Functional profiling of metagenomic short reads: How complex are complex microbial communities?
|
|
|
- Heather Hardy
- 5 years ago
- Views:
Transcription
1 Functional profiling of metagenomic short reads: How complex are complex microbial communities? Rohita Sinha Senior Scientist (Bioinformatics), Viracor-Eurofins, Lee s summit, MO


2 Understanding reality, mostly doesn t hurt
3 State of in silico prediction of protein function BLAST Performance of top 10 protein function prediction programs against (Left panel) Easy targets (had a 60% or higher sequence identity with any experimentally annotated protein) (Right panel) Hard cases Nature Methods 10, (2013) doi: /nmeth.2340
4 State of in silico prediction of protein function 1- The simplest method of function assignment (BLAST) gives reasonable results when homologs are present. 2- But more sophisticated methods are needed to increase the precision. 3- For the hard targets, a simple method like (BLAST) is not very reliable. Additional information: 1- Most methods primarily use sequence similarity. 2- More than half the methods used additional data, such as types of evolutionary relationships, proteinprotein interactions or gene expression data. 3- Some methods used literature mining. 4- Many of these methods used machine learning principles. 5- Use threading protocols to predict structure and structure alignment to predict functions.
5 Nature of the system we are dealing with Metagenomic approach Millions of DNA fragments Majority is originating from microbial genes In proteins space (millions of short peptides) For functional annotation Assembly based annotation It has its own pros & cons, we can debate it; but later! Can we accurately assign functions to such small peptides (10-80aa)?? Assembly free annotation (eg: MEGAN, MG-Rast, HUMAnN Here we try to assign function to each metagenomic read (by exploiting their evolutionary relationship with well characterized proteins)
6 What did we discuss so far? The Central Dogma of Genomics derives from structural biology!! Protein Sequences > Protein Structure >Protein Function 1- Function prediction methods are getting better as compared to base methods like (BLAST) 2- This requires to go beyond the central dogma of genomics (Seq---Str---Fun) 3- Easy proteins targets are more tractable with existing methods compared to hard cases 4- Computational protocols to predict protein functions are "optimized for full length proteins" In contrast, a metagenomic sample is: 1- Comprised of short peptide fragments (originating from full length proteins) 2- A mixture of Easy & Hard cases Genome Biol. 2000; 1(2): comment comment



7 Commonly used methods & applied concepts BMC Bioinformatics. 2011; WebMGA: BMC Genomics. 2011
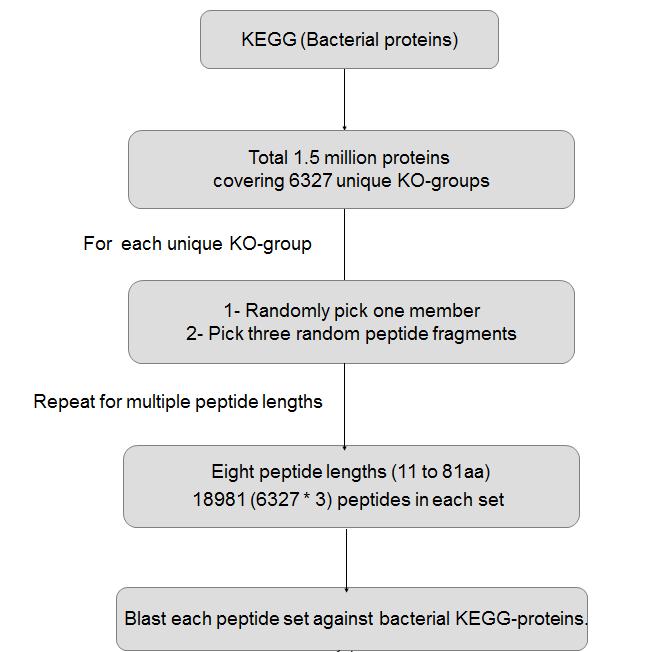

