Molecular Docking. Chao-Sheng Cheng. Department of Life Science, National Tsing Hua University
|
|
|
- Charles Randall
- 5 years ago
- Views:
Transcription

1 Molecular Docking Chao-Sheng Cheng Department of Life Science, National Tsing Hua University


2 Computational ligand design Target Structure unknow known Ligand-based approaches (Pharmacophore + QSAR) Structure-based approaches (Docking; De novo design) 2
3 Computational ligand design Two different strategies: Ligand-based (analog-based) design Relies on a set of known ligands and is particularly valuable if no structural information about the receptor is available. Structure-based (target-based) design Usually starts with the structure of a receptor site, such as the active site in a protein. This structure can be generated from direct experimentation or can be deduced from experimental structures through homology modeling 3
4 Structure-based (target-based) design (Krumrine et al., 2003) 4

5 Definition of Docking Docking is an energy-based operation for exploring the binding modes of two interaction molecules. Give the 3D structure of a protein target, compuonds can be designed to fit in a cavity, which is called "docking". The treatment ends when a minimum of energy is obtained for the complex. 5
6 Docking Goal 1. To build the binding model between ligand and protein 2. To clarify the critical residues which involve in ligand binding 3. To engineer the protein by mutagenesis 4. To develop a drug from a compound database 6
7 Before Docking 此類方法首先先要確認目標位置 ( target site) 在廣大的蛋白質表面中, 要找到 bind site 蛋白質形狀 ( shape) 和表面特性, 如 hydrophobic sites (H), hydrogen bond donors (D) and acceptors (A) 與藥物分子特性 嵌合時支鏈是否可以旋轉 (Flexible) 等問題 7
8 Before Docking 基本的問題 : Where: 在這麼大的蛋白質中, 配體如何能找到一個適合它的位置 What: 當 docking 完後所得到的複合體結構, 它的構形 (conformation) 是什麼? How: 要如何評估結果的好壞?? 8
9 Docking Require Ligand structure Protein structure Docking software Ligand: XK-263 Protein: HIV-1 Protease (1hvr) Software: AutoDock version 2.4 9
10 Docking Methods Some common searching algorithms include Molecular dynamics Monte Carlo methods [AutoDock, ProDock, MCDOCK...] Genetic algorithms [GOLD, AutoDock, DARWIN...] Fragment-based methods [FlexX and DOCK] Point complementary methods [FTDOCK, FLOG...] Distance geometry methods [DockIt] Tabu searches [PRO LEADS] Systematic searches 10
11 Force Field Models Force fields are usually employed to generate accurate predictions to complex problems by interpolating and extrapolating from relatively simple experimental set of molecules. Classical force field models AMBER, CHARMM and CVFF. Second generation force field models CFF and COMPASS. Generalized force field models ESFF and UFF. 11
12 Docking Flowchart Input Limitation Ligand structure Protein structure Docking Output Autodock Dock Ludi Gramm Complex structure information Analysis ProSall, SWISS PDB Viewer Evaluation 12
 也必須")
13 Docking Flowchart Input Docking Output Limitation For Input Ligand Data: structure Protein structure Protein must be high resolution! 大部分的溶劑分子必須移除, 除非該溶劑和蛋白質作用機制有很大關係 Autodock Dock 若原子的 Ludi B factors 值很高, 也必須將其移除 Gramm 對於 NMR 的結構, 若在較少限制的區域 (Poorly Complex constrained structure information regions) 也必須 Analysis ProSall, SWISS PDB Viewer 移除 Evaluation 13
14 Docking Flowchart Input Docking Output Limitation For Output Ligand Data: structure Protein structure 嵌合計算完成後, 首先要知道 藥物或化學小分子所結合的活化區位置 (active Autodock sites) Dock 結合的幾何圖形 Ludi (Geometry), 幾何上兩個配對的結構 Gramm 結合的能量 勢能 (potential energy) Complex structure information Analysis ProSall, SWISS PDB Viewer Evaluation 14
 或其它方法如 QSAR (Quantitative Structure- Activity Relationship)")
15 Evaluation 利用實驗方式 先分別得到蛋白質和配體 (ligand) 的樣品, 再利用儀器進一步測試生物活性 (bioassay) 結合強度或親和力(affinity) 等 藉由電腦程式分析 主要有幾種方式 :scoring function 蛋白質和配體之間幾何分配和表面特性 原子和原子之間的作用情形 Root mean square deviation (RMSD) 或其它方法如 QSAR (Quantitative Structure- Activity Relationship) 15

")


16 Analysis Complex form (1UVC) with one lipid Complex form (1UVB) with two lipids
 72.")

17 Total volume (A 3 ) Vander Waals (A 3 ) Cavity Probe-accessible (A 3 ) Free form Complex form with one lipid Complex form with two lipids

18 Superposition of structures cholesterol-nsltp2 (red) and nsltp2 (blue) MolScript 1. The major difference in the two conformations is observed at the loop between helices I and II, where the structure is stretched out approximately 3.10 Å. 2. Residues with larger chemical shift perturbations were also represented with yellow color. RMSD: 1.57 Å

19 DS ViewerPro Ligplot Leu 8, Ile 15, Phe 39, Tyr 45, Tyr 48, Val 49, Pro 52, Ala 54, Val 58, Leu 65 and Pro Various residues involved in the sterol binding are colored in green. Most of these residues are located around helices I, IV and V. 2. Comparing the theoretical docked structure and experimental NMR results, the residues directly interacting with the ligand are Leu 8, Ile 15, Phe 39, Tyr 45, Tyr 48, Val 49 and Val 58.

