THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE
|
|
|
- Eustacia Webster
- 6 years ago
- Views:
Transcription

1 THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE Ali Mohamadi, M.D. Senior Director, Global Regulatory Patient Engagement and Policy BioMarin Pharmaceutical 30-July-2016
2 Goals of Today s Presentation Explain FDA s role in the review of new drugs and the key steps in the process Describe the increasing importance of the patient voice at the FDA, as well as entry points where advocates can be particularly influential Discuss ways in which all stakeholders in drug development can work together for the benefit of patients 2
3 How FDA Approves New Drugs To market a prescription drug in the United States, a manufacturer requires approval from the FDA Must demonstrate the drug s safety and effectiveness Must ensure that manufacturing plant passes inspection Must obtain approval for all written material about the drug, also known as labeling 3

4 Standard Process of Drug Approval Source: Congressional Research Service Report for Congress, July
5 IND Application Before testing in humans (clinical testing) the drug s sponsor must submit an Investigational New Drug (IND) application to the FDA IND includes information about the proposed clinical study design, completed animal test data, and the lead investigator s qualifications Application must include an Indication for Use section describing what the drug does, the clinical condition and intended population Must include the written approval of an Institutional Review Board (IRB) 5
6 IND Application FDA has 30 days to review an IND application Unless FDA objects, a manufacturer may then begin clinical testing Focus of IND review: safety 6
7 Clinical Trials Once an IND has been approved, researchers can begin clinical trials Phase I: Small number of volunteers to determine dosing and identify side effects Phase II-III: Larger number of individuals with particular characteristic, condition, or disease of interest of interest to gather evidence of the drug s efficacy, effectiveness, and safety 7
8 Submission of Marketing Application Once a manufacturer completes the clinical trials (usually takes 3 to 7 years), it submits the marketing application to FDA These applications contain not only the clinical trial results, but also information about the manufacturing process and facilities, including quality control procedures 8
9 Marketing Application Content The applications include: Product description Indication Labeling Manufacturing description 9
10 FDA Review FDA considers three primary questions in its review of applications: Whether the drug is safe and effective in its proposed use and whether the benefits of the drug outweigh the risks ( substantial evidence of safety and effectiveness ); Whether the drug s proposed labeling is appropriate, and what it should contain; Whether the methods used to manufacture the drug and the controls used to maintain the drug s quality are adequate 10
11 FDA Review Team Multidisciplinary group including representatives from: Clinical Chemistry, Manufacturing, and Controls Pharmacology Statistics Pharmacology 11

12 NDA/BLA Review Timeline Source: CDER 21 st Century Review Process Desk Reference Guide 12
13 FDA Decision Approval: may include specific conditions, such as required post-approval studies, restrictions on distribution, or required labeling disclosures Major amendment: extension of review clock (typically by 3 months) for applicant to address ongoing questions/issues Complete response: aka rejection FDA sends letter to applicant describing specific deficiencies and recommending ways to make the application viable, as appropriate 13
14 Post-Approval Procedures FDA s role in ensuring a drug s safety and efficacy continues after approval (postmarket regulatory procedures) Companies must report all serious and unexpected adverse reactions (clinicians and patients may do so as well) FDA oversees surveillance, studies, labeling changes, and information dissemination 14

15 The Voice of the Patient at FDA Image source: JDRF/University of Colorado-Anschutz 15
16 Why Should FDA Value Patient Input? Patients who live with a disease have a direct stake in drug review process and are in a unique position to contribute to drug development FDA reviewers could benefit from systematic approach to obtaining patient perspective on disease severity or unmet medical need Recent legislation empowers patients in the drug development process more than ever 16
17 Recent Patient-Focused Legislation Food and Drug Administration Safety and Innovation Act (FDASIA) signed into law in July, 2012 Section 1137 contains specific language aimed at: Developing and implementing strategies to solicit the views of patients during the drug development process Consider patient perspectives during regulatory discussions Upcoming reauthorization of the Prescription Drug User Fee Act (PDUFA VI) will MAY include statutes to further strengthen patient voice at the FDA 17

18 Post-FDASIA Role of Patients in Drug Development and Review 18

19 FDA: Points of Entry for Patients Before and during clinical trials: Benefit-risk assessment Patient focused drug development During FDA review: Patient representative program Advisory committee participation 19
20 CDER s Benefit-Risk Framework With FDASIA, CDER identified the need for a more structured Benefit-risk assessment in the review process to: Better communicate the reasoning behind its decisions Ensure the big picture is kept in mind during a complex, detailed review 20

21 CDER s Benefit-Risk Framework 21

22 CDER s Benefit-Risk Framework 22

23 B-R Assessment in FDA Review 23
24 From Benefit-Risk to PFDD Patients are uniquely positioned to help FDA understand the therapeutic context Current mechanisms for obtaining patient input are often limited to discussions related to specific applications under review Patient Focused Drug Development (PFDD) is an FDA commitment under FDASIA CDER and CBER will convene at least 20 meetings on specific disease areas FY (19 already) Meetings can help advance systematic approach to gathering input 24
25 Identifying Disease Areas: Criteria Disease areas that: Are chronic, symptomatic, and affect functioning and ADLs Have aspects that are not captured in clinical trials Have no or few therapies, or available therapies do not affect how a patient feels, functions, or survives Reflect a broad range of severity and affected populations 25
26 Meeting Format Meetings follow similar, but tailored design Take into account current state of drug development, specific interests of review divisions, needs of patient population Discussion elicits patients perspectives on their disease and treatment approaches Input is gathered in multiple ways Patient panel input and discussion with audience Interactive webcast and teleconference Polling questions to aid meeting discussion Federal docket to allow for more detailed comments 26
27 Samples of What FDA Asks Which symptoms have the most significant impact on your daily life?... On your ability to do specific activities? How well does your current treatment regimen treat the most significant symptoms of your disease? What specific things would you look for in an ideal treatment for your condition? What factors do you take into account when making decisions about using treatments?. Deciding whether to participate in a clinical trial? 27
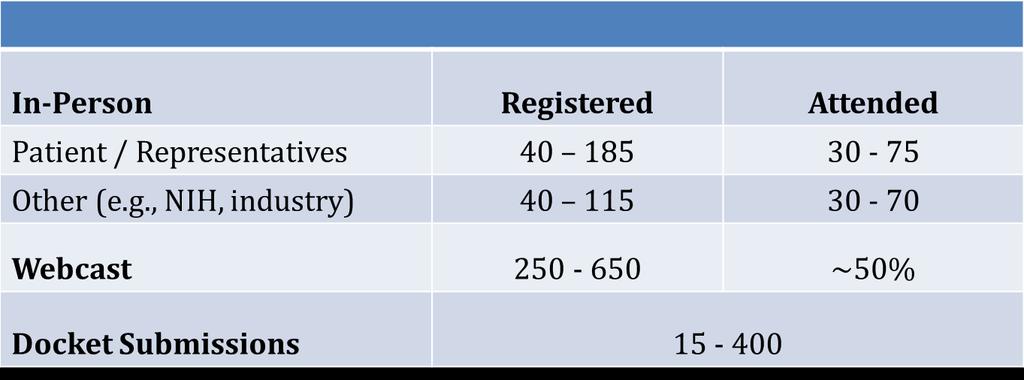
28 Participation Estimates 28
29 Preliminary Findings Patients with chronic serious disease are the real experts on what it s like to live with their condition For progressive degenerative diseases, many patients/parents feel an ideal treatment would at minimum stop progression of loss of function Patients chief complaints may not be factored explicitly into drug development plans, including measures of drug benefit planned in trials 29
30 Preliminary Findings Patients want to be as active as possible in the work to develop and evaluate new treatments They want their experience described using words that they consider to best describe how it feels They and their caregivers are able and willing to engage via the Internet, social media, and all other means at their disposal They don t expect FDA to address all the gaps in current treatment/approaches to drug development but do want FDA to help identify the most effective pathways for them to play major contributing role 30
