An introduction to RNA-seq. Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy
|
|
|
- Kristopher Carter
- 5 years ago
- Views:
Transcription

















1 An introduction to RNA-seq Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy
")

2 The central dogma Genome = all DNA in an organism (genotype) Transcriptome = all RNA (molecular phenotype?) Proteome = all protein (phenotype)

3 Why study the transcriptome? The transcriptome is the ACTIVE part of the genome Gene expression is dynamic, and varies over time, environment, pathology, tissue, etc. The transcriptome is a proxy for the proteome/phenotype Transcriptional changes precede phenotypical changes











4 The transcriptome is complex TSS TSS TSS Transcriptional complexity Note that DNA is double stranded, and RNA is single stranded, therefore RNA has direction (is stranded)! TSS tirna PASR mirna TASR genomic DNA micrornas spliced intron TSS transcription start site protein coding regions polyadenylation signal translation start site non-coding regions polyadenylation
5 Usually a blatant lie, should be cdna Abbreviation for sequencing RNA-seq Has it really been 10 years already? It depends on who you ask and how you define RNA-seq
6 Usually a blatant lie, should be cdna Abbreviation for sequencing RNA-seq Using ultra-deep cdna sequencing to measure and compare transcriptional complexity without requiring a priori knowledge of that complexity.
7 Structure of a Ribonucleic Acid Holley et al., Science 19 Mar 1965: 147(3664): Not Quantitative Oligo fragments reported as isolated in 1961, sequenced in 1964, and assembled (left) in 1965 First RNA sequencing (that I could find), but is it the first RNA-seq? Definitely the first de novo transcriptome assembly.














8 Digital Gene Expression TSS TSS TSS TSS Quantitative, but Does not capture structure 3 SAGE 5 SAGE MPSS di-tag/mate-pair
9 Shotgun Sequencing Captures Complexity TSS TSS TSS TSS
10 Gene Expression on the shoulders of giants First RNA sequenced in 1965 Reverse transcriptase discovered in 1970 Maxam-Gilbert and Sanger sequencing methods published in 1977 Expressed Sequence Tags (ESTs) Differential Display in 1992 Not (easily) quantitative cdna microarrays in 1995, oligonucleotide arrays in 1996 SAGE 1996, MPSS 2000, CAGE 2003 Does not capture complexity 454-based RNA sequencing in 2006 Did not explore DGE (price), not stranded Illumina/SOLiD-based RNA-seq in 2008 Illumina (not stranded), SOLiD (stranded) Single cell sequencing in 2009





11 I m getting my RNAseq data back today! How are you planning to do the analysis? Oh. Analysis. Right. If your plan is to hand the data to a bioinformatician to sort out the details, you are not doing your job. You can t analyse or interpret your data until you understand your data.
12 Things to consider when analysing RNAseq Is your data stranded or unstranded? Paired-end or single-end? Length?

















13 RNAseq protocol I LEGenD protocol Step 1: pre-process RNA Step 2: Adaptor Ligation NN RDV NN RDV NN RDV NN RDV RNA protocols can generate stranded short-tags AA NN RDV NN RDV Step 4: PCR amplification Step 3: 1 st Strand cdna RDV NN RDV RDV NN RDV RDV NN RDV RDV NN RDV RDV NN RDV RDV NN RDV
14 RNAseq protocol II Step 1: pre-process RNA Step 2: 1 st and 2 nd strand cdna NNNNNN RNAseq protocols can generate unstranded short-tags AA NNNNNN AA NNNNNN Step 4: PCR amplification Step 3: Adaptor Ligation RDV NNNNNN RDV RDV NNNNNN RDV RDV RDV NNNNNN RDV RDV NNNNNN NNNNNN RDV RDV NNNNNN RDV RDV
15 Why does strand matter? TSS TSS TSS TSS
16 Paired-end data vs single-end data F adapter R1 R2 Note that these are not independent measurements for gene counting use only RNA fragment R adapter fragments or one read only TSS TSS TSS
17 When Length Matters R1 R2 F adapter RNA fragment R adapter For counting, choose depth over length. For structure, choose length over depth.
18 Things to consider when analysing RNAseq Is your data stranded or unstranded? Paired-end or single-end? Length? For projects that involve alignment to a reference genome: Choosing the best aligner
19 Which is the best aligner? No such thing. Will not be discussing individual alignment algorithms, but a series of things to consider when trying to interpret results from an RNAseq (or an NGS!) experiment.


