The Revolution in Human Genetics: Deciphering Complexity. David Galas Institute for Systems Biology, Seattle, WA
|
|
|
- Nancy Davis
- 5 years ago
- Views:
Transcription




1 The Revolution in Human Genetics: Deciphering Complexity David Galas Institute for Systems Biology, Seattle, WA


2 Genetics and Environment integration is key to future medicine Genome Blood proteins, mirna, mrna, Cell Read-out Predictive, Personalized Diagnostics Disease & Health Complex biological Networks Environment
3 Summary Technological transformation of human genetics Whole genome sequencing Family of four project what have we learned? Next projects. Challenges Reference genome problem Complex genetic models Integration of data types: new methods


4 History of Genetic Marker Density Human Genetic Markers Type of marker No. of loci sampled Recombination Inference Blood groups ~20 N/A Electrophoretic ~30 N/A HLA type 1 N/A RFLPs >10 5 N/A VNTRs >10 4 minimal Microsatellites >10 5 minimal SNP Genotypes >10 6 probabilistic Exomes 1% marginal Genomes All exact
 Genetic-environment deconvolution Missing heritability")
5 Family Sequences and the Future of human genetics Traditional (common SNP, additive effects) GWAS has been a gene collection method reaching asymptotes for known phenotypes Inferences from family sequences plus populations-based studies Constrained analysis using: biological understanding of networks, additional data (e.g. population-based information) Genetic-environment deconvolution Missing heritability

6 Family genomics in perspective
7 Current technical issues Sources of DNA Library biases Inherent accuracy of reads Read lengths currently 20 to 70 Maps to reference variation file Coverage and depth (~40 fold) Assembly and phase Error rates types
8 Pilot project: Complete Genome Sequences of a Family of Four Parents healthy kids both have two genetic diseases Power of a family sequence permits: Noise reduction : Very low error rate <1/100,000 Discovery of ~230,000 new single base variants First full recombination map of a family First determination of inter-generational mutation rates Identification of disease gene candidates for rare genetic diseases (two in this case) Roach et al., Science, April 30, 2010

9 Whole Genome Sequencing of Family Unaffected parents Children with craniofacial And limb malformation (Miller Syn.) and lung disease (cilliary dyskenesis)




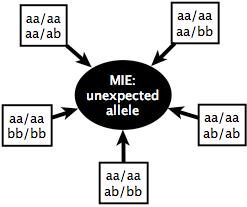


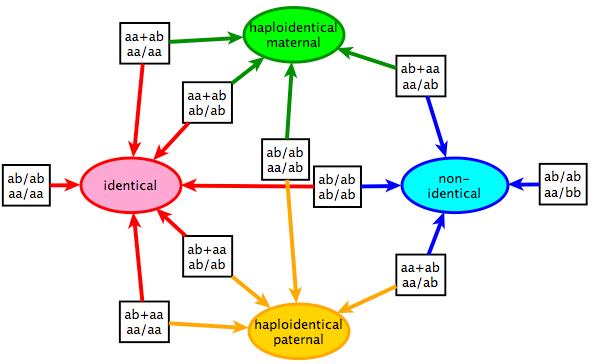

10 Inheritance state vector: family of four Allele Assortment Inheritance States haploidentical paternal identical haploidentical maternal nonidentical Larger family higher dimension





11 QuickTime and a decompressor are needed to see this picture. QuickTime and a decompressor are needed to see this picture. Sequencing a nuclear family yields: high resolution crossover sites and haplotypes Inheritance patterns in kids (0.5Mb bins) Dad Mom haploidentical paternal identical haploidentical maternal Inheritance vector is 4 dimensional nonidentical

12 Family Allele Inheritance Patterns
 10 4 10 3 A All SNPs, possibly detrimental 10 2 10 B C D All SNPs, probably detrimental Very rare SNPs, possibly detrimental Very rare SNPs,")
13 Simple recessive (SNPs) Utility of Constraints & Error Reduction A B C D Constraints: 1. recessive 2. twin states 3. very rare 4. detrimental Compound heterozygous (genes) A All SNPs, possibly detrimental B C D All SNPs, probably detrimental Very rare SNPs, possibly detrimental Very rare SNPs, probably detrimental 1 Four gene candidates!
14 Genomes of kids X ZNF721 DNAH5 DHODH KIAA0556 Miller s gene Ciliary dyskenesis gene centromere heterochromatin error region CNV candidate gene haploidentical maternal paternal recombination identical maternal recombination haploidentical paternal nonidentical Sibling genomes are identical across ~25% of their length (23.2% here)

15 VAAST FPT results for 2 MILLER Syndrome patients DHODH DNAH5 P gene P < 1e -3 P < 1e -7 P < 1e -8 P genome P > 0.1 P > 0.05 P < 5e -3 DHODH ranked 3 rd DNAH5 ranked 11 th out of 21,902 genes Mark Yandell, Univ of Utah


