NEXT GENERATION SEQUENCING. Farhat Habib
|
|
|
- Emerald Fleming
- 5 years ago
- Views:
Transcription
1 NEXT GENERATION SEQUENCING
2 HISTORY
3 HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites
4 HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites Next Generation Sequencing Much shorter reads, 25 to 300 bp Higher throughput Cheaper cost per Mb Single molecule sequencing (no cloning step) Since Jan 2008 more DNA sequenced than all previous years
")
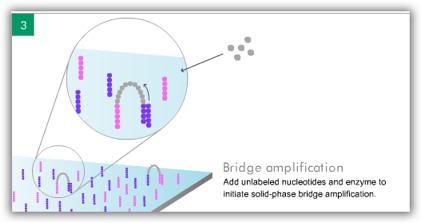
5 ILLUMINA (SOLEXA)
")
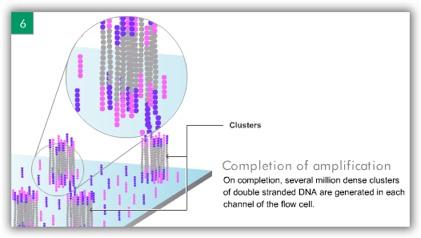
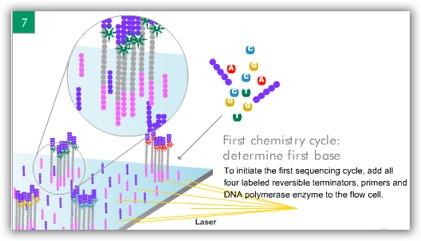
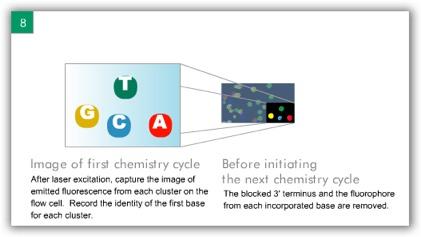
6 ILLUMINA (SOLEXA) Computational Biology Research Group
")
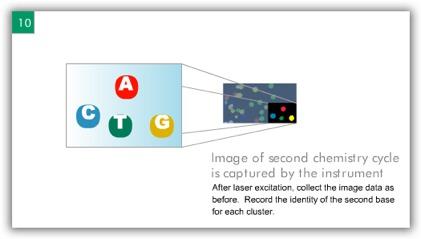
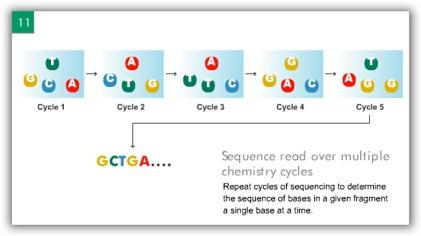
7 ILLUMINA (SOLEXA) Computational Biology Research Group
8 PAIRED END SEQUENCING The two ends of the fragments get different adapters Hence, one can sequence from one end with one primer, then repeat to get the other end with the other primer. This yields pairs of reads, separated by a known distance This provides additional information while aligning reads allowing better resolution in repeat areas
9 SOLID SEQUENCING
10 SOLID SEQUENCING
11 NGS APPLICATIONS
12 NGS APPLICATIONS Resequencing Characterise different related species or strains
13 NGS APPLICATIONS Resequencing Characterise different related species or strains Transcriptome analysis Analysis of the entire transcribed part of an organism s genome
14 NGS APPLICATIONS Resequencing Characterise different related species or strains Transcriptome analysis Analysis of the entire transcribed part of an organism s genome Examine chromatin modifications Quantify in vivo protein-dna interactions using the combination of chromatin immunoprecipitation and sequencing (ChIP-Seq)
15 NGS APPLICATIONS Resequencing Characterise different related species or strains Transcriptome analysis Analysis of the entire transcribed part of an organism s genome Examine chromatin modifications Quantify in vivo protein-dna interactions using the combination of chromatin immunoprecipitation and sequencing (ChIP-Seq) de novo genome assembly
16 ILLUMINA DATA
17 ILLUMINA DATA Generates short reads (~35-100bp)
18 ILLUMINA DATA Generates short reads (~35-100bp) Good for resequencing
19 ILLUMINA DATA Generates short reads (~35-100bp) Good for resequencing Can be used for de novo sequencing with paired end reads
20 READS
21 READS Acquire and process images and convert to FASTQ
22 READS Acquire and process images and convert to FASTQ Get data
23 READS Acquire and process images and convert to FASTQ Get data Quality control
24 READS Acquire and process images and convert to FASTQ Get data Quality control Map to genome
25 READS Acquire and process images and convert to FASTQ Get data Quality control Map to genome Visualisation
26 READS Acquire and process images and convert to FASTQ Get data Quality control Map to genome Visualisation Post Processing Peak Finding SNP Calling
27 FASTQ FORMAT
28 FASTQ
29 FASTQ TATACAATGCACTTAGTCATCCGCGTATCACTTTAT
30 FASTQ TATACAATGCACTTAGTCATCCGCGTATCACTTTAT +
31 FASTQ TATACAATGCACTTAGTCATCCGCGTATCACTTTAT + IIIIIIIIIIIIIIIIIIGIIIIIIIIII4IIII:I
32 FASTQ TATACAATGCACTTAGTCATCCGCGTATCACTTTAT + IIIIIIIIIIIIIIIIIIGIIIIIIIIII4IIII:I #0 index number for a multiplexed sample (0 for no indexing) /1 the member of a pair, /1 or /2 (paired-end or mate-pair reads only)
33 FASTQ SCORING Q phred = -10log10(e); Q solexa = 10log10(p(X)/(1-p(X))); e is the estimated probability of a base being wrong p(x) = probability of called base X being right Currently, the FASTQ format encodes a Phred quality score from 0 to 62 using ASCII 64 to 126 (although in raw read data Phred scores from 0 to 40 only are expected).
 Phred score is: Q Phred = -10 log 10 e Score written with character ascii code Q + 64.")
34 BASE CALL QUALITY STRINGS If p is the probability that the base call is wrong, the (standard Sanger) Phred score is: Q Phred = -10 log 10 e Score written with character ascii code Q + 64.
35 READ ALIGNMENT For applications such as resequencing, SNP calling, and many others the reads need to be aligned to a reference genome Number of reads is in million or more optimal alignment algorithms are too time intensive O(mn)
36 ADDITIONAL CHALLENGES Read errors Dominant cause for mismatches Detection of substitutions? Importance of base-call quality Repetitive regions/accuracy ~20% of human genome is repetitive for 32bp reads Use paired-end information
37 ALIGNMENT ALGORITHM APPROACHES Hashing (seed-and-extend paradigm, k-mers + Smith-Waterman) The entire genome Straightforward, easily parallelized but large memory The read sequences Flexible memory footprint, harder to parallelize Indexing by Burrows-Wheeler Transform Pros: fast and relatively small memory Cons: decrease in performance for longer reads