31 Potential PFDD Outcomes Each meeting results in a Voice of the Patient report that captures patient input Summarizes participants experiences and perspectives, in their own voices Meeting input may be incorporated into the benefit-risk framework This input can help FDA staff to: Conduct benefit-risk assessments for other products under review Advise sponsors on their development programs 31
32 Potential PFDD Outcomes Input could support drug development more broadly: Help identify specific areas of unmet need in patient population Stimulate longer-range development of new patientfocused outcome measures Incorporate the voice of the patient earlier in drug development programs 32


33 Potential PFDD Outcomes 33
34 Externally-led PFDD Meetings There has been interest in expanded efforts to gather patient input in support of drug development and evaluation Meetings conducted by patients and their advocates provide an opportunity to expand the benefits of PFDD Mechanisms have included: Public meetings conducted within D.C. area Small meetings at FDA/online with patients Patient surveys and/or written submissions to a public docket 34
35 Patient Representative Program FDA recruits patient representatives to provide the unique perspective of patients and family members directly affected by serious or life threatening disease Representatives bring a personal viewpoint to the process and communicate a collective patient perspective A patient perspective is created when a person goes through personal experience with the disease A collective patient perspective is created when the person has knowledge of others disease experiences 35
36 Patient Representative Program Criteria for becoming an FDA patient representative: U.S. citizen at least 18 years of age Personal experience with the disease as a patient or caregiver Ability to be objective while representing the concerns of other patients Willingness to communicate their views Knowledge about treatment options and research No financial or ethical conflicts of interest 36
37 Patient Representative Program Serve in several ways, including: On advisory committee panels, where they offer the patient perspective, ask questions, and give comments to assist the committee in making recommendations As consultants for drug review As presenters at FDA meetings and workshops on disease specific or regulatory/health policy issues FDA conducts training for patient representatives Individual FDA 101 training Monthly webinars Annual workshop for newly recruited patient representatives 37

38 FDA Advisory Committees 38
39 FDA Advisory Committees The purpose of an FDA Advisory Committee is to provide the Agency with external expert advice during the review Typically the FDA notifies the company no later than 74 days of receiving the application if an advisory committee will be convened Typically an advisory committee is convened 8-12 weeks prior to FDA having to make a decision on whether to approve or reject an application 39
40 FDA Advisory Committees May be convened if: 1. The application is a new molecular entity (NME) 2. The clinical trial design used novel clinical or surrogate endpoints 3. There are significant issues regarding safety and/or effectiveness of the drug or biologic 4. The application raises significant public health questions regarding the role of the drug or biologic in the treatment or prevention of a disease 40
41 Open Public Hearing Every advisory committee includes an open public hearing (OPH) Interested persons may present information or views orally or in writing Minimum of 60 minutes per meeting If there is overwhelming interest, the committee chair may extend the OPH While there is a patient representative on the advisory committee panel, the OPH is the opportunity for broader input from patients and caregivers 41
42 Open Public Hearing Speakers Have a personal story to share While the majority of the advisory committee meeting is devoted to evaluating scientific questions, the OPH provides an opportunity for individuals to discuss unmet medical need, to describe their risk tolerance, and describe their own experience in their own words Less common for speakers to weigh in on review-related issues that have arisen over the course of sponsor and FDA presentations and advisory committee deliberations 42
43 Open Public Hearing Speakers A patient or caregiver can provide their personal, real life experiences and describe how their life would be impacted if the investigational product were/were not approved A clinical trial participant can share their experience while on the investigational drug, describing the benefits they experienced and how that compared with any adverse events When there is uncertainty with the science, the patient voice can help committee members better understand the key issues affecting patients 43
44 Open Public Hearing Speakers In addition to patients/caregivers, patient organization representatives can provide comments to support the needs of patients Large umbrella organizations can provide context to the overall unmet need for new therapies across diseases Disease-specific organizations can speak to the needs of patients and the input they have received from them 44


45 What s Next? Image source: NPKUA 45
46 Looking to the Future Patients, industry, scientists, and the FDA must work together to advance the science of patient input, discussing: How to best proceed in obtaining patients reports, assessments, and preferences, to inform patientcentered development and benefit-risk assessment Patient preferences for treatment impacts and tolerance of uncertainty about meaningful, significant potential benefits versus harms Approaches to bridge from initial patient-focused meetings to more systematic collection of patients experience living with a particular disease 46
47 Initial Steps to Consider 1. Organizing and uniting the patient community for specific disease states The patient voice is represented by a wide range of individuals and organizations Representatives from the patient community (e.g., patients, caretakers, advocates, physicians) can unite to ensure that their respective voices are heard 47
48 Goals to Consider 2. Defining meaningful engagement Patient engagement can represent a range of activities, from passive engagement (e.g., clinical trial participation) to more active participation (e.g., research development) Across this spectrum of activities, the elements that constitute or can most effectively help achieve meaningful results have yet to be clearly defined 48
49 Future Goals 3. Identifying and removing barriers to meaningful engagement Stakeholders should work together to determine which barriers exist and ways to remove them to ensure that FDA, industry, scientists, and patients can work together to better understand each others needs 49
50 Future Goals 4. Promoting active, ongoing collaboration For patient engagement to move forward on a large scale, all parties must be active collaborators FDA, industry and patients/advocates must work together to collect information on: The impact of a disease or condition on quality of life A better understanding of individual experiences with treatment regimens What aspects of treatment or symptom relief are most important to patients 50

51 51

52 Contact: THANK YOU Ali Mohamadi, M.D. 52
Advancing the Science of Patient Input
 Patient-Focused Drug Development Advancing the Science of Patient Input Sara Eggers, PhD Office of Strategic Programs FDA CDER EveryLife Foundation for Rare Diseases Annual Rare Disease Scientific Workshop
Patient-Focused Drug Development Advancing the Science of Patient Input Sara Eggers, PhD Office of Strategic Programs FDA CDER EveryLife Foundation for Rare Diseases Annual Rare Disease Scientific Workshop
December 4, Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm Rockville, MD 20852
 Food and Drug Administration 5630 Fishers Lane, Rm Rockville, MD 20852") December 4, 2014 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA 2014 N 1698: Food and Drug Administration Activities