20 SWISS PDB Viewer 1. Distance/angle 2. Ribbon 3. Ramachandran plot 4. Mutation/rotamer 5. Superimpose 6. Compute H-bond/energy 7. Energy minimization 8. Cavity

21 Docking Software 21
22 Molegro

23
24 Molegro Virtual Docker - Overview Overview Molegro Virtual Docker is an integrated platform for predicting protein - ligand interactions. Molegro Virtual Docker handles all aspects of the docking process from preparation of the molecules to determination of the potential binding sites of the target protein, and prediction of the binding modes of the ligands. The Molegro Virtual Docker (MVD) has been shown to yield higher docking accuracy than other state-of-the-art docking products (MVD: 87%, Glide: 82%, Surflex: 75%, FlexX: 58%). Molegro Virtual Docker provides: High docking accuracy: the docking engine has been proven to correctly identify binding modes with high accuracy. Molegro Virtual Docker has been shown to outperform other docking programs with regard to identification of correct binding modes (see the technology pages for more information). Easy-to-use interface: the built-in wizards enable the user to easily setup and perform docking runs. Advanced visualization and analysis tools are provided to examine ligand-receptor interactions and fine-tune found docking solutions. Cross-platform: supported on Linux, Windows and Mac, allowing easy interoperability between platforms.

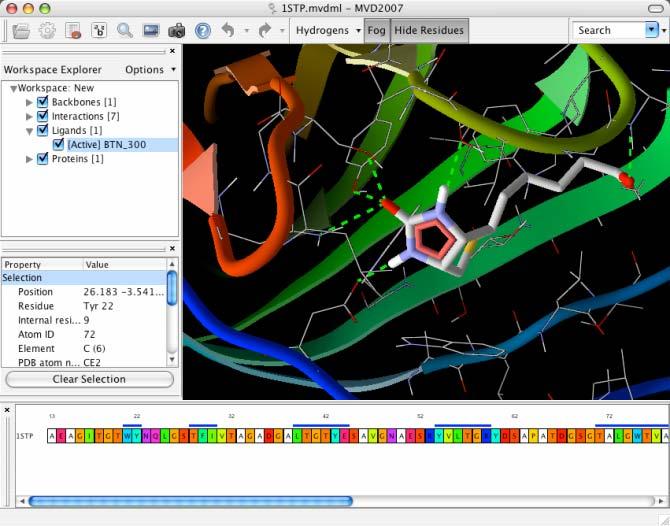


25 The main user interface
26 Basic Tutorials Tutorial 1: A Simple Docking Run Tutorial 2: Inspecting the Docking Results Tutorial 3: Visualization in MVD
27 Tutorial 1: A Simple Docking Run Import molecules into Molegro Virtual Docker (MVD). Detect potential binding sites and setup the search space. Run a docking simulation using the Docking Wizard. Inspect the docking results using the Pose Organizer.
28 Import molecules into MVD Require: 3D structure of a receptor 3D structure of one or more ligands Formats: PDB, Mol2, and SDF The exact 3D conformation of the ligands is not important - the torsional angles in the ligand will be determined during docking (but needs the ligand structure with proper bond lengths & bond angles)



29

30 In order for a docking simulation to succeed, various properties of the molecules must be assigned. How the atoms are charged The order and type of the bonds

31 This tab shows warnings from the molecule parsing. Careful to inspect the warnings Some warnings are harmless: Some proteins are cropped, causing MVD to warn about residues having an insufficient number of atoms 1HVR ok


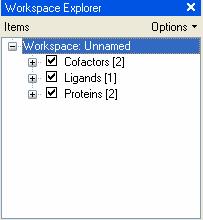
32 workspace 1. Turn on/off using checkbox

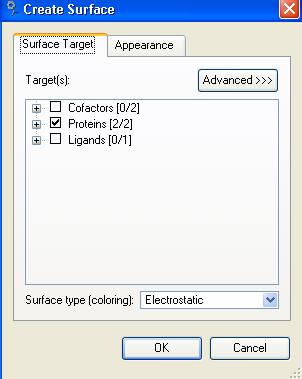

33 Create protein surface



34 Pay attention on our ligand Some bonds in the ligand are colored green flexible during the docking Red held fixed during the simulation (ring conformations are not changed during docking)





35



36 Detect potential binding sites and setup the search space 2 3 1


37 Run a docking simulation using the Docking Wizard Reference ligand- allow us to monitor the RMSD to the best found psoe while docking



