Innovative treatments in neuromuscular disorders
|
|
|
- Cleopatra Harper
- 5 years ago
- Views:
Transcription


1 Innovative treatments in neuromuscular disorders Great Ormond Street Hospital for Children NHS Foundation Trust Practical Neurology Study Days 2018 Francesco Muntoni Dubowitz Neuromuscular Centre UCL Institute of Child Health & Great Ormond Street Hospital London, UK
2 Francesco Muntoni: disclosures Duchenne trials CI of 3 AON clinical trials with AVI / Sarepta. PI of three Prosensa / Biomarin sponsored AON trials PI of PTC phase II and III sponsored trials. PI of Pfizer phase II clinical trial (myostatin inhibition) CI of Summit Phase I and II trials. CI of Esperare Phase I trial Spinal Muscular Atrophy trials PI of Trophos SMA III trial PI of Ionis/ Biogen AON Phase III study Other Avexis, Biogen, Sarepta, Pfizer, PTC, Roche, Summit, Wave ad-hoc SAB
3 Dramatic impact of novel therapies for neuromuscular disorders Genetic therapies Dealing with mutant RNA: - nonsense mutations for Duchenne muscular dystrophy (DMD) - exon skipping for out-of-frame DMD deletions - exon inclusion in Spinal muscular atrophy (SMA) Pharmacological therapies - Utrophin upregulation (DMD) - Increasing muscle mass (DMD - Dissociative steroids (DMD) - Pharmacological splicing modulation (SMA) Replacing genes with AAV gene therapy - AAV gene therapy for SMA type I - AAV gene therapy for Myotubular Myopathy - AAV gene therapy for DMD
4 Small molecules and antisense oligonucleotides to modify splicing or processing of mutant RNA Duchenne muscular dystrophy ~ 10-15% nonsense mutations in DMD exons ~ 65% out-of-frame DMD exonic deletions translarna eteplirsen (15%) golodirsen (8%) other AONs





5 Duchenne MD X linked recessive disorder 1 in 5,000 male births De novo mutations hamper genetic efforts to reduce incidence NH 2 CYS COOH Full length NH 2 CYS COOH Dp260 NH 2 CYS COOH Dp140 NH 2 CYS COOH Dp116 NH 2 CYS COOH Dp71
6 Nonsense Mutation Suppression Translarna (~13% all DMD) Nonsense (Premature Stop) Codon Normal Stop Codon Incomplete protein Nonsense suppression aims to induce full-length protein production Nonsense (Premature Stop) Codon Normal Stop Codon YIELD mrna Full-length protein
7 Mean change in 6MWD from baseline, m Phase III study : primary efficacy endpoint over 48 weeks (6MWT: ITT population) 15 m p =

8
9 Mean change in 6MWD from baseline, m Phase III study: primary efficacy endpoint over 48 weeks (6MWT: pre-specified 300 to < 400 m baseline 6MWD subgroup) 47 m p =
10 Treatment effects across primary and secondary endpoints Decline Metres, mean (95% CI) M e te r s, m e a n a n d 9 5 % C I Improvement MWT Meta-analysis (N=291) ACT DMD ITT population (N=228) Phase 2b ADP subgroup (N=63)* 10 m walk/run 4-stair ascent 4-stair descent S e c o n d s, m e a n a n d 9 5 % C I 10
11 Pulmonary function in Translarna -treated patients compared to an untreated natural history cohort CINRG cohort: Log FVC vs age Extension study 019: Log FVC vs age Predicted results show slope change at 12.5 years Predicted results show slope change at 16.5 years Patients preserved lung function by 13.8%** (p = 0.005) compared with those in natural history cohorts 11
12 Antisense oligonucleotides: DMD gene reading frame 65% of DMD boys have out-of-frame deletions Antisense oligonucleotides to induce exon skipping 52

13 Exon-Skipping Approach ~ Normal Dystrophin mrna ~ Normal Dystrophin Protein ~ Exon Deletion(s) Disrupts Reading Frame Ribosome ~ Unstable/No Dystrophin Protein: DMD Skipping Adjacent Exon Restores Reading Frame ~ AON ~ Intended to Produce Shortened Dystrophin Protein


14 Becker MD: in frame DMD gene deletions In frame deletion

:")
 eteplirsen Ann Neurol.")
15 AON exon 51 skipping therapy progress Clinical Trials Omethyl AON (Prosensa/GSK /Biomarin): drisapersen Morpholino PMO AON (Sarepta) eteplirsen Ann Neurol. 2013;74:

:618-623. Pane et al. PLoS One.")
16 Eteplirsen Compared to 6MWT natural history Morpholino (PMO) AON MEAN 6MWT SCORE CHANGE FROM BASELINE McDonald CM, et al. Muscle Nerve. 2010;42(6): Mazzone ES, et al. PLoS One. 2013;8(1):e Goemans N, al. Neuromuscul Disord. 2013;23(8): Pane et al. PLoS One. 2014; 9(10): e