December 4, 2014 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA 2014 N 1698: Food and Drug Administration Activities
Speed your time to market with FDA s expedited programs
 Regulatory Sciences Expediting drug approval Speed your time to market with FDA s expedited programs The faster way to marketing submission and drug approval for serious conditions and rare diseases In
Regulatory Sciences Expediting drug approval Speed your time to market with FDA s expedited programs The faster way to marketing submission and drug approval for serious conditions and rare diseases In
Annie Kennedy, Parent Project Muscular Dystrophy (PPMD) Testimony FDA PDUFA Stakeholder Meeting White Oak ~ December 17, 2015
 Testimony FDA PDUFA Stakeholder Meeting White Oak ~ December 17, 2015") Annie Kennedy, Parent Project Muscular Dystrophy (PPMD) Testimony FDA PDUFA Stakeholder Meeting White Oak ~ December 17, 2015 On behalf of Parent Project Muscular Dystrophy (PPMD), we are most grateful
Annie Kennedy, Parent Project Muscular Dystrophy (PPMD) Testimony FDA PDUFA Stakeholder Meeting White Oak ~ December 17, 2015 On behalf of Parent Project Muscular Dystrophy (PPMD), we are most grateful
PDUFA VI Public Meeting Remarks of Cynthia A. Bens Alliance for Aging Research. August 15, 2016
 1 PDUFA VI Public Meeting Remarks of Cynthia A. Bens Alliance for Aging Research August 15, 2016 Panel 1: Pre-Market Review and Post-Market Safety Good morning everyone and thanks to the FDA for inviting
1 PDUFA VI Public Meeting Remarks of Cynthia A. Bens Alliance for Aging Research August 15, 2016 Panel 1: Pre-Market Review and Post-Market Safety Good morning everyone and thanks to the FDA for inviting
D. Enhancing Regulatory Science and Expediting Drug Development:
 August 22, 2016 Division of Dockets Management (HFA-305) U.S. Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: Docket No. FDA-2016-N-1895-0001: Prescription Drug User Fee
August 22, 2016 Division of Dockets Management (HFA-305) U.S. Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: Docket No. FDA-2016-N-1895-0001: Prescription Drug User Fee
Chin Koerner Executive Director US Regulatory and Development Policy
 Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
Sec Short title; finding. Sec Authority to assess and use drug fees. Sec Reauthorization; reporting requirements.
 H.R. 2430, FDA Reauthorization Act of 2017 Section 1. Short Title. This Act may be cited as the FDA Reauthorization Act of 2017. Section 2. Table of Contents Table of Contents TITLE I: FEES RELATING TO
H.R. 2430, FDA Reauthorization Act of 2017 Section 1. Short Title. This Act may be cited as the FDA Reauthorization Act of 2017. Section 2. Table of Contents Table of Contents TITLE I: FEES RELATING TO
Structure and Mandate of FDA
 Structure and Mandate of FDA Leonard Sacks, M.D. Office of Medical Policy Center for Drug Evaluation and Research FDA FDA Clinical Investigator Training Course November 13, 2018 Mission of regulatory agencies
Structure and Mandate of FDA Leonard Sacks, M.D. Office of Medical Policy Center for Drug Evaluation and Research FDA FDA Clinical Investigator Training Course November 13, 2018 Mission of regulatory agencies
Compassionate Use Navigator Information for Physicians
 Compassionate Use Navigator Information for Physicians Contact: Elena Gerasimov, Program Director, Elena@kidsvcancer.org. As a physician, you must have wished there would be more treatment options for
Compassionate Use Navigator Information for Physicians Contact: Elena Gerasimov, Program Director, Elena@kidsvcancer.org. As a physician, you must have wished there would be more treatment options for
US FDA Expedited Programs and Expanded Access
 US FDA Expedited Programs and Expanded Access Ke Liu, MD, PhD Chief, Oncology Branch Division of Clinical Evaluation, Pharmacology and Toxicology Office of Tissues and Advanced Therapies Center for Biologics
US FDA Expedited Programs and Expanded Access Ke Liu, MD, PhD Chief, Oncology Branch Division of Clinical Evaluation, Pharmacology and Toxicology Office of Tissues and Advanced Therapies Center for Biologics
Docket #: FDA-2018-D-3268
 Subject: Comment on FDA Draft Guidance for Industry Titled Rare Diseases: Early Drug Development and the Role of Pre-Investigational New Drug Application Meetings Docket #: FDA-2018-D-3268 ARM is an international
Subject: Comment on FDA Draft Guidance for Industry Titled Rare Diseases: Early Drug Development and the Role of Pre-Investigational New Drug Application Meetings Docket #: FDA-2018-D-3268 ARM is an international
Managing Risk and Uncertainty Through the Drug Life cycle. Recent FDA Initiatives. Theresa Mullin, PhD
 Managing Risk and Uncertainty Through the Drug Life cycle Recent FDA Initiatives Theresa Mullin, PhD Director Office of Strategic Programs US FDA Center for Drug Evaluation and Research October 14, 2014
Managing Risk and Uncertainty Through the Drug Life cycle Recent FDA Initiatives Theresa Mullin, PhD Director Office of Strategic Programs US FDA Center for Drug Evaluation and Research October 14, 2014
March 2, Dear Chairman Upton, Ranking Member Pallone, and Representative DeGette:
 March 2, 2015 The Honorable Fred Upton Chairman Energy and Commerce Committee House of Representatives Washington, DC 20515 The Honorable Frank Pallone Ranking Member Energy and Commerce Committee House
March 2, 2015 The Honorable Fred Upton Chairman Energy and Commerce Committee House of Representatives Washington, DC 20515 The Honorable Frank Pallone Ranking Member Energy and Commerce Committee House
Quality Risk Management and Submission Strategies for Breakthrough Therapies
 Quality Risk Management and Submission Strategies for Breakthrough Therapies IFPAC/Washington DC January 22, 2014 Sarah Pope Miksinski, Ph.D. Acting Director, Division of New Drug Quality Assessment 2
Quality Risk Management and Submission Strategies for Breakthrough Therapies IFPAC/Washington DC January 22, 2014 Sarah Pope Miksinski, Ph.D. Acting Director, Division of New Drug Quality Assessment 2
CDER s Advisors & Consultants Staff (ACS) Perspective on Preparing for Advisory Committee Meetings. Igor Cerny, Pharm.D. Director, ACS CDER/FDA
 Perspective on Preparing for Advisory Committee Meetings. Igor Cerny, Pharm.D. Director, ACS CDER/FDA") CDER s Advisors & Consultants Staff (ACS) Perspective on Preparing for Advisory Committee Meetings Igor Cerny, Pharm.D. Director, ACS CDER/FDA Key Points That Will Be Discussed Today s Perspective is CDER
CDER s Advisors & Consultants Staff (ACS) Perspective on Preparing for Advisory Committee Meetings Igor Cerny, Pharm.D. Director, ACS CDER/FDA Key Points That Will Be Discussed Today s Perspective is CDER
Rare Diseases Drug Development and Patient Perspective Initiatives at FDA
 Rare Diseases Drug Development and Patient Perspective Initiatives at FDA Jonathan C. Goldsmith, MD, FACP Associate Director Rare Diseases Program/OND/CDER/FDA EveryLife Foundation for Rare Diseases Annual
Rare Diseases Drug Development and Patient Perspective Initiatives at FDA Jonathan C. Goldsmith, MD, FACP Associate Director Rare Diseases Program/OND/CDER/FDA EveryLife Foundation for Rare Diseases Annual
Health Industry Alert
 Health Industry Alert August 4, 2017 Senate Passes Long-Awaited FDA User Fee Package Following nearly two years of negotiations and hearings examining the Generic Drug User Fee Amendments (GDUFA) and the
Health Industry Alert August 4, 2017 Senate Passes Long-Awaited FDA User Fee Package Following nearly two years of negotiations and hearings examining the Generic Drug User Fee Amendments (GDUFA) and the
Fast track Approval process- Ethical considerations
 Fast track Approval process- Ethical considerations Sara Ingersoll, MS Program Assistant, Health Sciences Institutional Review Board Rutgers, The State University of New Jersey New Brunswick/Piscataway
Fast track Approval process- Ethical considerations Sara Ingersoll, MS Program Assistant, Health Sciences Institutional Review Board Rutgers, The State University of New Jersey New Brunswick/Piscataway
New Drug Division. Consultant. Chemistry. Biostatistics. Scientific Investigations. Medical Team. Consultant. Project Managers. Inspection.
