G4120: Introduction to Computational Biology
|
|
|
- Fay Kathryn Walker
- 5 years ago
- Views:
Transcription
1 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Lecture 3 February 13, 2003 Copyright 2003 Oliver Jovanovic, All Rights Reserved.
2 Bioinformatics and Computational Biology Internet Resources National Center for Biotechnology Information (NCBI) PubMed, PubMed Central, Books and other reference material GenBank, RefSeq, CDD, MMDB and other sequence and structure databases Prokaryotic genome data and browsers (over 100 microbial genomes, over 1,000 viruses, over 300 plasmids) Eukaryotic genome data and browsers (9 complete eukaryotic genomes, additional maps and partial sequences) BLAST, PSI-BLAST and VAST search tools. Ensembl (EMBL-EBI/Sanger Institute) Eukaryotic genome data and browsers (human, mouse, rat,fugu, zebrafish, mosquito, Drosophila, C. elegans, and C. briggsae). UCSC Genome Bioninformatics Eukaryotic genome data and browsers (human, mouse, rat). European Bioinformatics Institute Sequence analysis tools and databases Expert Protein Analysis System (Expasy) Protein analysis and biochemical information, links to useful computational biology tools, software and references. Protein Data Bank Worldwide repository for 3D protein structure data and tools.
3 Macintosh Bioinformatics and Computation Biology Software Sources IU Bio-Archive (Macintosh, Unix and Java Molecular Biology Software) Pasteur Institute Macintosh Bioinformatics Archive ftp://ftp.pasteur.fr/pub/gensoft/macintosh/ European Bioinformatics Institute Biology Software Directory Apple Computer Bioinformatics Ports to Mac OS X European Molecular Biology Open Software Suite (EMBOSS) BioTeam, Inc. Bioinformatics Tools Ports to Mac OS X Fink Scientific Tool Ports to Mac OS X SourceForge
4 Databases Flat File Database (FFDB) A collection of similar files made useful by ordering and indexing. All the information about one sequence would be stored in one structured text file, and you generally examine one file at a time. Examples: GenBank, FileMaker Pro Relational Database (RDB) All data is stored inside one or more tables of rows and column, with all operations done on the tables themselves or producing other tables as the result. All the information about one sequence would be stored in a collection of tables with other data, so you can easily look at just the information relating to that sequence, or how it relates to the database as a whole. Structured Query Language (SQL) is used to access data in a relational database. Examples: msql, MySQL, PostgreSQL, Microsoft SQL Server, Oracle Object Oriented Databases (OODB) Data is stored and retrieved in an fashion consistent with object oriented programming principles (based on languages such as Smalltalk, C++ or Java). They generally handle complex structures and concurrent interaction by multiple clients well. Many relational databases have or are acquiring object oriented database features. Examples: PDB, Versant VDB, Gemstone GemFire
5 Searching Sequence Databases Needleman-Wunsch Needleman-Wunsch gives you the optimal global alignment of two sequences. This is best for comparing closely related sequences of similar lengths. Examples: GCG Gap, EMBOSS Needle Smith-Waterman Smith-Waterman gives you the optimal local alignment of two sequences. This is better for comparing distantly related sequences (where non-functional regions may have diverged). Examples: GCG BestFit, EMBOSS Water BLAST BLAST gives a fast approximation of Smith-Waterman, from times faster, but will not necessarily find optimal local alignments. Examples: NCBI BLAST, WU-BLAST
6 Definitions Identity - the extent to which two sequences are invariant. Similarity - The extent to which sequences are related, based on sequence identity and/or conservation. Conservation - changes in an amino acid sequence that preserve the biochemical properties of the original residue. This is measured in most sequence comparison algorithms by substitution matrices in which scores for each position are derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins. Homology - similarity attributed to descent from a common ancestor. It may or may not result in similar function. Orthologous - homologous sequences in different species that arose from a common ancestral gene. Paralogous - homologous sequences within a single species that arose by gene duplication.