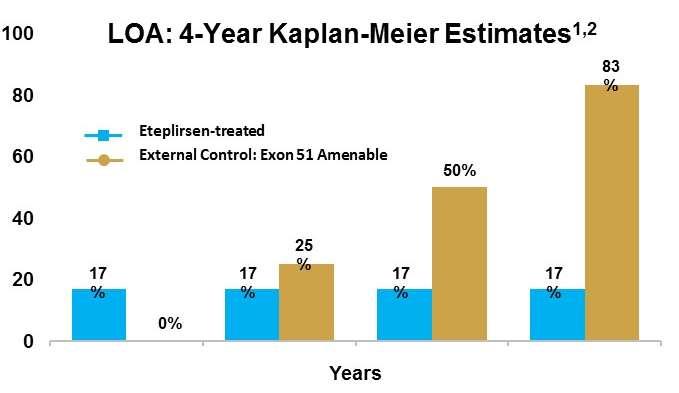
17


18 Eteplirsen 30 mg/kg is FDA Approved in the US for the Treatment of Patients with Exon 51 Skippable DMD Treated patients decrease 2.3% annually for FVC% predicted Natural history data demonstrated a 4.1% decline in FVC% predicted.

19 PMO for Duchenne muscular dystrophy
20 SKIP-NMD Consortium INTERNATIONAL COLLABORATION TO IDENTIFY & DEVELOP THERAPY FOR EXON 53 AMENABLE DMD SKIP-NMD consortium: EU FP7 Grant ( 5,512,424) Objectives Design a PMO that skips exon 53 and efficiently induces dystrophin production Assess efficacy (in vitro) Assess safety (in vivo) Perform a clinical trial in eligible DMD boys Develop outcome measures and non-invasive biomarkers to monitor efficacy Consultants for Research in Imaging & Spectroscopy 20
21 Next generation AON for DMD Wave stereopure PPMOs

22 PPMO

 - exon skipping for out-of-frame DMD deletions - exon inclusion in Spinal muscular atrophy")
23 Dramatic impact of new technologies for neuromuscular disorders Replacing genes with AAV gene therapy - AAV gene therapy for SMA type I - AAV gene therapy for Giant Axonal Neuropathy - AAV gene therapy for DMD - AAV gene therapy for Myotubular Myopathy Dealing with mutant RNA: - nonsense mutations for Duchenne muscular dystrophy (DMD) - exon skipping for out-of-frame DMD deletions - exon inclusion in Spinal muscular atrophy (SMA)



24 DMD AAV gene therapy programs DMD Single IV administration Highly internally deleted dystrophins (AAV capacity 4.7Kb max)

25 Different therapeutic strategies in Clinical Trials for DMD DMD Substituting/co rrecting primary protein defect Limiting the progression of dystrophic process Exon skipping Nonsense suppression Utrophin upregulation Gene therapy Stem cell therapy Eteplirsen Ataluren Prevent muscle necrosis and fibrosis Antiinflammatories Idebenone VBP15 Vamorolone Rimeporide Increase muscle regeneration Growth factors IGF Myostatin inhibition
 X-linked Myotubular Myopathy ASPIRO Phase 1/2 Clinical Study Planned doses for clinical study: 1.0, 3.0 and 5.")
26 Other AAV gene therapy programs Giant Axonal Neuropathy scaav9-jet-gan 3 different doses 3.5 x vg/ 10.5 ml (N=2) 1.2 x vg/ 10.5 ml (N=2) 1.8 x vg/ 10.5 ml (N=2-4) X-linked Myotubular Myopathy ASPIRO Phase 1/2 Clinical Study Planned doses for clinical study: 1.0, 3.0 and 5.0 x vg/kg Orphan Drug Designation received from FDA and EMA IND active
27 Other AAV gene therapy programs ASPIRO Phase 1/2 Clinical Study X-linked Myotubular Myopathy Planned doses for clinical study: 1.0, 3.0 and 5.0 x vg/kg Orphan Drug Designation received from FDA and EMA IND active
28 Improvement in respiratory function
29 Genetic therapies for other neuromuscular disorders
30 Concluding remarks Outstanding progress in the therapeutic approaches for neuromuscular disorders in the last decade Second generation therapies now entering the clinic (i.e. AON for DMD), and novel AON capable of crossing the blood brain barrier in advanced preclinical development Chronic phenotypes will emerge, and in the following years we will need to characterise and manage these novel phenotypes
31 Concluding remarks (2) While success and tolerability of these approaches encourages their continuing development, both their long term safety, and cost/ benefit will require careful analysis The cost of some of these intervention is very considerable, and this might preclude their access to our patients Timely diagnose children with these disorders, and be competent in complex novel therapy management While cautious and measured counselling to families is imperative, there are reasons to change from pessimism to optimism for these patients, although there is a lot more work to do




32 Acknowledgment Clinical and research team at GOSH/ ICH Carsten Bonnemann, NIH Matthew Wood, Oxford George Dickson, RHUL