 PDUFA 4 in Context FDA/Industry Conference Temple University May 2008 RADM Sandra L. Kweder, M.D. Deputy Director, Office of New Drugs Objectives & financial disclaimer Promise of FDAAA PDUFA Good Review
PDUFA 4 in Context FDA/Industry Conference Temple University May 2008 RADM Sandra L. Kweder, M.D. Deputy Director, Office of New Drugs Objectives & financial disclaimer Promise of FDAAA PDUFA Good Review
The Future of Drug Safety
 The Future of Drug Safety IOM Committee on the Assessment of the US Drug Safety System R. Alta Charo Warren P. Knowles Professor of Law & Bioethics University of Wisconsin Charge to the Committee Examine
The Future of Drug Safety IOM Committee on the Assessment of the US Drug Safety System R. Alta Charo Warren P. Knowles Professor of Law & Bioethics University of Wisconsin Charge to the Committee Examine
Role of the FDA and PDUFA in Drug Development
 Role of the FDA and PDUFA in Drug Development Questions to be addressed 1. What is the Food and Drug Administration and why is it important? 2. Why was the Prescription Drug User Fee Act (PDUFA) created?
Role of the FDA and PDUFA in Drug Development Questions to be addressed 1. What is the Food and Drug Administration and why is it important? 2. Why was the Prescription Drug User Fee Act (PDUFA) created?
May 27, Prescription. lives. Since. treatments. The Food. particularly. condition and availability
 May 27, 2014 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: : Drug and Biological Products Approved Under the Accelerated Approval
May 27, 2014 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: : Drug and Biological Products Approved Under the Accelerated Approval
PRESCRIPTION DRUG USER FEE ACT PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2013 through 2017.
 NCCS CANCER POLICY ROUNDTABLE MEETING Madison Hotel, Washington, D.C. October 18-19, 2011 B A C K G R O U N D M A T E R I A L S A N D A R T I C L E S PRESCRIPTION DRUG USER FEE ACT PDUFA Reauthorization
NCCS CANCER POLICY ROUNDTABLE MEETING Madison Hotel, Washington, D.C. October 18-19, 2011 B A C K G R O U N D M A T E R I A L S A N D A R T I C L E S PRESCRIPTION DRUG USER FEE ACT PDUFA Reauthorization
Methods for Expediting Innovative Novel Drugs to Market
 1 Methods for Expediting Innovative Novel Drugs to Market Presented by: Ada Kung, ( 龔曉嘉博士 ) Ph.D. DABT, MBA akung@expedient-solutions.com www.expedient-solutions.com 2 FDA Guidance for Industry Expedited
1 Methods for Expediting Innovative Novel Drugs to Market Presented by: Ada Kung, ( 龔曉嘉博士 ) Ph.D. DABT, MBA akung@expedient-solutions.com www.expedient-solutions.com 2 FDA Guidance for Industry Expedited
Re: Docket No. FDA-2018-D-1895: Indications and Usage Section of Labeling for Human Prescription Drug and Biological Products Content and Format
 September 7, 2018 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2018-D-1895: Indications and Usage Section of Labeling
September 7, 2018 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2018-D-1895: Indications and Usage Section of Labeling
Rare Diseases and CDER: Challenges and Opportunities
 Rare Diseases and CDER: Challenges and Opportunities The Science of Small Clinical Trials November 27 & 28, 2012 Kathryn O Connell, MD PhD Medical Officer, Rare Diseases Program Office of New Drugs, CDER,
Rare Diseases and CDER: Challenges and Opportunities The Science of Small Clinical Trials November 27 & 28, 2012 Kathryn O Connell, MD PhD Medical Officer, Rare Diseases Program Office of New Drugs, CDER,
Regulatory Innovations in Neurological Disorder Therapies, including Spinraza and Exondys 51
 Regulatory Innovations in Neurological Disorder Therapies, including Spinraza and Exondys 51 ASENT 19 th Annual Meeting March 15, 2017 Frank Sasinowski, M.S., M.P.H., J.D Director, Hyman, Phelps & McNamara,
Regulatory Innovations in Neurological Disorder Therapies, including Spinraza and Exondys 51 ASENT 19 th Annual Meeting March 15, 2017 Frank Sasinowski, M.S., M.P.H., J.D Director, Hyman, Phelps & McNamara,
MANUAL: Administrative Policy & Procedure Manual POLICY:
 SJMHS Locations: St. Joseph Mercy Ann Arbor, St. Joseph Mercy Chelsea, St. Joseph Mercy Livingston, St. Mary Mercy Livonia MANUAL: Administrative Policy & Procedure Manual POLICY: Expanded access is the
SJMHS Locations: St. Joseph Mercy Ann Arbor, St. Joseph Mercy Chelsea, St. Joseph Mercy Livingston, St. Mary Mercy Livonia MANUAL: Administrative Policy & Procedure Manual POLICY: Expanded access is the
Implementing the 21 st Century Cures Act: Supporting Orphan Drug Development
 Implementing the 21 st Century Cures Act: Supporting Orphan Drug Development Rare Disease Congressional Caucus Briefing March 2, 2017 Frank Sasinowski, M.S., M.P.H., J.D. Director, Hyman, Phelps & McNamara,
Implementing the 21 st Century Cures Act: Supporting Orphan Drug Development Rare Disease Congressional Caucus Briefing March 2, 2017 Frank Sasinowski, M.S., M.P.H., J.D. Director, Hyman, Phelps & McNamara,
Summary of Provisions in 21 st Century Cures Act (H.R. 6) as passed by full House of Representatives, July 10, 2015
 as passed by full House of Representatives, July 10, 2015") Pediatric-Specific Provisions Summary of Provisions in 21 st Century Cures Act (H.R. 6) as passed by full House of Representatives, July 10, 2015 Requires the NIH to complete a strategic plan, and in the
Pediatric-Specific Provisions Summary of Provisions in 21 st Century Cures Act (H.R. 6) as passed by full House of Representatives, July 10, 2015 Requires the NIH to complete a strategic plan, and in the
FDA from a Former FDAer: Secrets and insights into regulatory review and drug development
 FDA from a Former FDAer: Secrets and insights into regulatory review and drug development Andrew E. Mulberg, MD, FAAP Vice-President, Global Regulatory Affairs; Former Division Deputy, DGIEP, U.S. FDA
FDA from a Former FDAer: Secrets and insights into regulatory review and drug development Andrew E. Mulberg, MD, FAAP Vice-President, Global Regulatory Affairs; Former Division Deputy, DGIEP, U.S. FDA