20 Different aligners give different results The patterns are largely the same so don t panic unless you re doing RNAseq Koehler et al Bioinformatics (2):
21 Things to consider when analysing RNAseq Is your data stranded or unstranded? Paired-end or single-end? Length? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags?
 Sanetra et al.")

22 Genome Redundancy (Repeats & Duplication) Sanetra et al. Frontiers in Zoology :15 Margulies E H et al. PNAS 2005;102(9):
23 Multi-mapping tags How does it treat tags with multiple matches to the reference genome (multimapping tags)? report all positions (huge storage requirements) discard all positions (huge information loss) report first alignment only (information loss/misleading) report random alignment only (information loss/misleading) report best alignment and metric (information loss/misleading) report first X alignments and flag read as multi-mapping (reasonable comprimise) Does the method selected address my biological question?
24 Things to consider when analysing RNAseq Is your data stranded or unstranded? Paired-end or single-end? Length? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags? Gapped or ungapped alignment?
25 Gapped or un-gapped alignment? Fastest methods are un-gapped, but what about micro-indels, structural vaiants or exon-exon junctions? TSS AUG Does the method selected address my biological question?
26 Things to consider when analysing RNAseq Is your data stranded or unstranded? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags? Gapped or ungapped alignment? Other practicalities
27 Other practicalities What are the resource restrictions? What resources are available to me? Laptop, shared or dedicated cluster, web server? Efficiency using RAM, CPU, and HDD choose any two How long can I store and access raw/mapped data? is the aligner fast enough to make my analyses viable? What are the input and output formats? now compare that to your data AND to any downstream programs you want to use What do my colleagues know and use? Every algorithm has quirks, and often these are not documented experience in alignment tools is valuable is the tool still supported by the authors? BEWARE of zealots don t restart the Holy Wars
28 Things to consider when analysing RNAseq Is your data stranded or unstranded? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags? Gapped or ungapped alignment? Other practicalities For projects that involve de novo transcriptome assembly: Selecting a good method








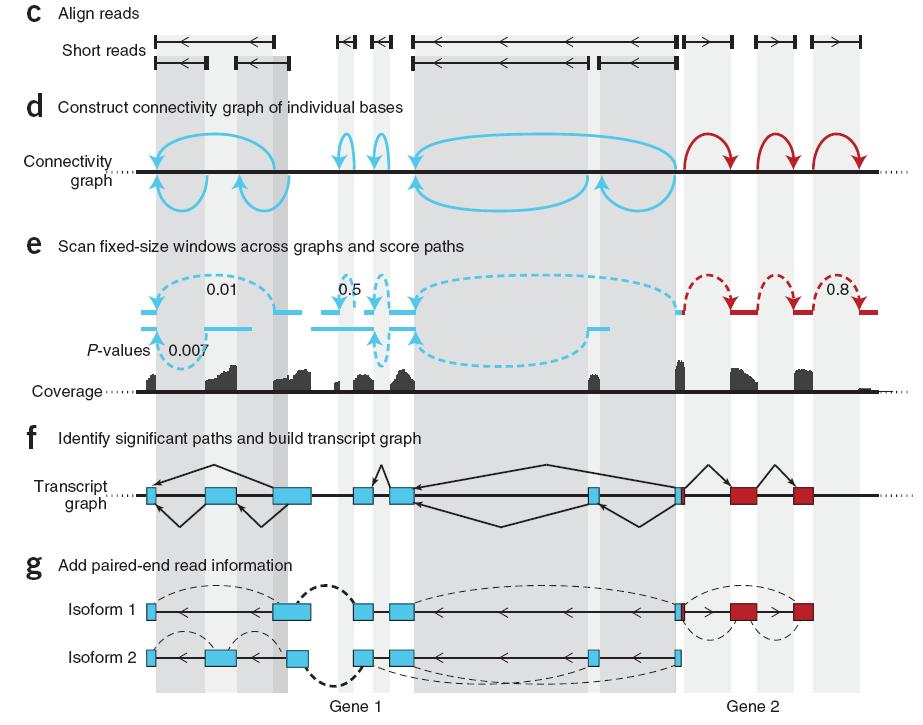
29 Reference assisted transcript assembly Scripture Cufflinks Guttman et al., Nat Biotech (5):503-10