8 Understanding alignment behavior of short peptides

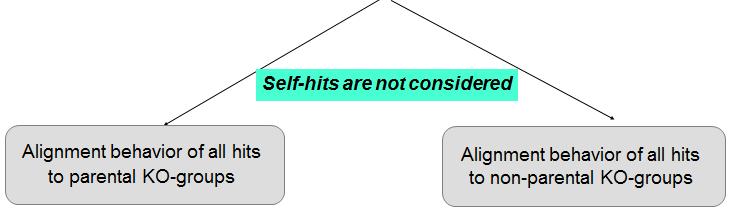
9 Understanding alignment behavior of short peptides Comparison of alignment behavior (Left panel) when the short peptides align to members of their parent KO-groups (Right panel) when short peptides align to members of their non-parent KO-groups. Hits to same KO-group members have significant identity above 80%. But a major fraction is in the range of 40-80% (for both left & right panel) and makes it difficult to discriminate between true and false positive hits. Hexbin colors within the graph are proportional to their frequency or members within the bin. Member frequency and color relationship is depicted in the arrow-headed color bar

10 State of in silico prediction of protein function
 Total number of hits to non-parental KOgroup for which we could get EC-numbers of both query and subject sequences.")
11 Understanding alignment behavior of short peptides Recursive circular diagrams (for peptide lengths of 51-81) Showing proportion of hits to non-parental KOgroups that share Enzyme Commission number at different hierarchy levels. (A) Total number of hits to non-parental KOgroup for which we could get EC-numbers of both query and subject sequences. (B) Total number (fraction) of those hits sharing same 1 st level EC group. (C) Total number (fraction) of those hits sharing same 1 st and 2 nd level EC group. (D) Total number (fraction) of those hits sharing same 1 st, 2 nd and 3 rd level EC groups. (E) Total number (fraction) of those hits sharing same EC-number (all 4 groups).

12 Short peptides carry enough structural signature to avoid random alignments Majority of short peptides, either align to same KO groups (parental) or to non-parental KO-groups with similar Enzyme Commission number. We know that protein fold space is limited and the same folds are repeatedly used to perform different biochemical functions. In contrast, convergence of unique protein folds to execute the same function (and share the same EC number) has occurred in only a small number of cases (7.5% of all known EC nodes). This clearly indicates that short peptides are actually carrying structural signatures, which are sufficient enough to restrict their alignments to either same KO-groups proteins or to KO-groups with similar Enzyme Commission number

13 Frequency weighted abundance profiler

14 Remember!! There are also reads originated from hard targets (reads originated from uncharacterized proteins, with no homologs in databases)
15 Remember!! There are also reads originated from hard targets (reads originated from uncharacterized proteins, with no homologs in databases) Only ~35% of these peptides align to database proteins. Same figure for characterized peptides was ~99%. Per peptide hits are ~3.0 as compared to 200 of characterized peptides. 3rd quartile of the best alignment of uncharacterized peptides is equal to median of peptides from characterized proteins
16 Frequency weighted abundance profiler (with ability to filter reads from uncharacterized proteins)

17 KO groups with more members Vs less populated KO groups KO groups with more members actually cover longer evolutionary space, hence produce multiple low identity alignments. It brings down the median value of all the true alignments within that KO group. Apparently future addition of new proteins in each group, would continue this trend.


 revealed a generally consistent pattern regardless of")
18 Some intriguing comparisons of taxonomic & functional diversities Taxonomic Front. Microbiol., 22 May 2017 Functional Although all of the 18 gut microbiomes surveyed showed a high level of b-diversity with respect to the relative abundance of bacterial phyla (Fig. 3a), analysis of the relative abundance of broad functional categories of genes and metabolic pathways (KEGG) revealed a generally consistent pattern regardless of the sample surveyed A core gut microbiome in obese and lean twins. Nature Dec. 2008
19 Summary/conclusions 1. Short reads tend to align to similar domains and hits to non-parental KO-groups cannot be distinguished just by sequence-similarity measures. 2.Rather than trying to infer parental KO, if all hits are considered as non-random and informative, normalizing their alignment weight by the total number of hits gives more weight to unique peptides. 3.Weighting reads by their uniqueness gives a precise functional-abundance profile of microbial KO-group proteins. 4- Peptides from uncharacterized proteins have distinct alignment behavior, which can be used to discard hits from such peptides. 5- Existing and commonly used methods tend to make predictions based on similarity of short-peptides (translated short NGS reads) to well characterized reference proteins. But this process is error prone.
20 Acknowledgements Dr. Andrew K. Benson Dr. Jennifer Clarke