Food and Drug Administration Reauthorization Act of 2017
 L A W O F F I C E S HYMAN, PHELPS & MCNAMARA, P.C. 7 0 0 T H I R T E E N T H S T R E E T, N.W. S U I T E 1 2 0 0 W A S H I N G T O N, D. C. 2 0 0 0 5-5 9 2 9 ( 2 0 2 ) 7 3 7-5 6 0 0 F A C S I M I L E (
L A W O F F I C E S HYMAN, PHELPS & MCNAMARA, P.C. 7 0 0 T H I R T E E N T H S T R E E T, N.W. S U I T E 1 2 0 0 W A S H I N G T O N, D. C. 2 0 0 0 5-5 9 2 9 ( 2 0 2 ) 7 3 7-5 6 0 0 F A C S I M I L E (
Orphan Drug Topics: 1. Discovery of FDA s Flexibility 2. Evolving Voice of the Patient in Drug Review. February 21, 2018
 Orphan Drug Topics: 1. Discovery of FDA s Flexibility 2. Evolving Voice of the Patient in Drug Review ISCTM 14 th Annual Scientific Meeting February 21, 2018 Frank Sasinowski, M.S., M.P.H., J.D fjs@hpm.com
Orphan Drug Topics: 1. Discovery of FDA s Flexibility 2. Evolving Voice of the Patient in Drug Review ISCTM 14 th Annual Scientific Meeting February 21, 2018 Frank Sasinowski, M.S., M.P.H., J.D fjs@hpm.com
1201 Maryland Avenue SW, Suite 900, Washington, DC ,
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org June 19, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org June 19, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC ,
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org May 31, 2012 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville, MD
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org May 31, 2012 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville, MD
Developing Drugs for Rare Diseases: Patient Advocacy s Perspective. Kristina Bowyer Executive Director, Patient Advocacy
 Developing Drugs for Rare Diseases: Patient Advocacy s Perspective Kristina Bowyer Executive Director, Patient Advocacy February 5, 2018 An Advocacy Perspective Why develop drugs for Rare Disease? What
Developing Drugs for Rare Diseases: Patient Advocacy s Perspective Kristina Bowyer Executive Director, Patient Advocacy February 5, 2018 An Advocacy Perspective Why develop drugs for Rare Disease? What
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited.
. All rights reserved. Further dissemination without express written consent strictly prohibited.") Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited. Special Review Designations & Approval Pathways Special Designations
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited. Special Review Designations & Approval Pathways Special Designations
PFF DISEASE EDUCATION WEBINAR SERIES. Welcome!
 PFF DISEASE EDUCATION WEBINAR SERIES Welcome! PFF DISEASE EDUCATION WEBINAR SERIES David J. Lederer, MD, MS PFF Senior Medical Advisor, Patient Communications Harold Collard, MD PFF Medical Advisory Board
PFF DISEASE EDUCATION WEBINAR SERIES Welcome! PFF DISEASE EDUCATION WEBINAR SERIES David J. Lederer, MD, MS PFF Senior Medical Advisor, Patient Communications Harold Collard, MD PFF Medical Advisory Board
CDER 2016 Actions and 2017 Priorities. Richard Moscicki Deputy Center Director for Science Operations, CDER, FDA
 CDER 2016 Actions and 2017 Priorities Richard Moscicki Deputy Center Director for Science Operations, CDER, FDA Disclosure My comments today are mine and do not necessarily represent the views of the US
CDER 2016 Actions and 2017 Priorities Richard Moscicki Deputy Center Director for Science Operations, CDER, FDA Disclosure My comments today are mine and do not necessarily represent the views of the US
GUIDELINES FOR CONDUCTING CLINICAL TRIALS.
 GUIDELINES FOR CONDUCTING CLINICAL TRIALS. FOREWORD The purpose of Drug Registration is to ensure that a pharmaceutical product has been adequately tested and evaluated for safety, efficacy and quality,
GUIDELINES FOR CONDUCTING CLINICAL TRIALS. FOREWORD The purpose of Drug Registration is to ensure that a pharmaceutical product has been adequately tested and evaluated for safety, efficacy and quality,
A Life-cycle Approach to Dose Finding Studies
 A Life-cycle Approach to Dose Finding Studies Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Center for Drug Evaluation and Research, USFDA This presentation reflects the views of the author
A Life-cycle Approach to Dose Finding Studies Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Center for Drug Evaluation and Research, USFDA This presentation reflects the views of the author
Utilizing Innovative Statistical Methods. Discussion Guide
 Utilizing Innovative Statistical Methods and Trial Designs in Rare Disease Settings Discussion Guide Background Rare diseases are a complex and diverse set of conditions which, when taken together, affect
Utilizing Innovative Statistical Methods and Trial Designs in Rare Disease Settings Discussion Guide Background Rare diseases are a complex and diverse set of conditions which, when taken together, affect
FDA Guidance, Clinical Pharmacology, and Regulatory Science
 Principles of Clinical Pharmacology NIH, April 25, 2013 FDA Guidance, Clinical Pharmacology, and Regulatory Science Carl Peck, MD UCSF Center for Drug Development Science Washington DC and San Francisco
Principles of Clinical Pharmacology NIH, April 25, 2013 FDA Guidance, Clinical Pharmacology, and Regulatory Science Carl Peck, MD UCSF Center for Drug Development Science Washington DC and San Francisco
Volunteering for Clinical Trials
 Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Re: Docket No. FDA 2005-D-0339: Draft Guidance on Drug Safety Information FDA's Communication to the Public
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org May 8, 2012 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org May 8, 2012 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville,
Topics. Safety First Initiative. Drug Safety Communications DRUG SAFETY INITIATIVES AT THE FOOD AND DRUG ADMINISTRATION
 DRUG SAFETY INITIATIVES AT THE FOOD AND DRUG ADMINISTRATION Presentation to the FDA/Industry Conference sponsored by the School of Pharmacy at Temple University May 6, 2008 Topics Safety First initiative
DRUG SAFETY INITIATIVES AT THE FOOD AND DRUG ADMINISTRATION Presentation to the FDA/Industry Conference sponsored by the School of Pharmacy at Temple University May 6, 2008 Topics Safety First initiative