30 Reference free alignment - de novo assembly Gene Symbol: MGAT5 Trinity Oases Abyss Gene Symbol: RAN


















31 The Jigsaw Puzzle Analogy















 Some")


 Some")



32 There s more than one solution (alternative splicing) There s no way to know if you are correct There s no box with a picture There are no colours More than 100 million pieces Many pieces are redundant (but not equally redundant, expression differs) Many pieces fit in multiple places There are no edge or corner pieces All pieces are different sizes The puzzle is double sided (and you are working on both at the same time) Some pieces are fused to other pieces (but they shouldn t be, and you can t tell which ones) Some pieces are from another puzzle (but you can t tell which ones by looking) Some pieces are broken (but you can t tell which ones by looking) Lots of missing pieces
33 Things to consider when analysing RNAseq Is your data stranded or unstranded? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags? Gapped or ungapped alignment? Other practicalities For projects that involve de novo transcriptome assembly: Selecting a good method Evaluating the alignment/assembly









34 Look at your data! Genomic context of expression Gene Symbol GRB7 Exon-exon junction usage Alternative splicing Single nucleotide resolution coverage plot Novel exons or novel transcripts Known gene structure (exons and introns)
35 Evaluating aligned data Check your data! visualization strategies (e.g. IGV or genome browsers) Check for enrichment of reads over exons, absence of evidence of DNA contamination Check your mapping statistics Look for % mapped?, % mapped at what length?, redundancy of tag start sites (complexity of libraries), etc. Make sure the controls are doing what they should be e.g. knocked down genes should have lower signal than the control sample, etc. Be careful with your interpretation! Remember the limitations and parameters of your alignment strategy! Eg. Variable alignment strategies that trim starts and ends of tags will overestimate the relative complexity of your library Eg. Discarding all tags that map to multiple regions will limit your ability to detect closely related gene families, or sequence motifs in repetitive/low complexity areas
36 Things to consider when analysing RNAseq Is your data stranded or unstranded? For projects that involve alignment to a reference genome: Choosing the best aligner How does it treat multi-mapping tags? Gapped or ungapped alignment? Other practicalities For projects that involve de novo transcriptome assembly: Selecting a good method Evaluating the alignment look at your data Counting your data
37 TSS TSS TSS TSS TSS TSS TSS TSS Counting your data
38 Uniqueome affects quantitation of RNAseq Correction for unique content improves correlation to microarrays Koehler et al Bioinformatics (2):
39 How to detect a transcript? A B C D protein coding regions non-coding regions polyadenylation spliced intron
40 How to detect a transcript? A B C D Different gene models will give different results from the same data. Accuracy relies on the quality of the gene models used. ~80% 92.6% known transcripts have diagnostic features (covers 99.8% of loci) diagnostic features covering individual transcripts from loci protein coding regions non-coding regions polyadenylation spliced intron
41 Differential Gene Expression You absolutely CANNOT answer this question without replicates All biological systems are variable it s an evolutionary NEED Understanding biological variations means no wasted time chasing false positives Your question is actually whether the variation between your groups is bigger than the variation within your groups You must have independent biological replicates Do not fall into the reducing variation trap Variation is your friend, not your enemy you want to MAXIMIZE your biological variation Using technical replicates makes your p-values WORTHLESS If you can t afford replicates then you can t afford the experiment.

42 Our Time is
43 Scaling to Library Depth TMM Trimmed Mean of M Values Raw Read Counts Counts per Million Raw Read Counts Counts per Million % % % 80% 70% 60% 50% 40% 30% 20% 10% % 80% 70% 60% 50% 40% 30% 20% 10% 0 Library 1 Library 2 0% Library 1 Library 2 0 Library 1 Library 2 0% Library 1 Library 2 mirna A mirna B mirna A mirna B mirna A mirna B mirna C mirna A mirna B mirna C Robinson & Oshlack Genome Biol 2010; 11(3):R25
44 RNAseq pros and cons Pros: Cons: you do not need to know the sequence before the assay begins you can profile any RNA from any organism you know the sequences of the genes you are measuring (can see mutations etc.) a lot of publicly available data potentially unlimited dynamic range (depending on how much you can spend) potentially unlimited sensitivity (depending on how much you can spend) equipment usually only available in specialist centres very expensive for most projects, limiting the number of replicates analysis techniques are still evolving. requires HPC to analyse analysis is time consuming