16 Schematic of VAAST Analysis of MILLER Kindred 1 using a single quartet : only two candidate genes DHODH DNAH5 P gene P < 1e -3 P < 1e -7 P < 1e -8 P genome P > 0.1 P > 0.05 P < 5e -3 DHODH ranked 1 st DNAH5 ranked 2 nd out of 21,902 genes Mark Yandell, Univ of Utah
17 Effective Error Rate (1-0.7) x (1.0x10-5 ) = 3x10-6 = % accurate Intergenerational mutation rate: 1.1x10-8 per position per haploid genome = 70 mutations in each diploid genome
18 Compression regions & reference genomes Sorting out the errors in the reference genome is essential: Variations vs false identifications A C Reality Reference Sequences would be called as A/C hets Thus, regions of high frequencies of hets in families, together with coverage anomaly implies compression error
19
20 Compression Blocks in the Human Genome (probable) Numbers indicate the number of markers in each compression block
21 Now & Future references Now Future Sequence Reads Sequence Reads Map onto Comparisons & Inference Chosen Reference Genome Sequence Simple, crude, error generating Set of high - accuracy Genome Sequences Complex, accurate








22 More complex genetics Larger families = high dimension inheritance vectors Need this to test more complex genetic models Iterative model testing Biology Other genetic data as priors Increase model complexity Genetic Model No of genes Types of variations Model of interactions Test against family data All gene variants Relationship in family Candidate model Hypothesis for testing
23 Family genome sequences Can provide: Accurate sequence Low FDR of rare SNPs Highly accurate phase & recombination points New approaches to testing complex genetic models
24 Follow-on Studies We expect to analyze 120 to 150 high accuracy genomes in next set: (almost all will be in families) As of October 2010 we have about 50 whole genomes done Huntington s disease: modifier genes Congenital heart defect: modifier genes Planned (focus on neurodegeneration): Parkinson s Spinal muscular atrophy others
25 Congenital Heart Defects D. Srivastava Gladstone Institute - Primary defect = GATA4 mutation - Identify modifiers


26 New Computational Approaches to Genetics: integrating networks and genetic data analysis Simple, additive genome-wide genetic studies for detection of genetic effects are powerful, but deeply flawed Beyond additive GWAS genetic effects, How to add biology: beyond genotype-phenotype correlations GWAS Systems genetics Additivity assumption Reality Problem: Can we constrain statistical analysis with complex biological knowledge (gene interactions, knowledge of networks, related phenotype components, GWAS data)
27 Approaches to the problem A framework for complex knowledge constraints Probabilistic graphical models (Bayesian nets, Markov nets, PBNs) Handle uncertainty and noise in the data Compactly represent probability distributions Use powerful algorithms for probabilistic reasoning + First-order logic Classes of objects Recursive, potentially infinite structures Relational data Probabilistic logic-based modeling Combining probabilistic and logic representations The best of both worlds can represent partial knowledge Relational Markov Networks, Loopy Logic, Markov Logic Networks, etc. The most general approach: Markov Logic Networks

 57, 14, 130, 20 new")
28 Yeast sporulation data analysis by MLN Detected informative markers: 71, 117, 160, 72, 116, 123, 57, 14 (130), 79, 20 Markers in red are confirmed by (Gerke,Lorenz,Cohen, 09) 57, 14, 130, 20 new informative loci
29 Genetic interactions inferred by MLN from Yeast Cross Sporulation phenotype Red: positive interaction Green: Negative interaction Intensity indicates strength of interaction Sakhanenko and Galas, J. Comp. Biol. (October 2010)
30 Next steps towards computational Systems Genetics More complex genetic models Testing on yeast data Family genome sequences of humans Trim down full first order logic propositions Adopt language for biological descriptions Integrate knowledge of network function
31 Summary Technological transformation of human genetics Whole genome sequencing Family of four project what have we learned? Next projects. Challenges Reference genome problem Complex genetic models Integration of data types: new methods




32 Acknowledgements Institute for Systems Biology: Jared Roach, Arian Smit, Gustavo Glusman, Paul Shannon, Lee Rowen, Robert Hubley, Lee Hood, Kai Wang, Nikita Sakhanenko, Ji-Hoon Cho, Alton Eldridge University of Utah Lynn Jorde, Chad Huff, Mark Yandell Gustavo Glusman, Jared Roach, Chad Huff, Arian Smit Funding ISB-Univ of Luxembourg Program, NIH, NSF- FIBR Program, DoD University of Washington Mike Bamshad, Jay Shendure University of Luxembourg, LCSB Rudi Balling, Antonio DelSol Complete Genomics: Rade Drmanac, Dennis Ballinger, Krishna Pant, Andrew Sparks