DRAFT GUIDANCE. This guidance document is being distributed for comment purposes only. Document issued on: July 14, 2011
 Draft Guidance for Industry and Food and Drug Administration Staff In Vitro Companion Diagnostic Devices DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Document issued
Draft Guidance for Industry and Food and Drug Administration Staff In Vitro Companion Diagnostic Devices DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Document issued
Investigational New Drug Development Steps for CRCs
 Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
CAMD ANNUAL REGULATORY SCIENCE WORKSHOP OCTOBER 19, 2016 A CASE FOR INNOVATION, PROGRESS, AND OPTIMISM
 CAMD ANNUAL REGULATORY SCIENCE WORKSHOP OCTOBER 19, 2016 A CASE FOR INNOVATION, PROGRESS, AND OPTIMISM ShaAvhrée Buckman-Garner, MD, PhD, FAAP Director, Office of Translational Sciences, CDER, FDA 1 OVERVIEW
CAMD ANNUAL REGULATORY SCIENCE WORKSHOP OCTOBER 19, 2016 A CASE FOR INNOVATION, PROGRESS, AND OPTIMISM ShaAvhrée Buckman-Garner, MD, PhD, FAAP Director, Office of Translational Sciences, CDER, FDA 1 OVERVIEW
Topics Covered. FDA s Role in Expediting the Development of Novel Medical Products. How a Regulatory Agency Comes into Existence 3/5/2018
 FDA s Role in Expediting the Development of Novel Medical Products Peter Marks, M.D., Ph.D. Director Center for Biologics Evaluation and Research Topics Covered Brief history of FDA Expediting product
FDA s Role in Expediting the Development of Novel Medical Products Peter Marks, M.D., Ph.D. Director Center for Biologics Evaluation and Research Topics Covered Brief history of FDA Expediting product
USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH
 Policy P.11 Written By: B. Laurel Elder Created: June 3, 2013 Edited Replaces 10-9-2009 Version P.11.4 USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH A. Procedure Contents this procedure
Policy P.11 Written By: B. Laurel Elder Created: June 3, 2013 Edited Replaces 10-9-2009 Version P.11.4 USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH A. Procedure Contents this procedure
Drug Safety and FDA Revitalization Legislation
 Drug Safety and FDA Revitalization Legislation Dan Kracov August 23, 2007 FDA Regulatory and Compliance Symposium House and Senate FDA Revitalization Bills Senate: S. 1082, Food and Drug Administration
Drug Safety and FDA Revitalization Legislation Dan Kracov August 23, 2007 FDA Regulatory and Compliance Symposium House and Senate FDA Revitalization Bills Senate: S. 1082, Food and Drug Administration
How Have User Fees Affected the FDA?
 HEALTH & MEDICINE How Have User Fees Affected the FDA? The 1992 FDA reform successfully reduced drug review times. Mary K. Olson Yale University D elay in the approval of new drugs has been a policy problem
HEALTH & MEDICINE How Have User Fees Affected the FDA? The 1992 FDA reform successfully reduced drug review times. Mary K. Olson Yale University D elay in the approval of new drugs has been a policy problem
Regulatory Perspective on the Value of Bayesian Methods
 American Course on Drug Development and Regulatory Sciences Substantial Evidence in 21st Century Regulatory Science Borrowing Strength from Accumulating Data April 21, 2016 Regulatory Perspective on the
American Course on Drug Development and Regulatory Sciences Substantial Evidence in 21st Century Regulatory Science Borrowing Strength from Accumulating Data April 21, 2016 Regulatory Perspective on the
FDA REVIEW PRINCIPLES EDMOND ROLAND, MD, FAHA AFSSAPS
 FDA REVIEW PRINCIPLES EDMOND ROLAND, MD, FAHA AFSSAPS FDA REVIEW PRINCIPLES «A well-managed review process for an NDA or BLA begins with interactions between the applicant and the Agency s therapeutic
FDA REVIEW PRINCIPLES EDMOND ROLAND, MD, FAHA AFSSAPS FDA REVIEW PRINCIPLES «A well-managed review process for an NDA or BLA begins with interactions between the applicant and the Agency s therapeutic
Type of Activity. Universal Activity Number L04-P
 Below are the pharmacy designated Universal Activity Numbers (UANs) and type of activity that is applicable for each of the following program offerings Session # Title 104 Impact of Biologics, Vaccines,
Below are the pharmacy designated Universal Activity Numbers (UANs) and type of activity that is applicable for each of the following program offerings Session # Title 104 Impact of Biologics, Vaccines,
Biomarker Utility and Acceptance in Drug Development and Clinical Trials: an FDA Regulatory Perspective
 Biomarker Utility and Acceptance in Drug Development and Clinical Trials: an FDA Regulatory Perspective Chris Leptak, MD/PhD OND Biomarker and Companion Diagnostic Lead CDER/Office of New Drugs, Immediate
Biomarker Utility and Acceptance in Drug Development and Clinical Trials: an FDA Regulatory Perspective Chris Leptak, MD/PhD OND Biomarker and Companion Diagnostic Lead CDER/Office of New Drugs, Immediate
Patient Focused Drug Development 2.0
 Patient Focused Drug Development 2.0 September 15, 2015 Pat Furlong, PPMD ParentProjectMD.org Understanding the Duchenne Muscular Dystrophy Environment 2 About Duchenne muscular dystrophy X-linked, pediatric
Patient Focused Drug Development 2.0 September 15, 2015 Pat Furlong, PPMD ParentProjectMD.org Understanding the Duchenne Muscular Dystrophy Environment 2 About Duchenne muscular dystrophy X-linked, pediatric
The federal food drug and cosmetic Act was originally passed in 1938 and required drugs be proved safe. (click)
") 1 2 The federal food drug and cosmetic Act was originally passed in 1938 and required drugs be proved safe. (click) In 1962, Congress amended the FD&C Act to add a requirement that, to obtain marketing
1 2 The federal food drug and cosmetic Act was originally passed in 1938 and required drugs be proved safe. (click) In 1962, Congress amended the FD&C Act to add a requirement that, to obtain marketing
Center for Drug Evaluation and Research. CDER Small Business and Industry Assistance. (CDER SBIA) and New Drug Review.
