at theend of B. When you do PCR (review that in a previous lecture) you amplify (thousands of times!) just the region BETWEEN the two primers.
58
59 Deletion Mutations Once you have your PCR product, which is just the DNA encoding A and B exons, then you cut it out, put the DNAin another expression vector, grow up the bacteria and then extract LOTS of your deletion mutation DNA and use it to transfect into mammalian cells and see what that gene does when it makes protein.
60
61
62 Sample exam question Example exam question for extra credit on your grade. If you do a point mutation of your gene of interest, describe the primers, and the steps you need to do to study the final product in mammalian cells. Why you would do this?