38 Define scoring function & search space Search algorithm




39




40 Best poses for the current ligand Best found score for the current run




41 Tutorial 2: Inspecting the Docking Results


42 Show all & dynamic update Select here


43

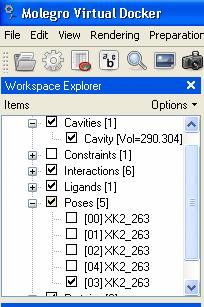



44 Right button of mouse
45 Tutorial 3: Visualization in MVD Navigating in the 3D view. Using Visualization Presets and Styles. Work with surfaces and backbone representations. Use Clipping Planes, Labels, and the Sequence Viewer.
46 Hydrogen bond interaction
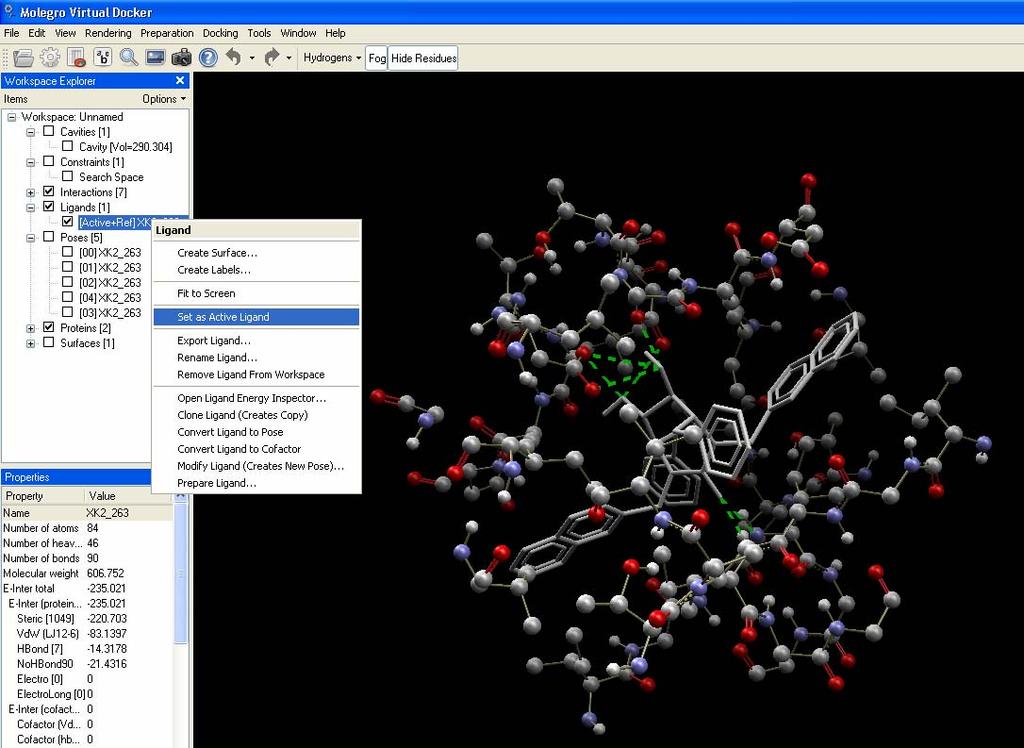
47 1 2
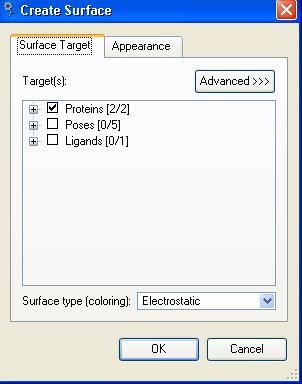
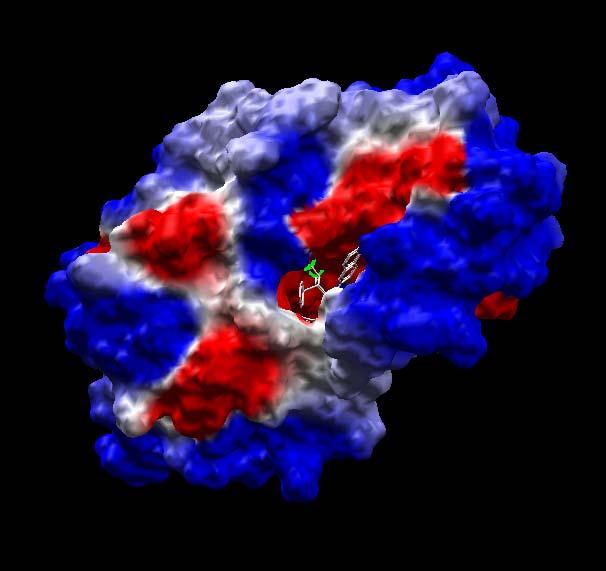
48 surface


49



50 Clipping plane
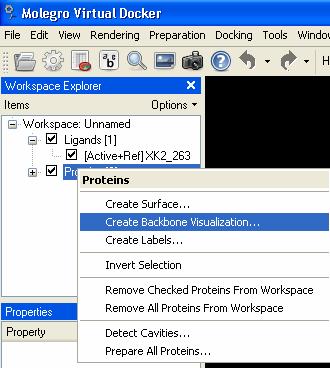




51 backbone



52 Show sequence Right mouse button to select a.a.


53 visualization