7 Rules of Thumb when running BLAST The shortest possible word size (2 for proteins, 7 for nucleotides) gives the most sensitivity, though the search may take more time. Note: A larger word size (3 for proteins, 11 for nucleotides) is the default setting for NCBI BLAST. You will have to change it manually. At least initially, run your search with the Low Complexity filter off. Then, if you appear to be getting spurious hits, or for comparison purpose, run it again with the filter on. Although it can be helpful, the filter can also filter out a significant match. Note: Filter on the default setting for NCBI BLAST. You will have to turn it off manually. PSI-BLAST can be useful for searching for very weak protein homologies. If searching with short DNA or protein sequences make sure you use the appropriate Search for short nearly exact matches BLAST page, or make sure to use those settings. BLAST is not the best tool to use for very short sequences.
8 Consequences of Matrix and Algorithm Choice The default BLOSUM 62 substitution scoring matrix is best for comparing moderately distant and relatively closely related proteins. When searching for distantly related proteins, try using the PAM 250 or BLOSUM 45 matrices as well. If comparing closely related proteins, try using the PAM 1 or BLOSUM 80 matrices as well. Keep in mind that BLAST is a heuristic version of Smith-Waterman, and may miss a significant alignment. The following examples, kindly provided by Christopher Dwan of The University of Minnesota Center for Computational Genomics and Bioinformatics, illustrate the consequences of choice of matrix and algorithm when searching for sequence alignments. The Arabidopsis Unigene database (91,331 unique sequences representing possible coding regions of the arabadopsis genome as of December 15, 2001) was run against Genpept (NCBI s nonredundant set of protein sequences as of December 15, 2001) using BLAST with two different matrices (BLOSUM 62 and PAM 250) or using two different algorithms (BLAST and Smith- Waterman).

9 Blosum 62 v. PAM 250
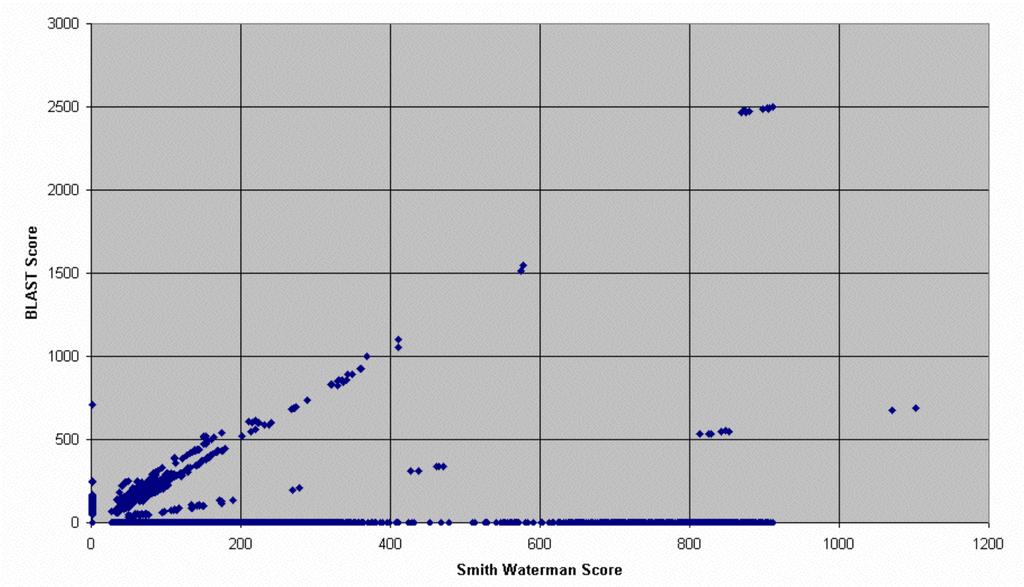
10 BLAST v. Smith-Waterman
11 Rules of Thumb for Significance of Protein Alignments Protein Identity Significance Under 20% Unlikely to be significant 20% to 30% "Gray zone" may or may not be significant Over 30% Likely to be significant Keep in mind that when searching GenBank with a protein sequence it is possible to get results with a stretch of amino acids with over 50% identity by chance alone. Identity throughout an entire protein is more likely to be significant, however, homologous proteins with a very low level of identity exist. Such distant relatives can be identified through comparison to other homologous proteins. Identity within known functional domains is more likely to be significant, and may suggest functional homology.