 and New Drug Review.") Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
Clinical Trial Methods Course 2017 Trials in Rare Diseases. Erika Augustine, MD, MS University of Rochester Medical Center August 10, 2017
 Clinical Trial Methods Course 2017 Trials in Rare Diseases Erika Augustine, MD, MS University of Rochester Medical Center August 10, 2017 Overview Challenges in studying rare diseases Strategies for trial
Clinical Trial Methods Course 2017 Trials in Rare Diseases Erika Augustine, MD, MS University of Rochester Medical Center August 10, 2017 Overview Challenges in studying rare diseases Strategies for trial
Acting Deputy Commissioner for Operations, U.S. Food and Drug Administration
 Available on FDA website at: http://www.fda.gov/newsevents/testimony/ucm113266.htm Pediatric Clinical Trials for Anti-depressant Drug Products Statement of Janet Woodcock, M.D. Acting Deputy Commissioner
Available on FDA website at: http://www.fda.gov/newsevents/testimony/ucm113266.htm Pediatric Clinical Trials for Anti-depressant Drug Products Statement of Janet Woodcock, M.D. Acting Deputy Commissioner
2015 Policy Landscape: A Biomedical R&D Planning Guide
 2015 Policy Landscape: A Biomedical R&D Planning Guide A FasterCures Webinar Jan. 29, 2015 Action FasterCures is an action tank driven by a singular goal to save lives by speeding up and improving the
2015 Policy Landscape: A Biomedical R&D Planning Guide A FasterCures Webinar Jan. 29, 2015 Action FasterCures is an action tank driven by a singular goal to save lives by speeding up and improving the
Pharmacovigilance and Drug Safety Compliance Update GAP Analysis: How We Get to Where We Want to Be
 Pharmacovigilance and Drug Safety Compliance Update GAP Analysis: How We Get to Where We Want to Be John P. Ford Sidley Austin LLP Core PV Concepts Risk identification, collection of information Risk assessment,
Pharmacovigilance and Drug Safety Compliance Update GAP Analysis: How We Get to Where We Want to Be John P. Ford Sidley Austin LLP Core PV Concepts Risk identification, collection of information Risk assessment,
Guidance for Industry
 Guidance for Industry Submission of Abbreviated Reports and Synopses in Support of Marketing Applications U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation
Guidance for Industry Submission of Abbreviated Reports and Synopses in Support of Marketing Applications U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation
Prescription Drug User Fee Act Patient-Focused Drug Development; Request for Comments
 This document is scheduled to be published in the Federal Register on 10/08/2014 and available online at http://federalregister.gov/a/2014-23965, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 10/08/2014 and available online at http://federalregister.gov/a/2014-23965, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
CADTH COMMON DRUG REVIEW. Procedure for the CADTH Common Drug Review
 CADTH COMMON DRUG REVIEW Procedure for the CADTH Common Drug Review AUGUST 2014 RECORD OF UPDATES TO THE PROCEDURE FOR THE CADTH COMMON DRUG REVIEW Update Original June 2003 1 January 2005 2 July 2005
CADTH COMMON DRUG REVIEW Procedure for the CADTH Common Drug Review AUGUST 2014 RECORD OF UPDATES TO THE PROCEDURE FOR THE CADTH COMMON DRUG REVIEW Update Original June 2003 1 January 2005 2 July 2005
Expanded Access. to Investigational Drugs & Biologics. for Treatment Use
 SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
Regulatory Issues and Strategies: U.S. FDA
 Regulatory Issues and Strategies: U.S. FDA Aliza Thompson, MD Division of Cardiovascular and Renal Products Center for Drug Evaluation and Research Food and Drug Administration United States Disclaimers
Regulatory Issues and Strategies: U.S. FDA Aliza Thompson, MD Division of Cardiovascular and Renal Products Center for Drug Evaluation and Research Food and Drug Administration United States Disclaimers
Unpacking Expanded Access to Investigational Drugs and Biologics
 Unpacking Expanded Access to Investigational Drugs and Biologics When All Else Fails Richard Klein Office of Health and Constituent Affairs Food and Drug Administration September 13, 2016 FDA Drug Approval
Unpacking Expanded Access to Investigational Drugs and Biologics When All Else Fails Richard Klein Office of Health and Constituent Affairs Food and Drug Administration September 13, 2016 FDA Drug Approval
Pediatric Exclusivity and Drug Development Requirements in the Overall Pediatric Population. Prepared by Beckloff Associates, Inc.
 and Drug Development Requirements in the Overall Pediatric Population 1 INTRODUCTION AND OVERVIEW Prepared by Beckloff Associates, Inc. Although children suffer from many of the same diseases as adults
and Drug Development Requirements in the Overall Pediatric Population 1 INTRODUCTION AND OVERVIEW Prepared by Beckloff Associates, Inc. Although children suffer from many of the same diseases as adults
Accelerated Approvals
 Accelerated Approvals An Industry Perspective Kumeshnie Padayachee University of Pretoria 09 September 2015 Table of Contents ZA Expedite Review Process Fast Track Overview of FDA Expedited Pathways: Focus
Accelerated Approvals An Industry Perspective Kumeshnie Padayachee University of Pretoria 09 September 2015 Table of Contents ZA Expedite Review Process Fast Track Overview of FDA Expedited Pathways: Focus
Clinical Trials A Closer Look
 The Food and Drug Administration (FDA) is the main consumer watchdog for numerous products: Drugs and biologics (prescription and over-the counter) Food Medical devices Animal feed and drugs Cosmetics
The Food and Drug Administration (FDA) is the main consumer watchdog for numerous products: Drugs and biologics (prescription and over-the counter) Food Medical devices Animal feed and drugs Cosmetics
Industry Perspective: The Challenges and Benefits in using Expedited Regulatory Pathways
 Industry Perspective: The Challenges and Benefits in using Expedited Regulatory Pathways Alan Poirier, Pfizer Inc. U.S. Regulatory Policy and Global Intelligence Worldwide Safety and Regulatory American
Industry Perspective: The Challenges and Benefits in using Expedited Regulatory Pathways Alan Poirier, Pfizer Inc. U.S. Regulatory Policy and Global Intelligence Worldwide Safety and Regulatory American
IND Development Process Published on ResearchGo UCLA (
 IND Development Process IND Development Overview The Pre-IND Process The IND Study Protocol Preparing the Initial IND Submission Filing the IND Maintaining the IND IND Templates, Education and Useful Links
IND Development Process IND Development Overview The Pre-IND Process The IND Study Protocol Preparing the Initial IND Submission Filing the IND Maintaining the IND IND Templates, Education and Useful Links
FDA approval of emergency expanded access use may be requested by telephone, facsimile, or other means of electronic communications.
 Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of an Individual Patient with an Investigational Drug or a REMS-restricted, Approved Drug Guidelines:
Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of an Individual Patient with an Investigational Drug or a REMS-restricted, Approved Drug Guidelines:
February 15, Re: Docket No. FDA-2017-D-6159: Expedited Programs for Regenerative Medicine Therapies for Serious Conditions
 February 15, 2018 Dockets Management Staff (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2017-D-6159: Expedited Programs for Regenerative Medicine
February 15, 2018 Dockets Management Staff (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2017-D-6159: Expedited Programs for Regenerative Medicine
Bipartisan Policy Center, Top Medical Innovation Priorities
 Bipartisan Policy Center, Top Medical Priorities FDA: Advancing Medical is a Bipartisan Policy Center initiative led by former Senator Bill Frist, MD, former Congressman Bart Gordon, and BPC Health Initiative
Bipartisan Policy Center, Top Medical Priorities FDA: Advancing Medical is a Bipartisan Policy Center initiative led by former Senator Bill Frist, MD, former Congressman Bart Gordon, and BPC Health Initiative
FDA EMA/FDA/MHLW-PMDA
 FDA Incentives John D. Milto, M.D. Medical Officer Office of Orphan Products Development (OOPD), FDA EMA/FDA/MHLW-PMDA Orphan Medicinal Product Workshop London March 10, 2014 Orphan Product Related Incentives
FDA Incentives John D. Milto, M.D. Medical Officer Office of Orphan Products Development (OOPD), FDA EMA/FDA/MHLW-PMDA Orphan Medicinal Product Workshop London March 10, 2014 Orphan Product Related Incentives
Human Subjects Protection Program Plan
 December 1, 2016 HRP-101 12/1/16 2 of 12 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research... 3 Human Research:... 4 Human Subject as Defined
December 1, 2016 HRP-101 12/1/16 2 of 12 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research... 3 Human Research:... 4 Human Subject as Defined
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
Expanded Access to Investigational Imaging Drugs
 Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
Clinical trial applications in the EU and US
 Clinical trial applications in the EU and US Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Symposium Strasbourg 17-18 March 2005 Overview of the presentation
Clinical trial applications in the EU and US Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Symposium Strasbourg 17-18 March 2005 Overview of the presentation
Streamlining IRB Procedures for Expanded Access
 Streamlining IRB Procedures for Expanded Access Marjorie A. Speers, Ph.D. Executive Director, WCG Foundation Richard Klein Director, FDA Patient Liaison Program Office of Health and Constituent Affairs
Streamlining IRB Procedures for Expanded Access Marjorie A. Speers, Ph.D. Executive Director, WCG Foundation Richard Klein Director, FDA Patient Liaison Program Office of Health and Constituent Affairs
How to put together an IND application
 How to put together an IND application Judit Milstein, Chief, Project Management Staff Judit.milstein@fda.hhs.gov Eithu Lwin, Regulatory Health Project Manager Eithu.Lwin@fda.hhs.gov Division of Transplant
How to put together an IND application Judit Milstein, Chief, Project Management Staff Judit.milstein@fda.hhs.gov Eithu Lwin, Regulatory Health Project Manager Eithu.Lwin@fda.hhs.gov Division of Transplant
Advocate Health Care Network. Human Research Protection Program. Plan
 Advocate Health Care Network Human Research Protection Program Plan Revised October 19, 2014 Page 1 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Clinical Trial... 3 Engaged in Human Research...
Advocate Health Care Network Human Research Protection Program Plan Revised October 19, 2014 Page 1 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Clinical Trial... 3 Engaged in Human Research...
External Defibrillator Improvement Initiative
 External Defibrillator Improvement Initiative November 2010 Center for Devices and Radiological Health U.S. Food and Drug Administration External Defibrillator Improvement Initiative Table of Contents
External Defibrillator Improvement Initiative November 2010 Center for Devices and Radiological Health U.S. Food and Drug Administration External Defibrillator Improvement Initiative Table of Contents
Safety Assessment for Investigational New Drug Safety Reporting; Public Workshop
 This document is scheduled to be published in the Federal Register on 11/27/2017 and available online at https://federalregister.gov/d/2017-25454, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 11/27/2017 and available online at https://federalregister.gov/d/2017-25454, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
Regulatory Issues Affecting Commercialization Michael Werner Executive Director, Alliance for Regenerative Medicine Partner, Holland & Knight, LLP
 Regulatory Issues Affecting Commercialization Michael Werner Executive Director, Alliance for Regenerative Medicine Partner, Holland & Knight, LLP Terrapin Conference, Sept. 2011 Alliance for Regenerative
Regulatory Issues Affecting Commercialization Michael Werner Executive Director, Alliance for Regenerative Medicine Partner, Holland & Knight, LLP Terrapin Conference, Sept. 2011 Alliance for Regenerative
What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials
 Vol. 14, No. 10, October 2018 Happy Trials to You What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials While multiple, concurrently accruing patient cohorts in first-in-human (FIH)
Vol. 14, No. 10, October 2018 Happy Trials to You What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials While multiple, concurrently accruing patient cohorts in first-in-human (FIH)
Maximize the Collection of Real- World Data in Expanded Access Programs
 Maximize the Collection of Real- World Data in Expanded Access Programs Jose Ricardo Perez, MD. Executive Medical Director Novartis Oncology East Hanover, NJ Disclaimer: The opinions expressed in this
Maximize the Collection of Real- World Data in Expanded Access Programs Jose Ricardo Perez, MD. Executive Medical Director Novartis Oncology East Hanover, NJ Disclaimer: The opinions expressed in this
DEFINING BEST PRACTICE FOR ADVERSE EVENT REPORTING WITH INDUSTRY-SPONSORED CLINICAL TRIAL MANUSCRIPTS
 11TH ANNUAL MEETING OF ISMPP DEFINING BEST PRACTICE FOR ADVERSE EVENT REPORTING WITH INDUSTRY-SPONSORED CLINICAL TRIAL MANUSCRIPTS April 28, 2015 Hyatt Regency Crystal City Arlington, VA, USA 11TH ANNUAL
11TH ANNUAL MEETING OF ISMPP DEFINING BEST PRACTICE FOR ADVERSE EVENT REPORTING WITH INDUSTRY-SPONSORED CLINICAL TRIAL MANUSCRIPTS April 28, 2015 Hyatt Regency Crystal City Arlington, VA, USA 11TH ANNUAL
1201 Maryland Avenue SW, Suite 900, Washington, DC ,
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org June 6, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville, MD
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org June 6, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville, MD
Re: Docket No. FDA-2014-D-1461: Rare Pediatric Disease Priority Review Vouchers
 February 13, 2015 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2014-D-1461: Rare Pediatric Disease Priority Review
February 13, 2015 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2014-D-1461: Rare Pediatric Disease Priority Review
Expanded Access and the Individual Patient IND
 Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry
 Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions
Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions
Re: Docket No. FDA-2015-D-4562: Draft Guidance Safety Assessment for IND Safety Reporting
 February 16, 2016 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2015-D-4562: Draft Guidance Safety Assessment for IND
February 16, 2016 Dockets Management Branch (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Docket No. FDA-2015-D-4562: Draft Guidance Safety Assessment for IND
Auditor General of Canada to the House of Commons
 2011 Report of the Auditor General of Canada to the House of Commons FALL Chapter 4 Regulating Pharmaceutical Drugs Health Canada Office of the Auditor General of Canada The Fall 2011 Report of the Auditor
2011 Report of the Auditor General of Canada to the House of Commons FALL Chapter 4 Regulating Pharmaceutical Drugs Health Canada Office of the Auditor General of Canada The Fall 2011 Report of the Auditor
Insights into the Rare Disease Drug Approval Landscape: Trends and Current Development
 Insights into the Rare Disease Drug Approval Landscape: Trends and Current Development Mike Lanthier Operations Research Analyst FDA Office of the Commissioner NORD Rare Diseases & Orphan Products Breakthrough
Insights into the Rare Disease Drug Approval Landscape: Trends and Current Development Mike Lanthier Operations Research Analyst FDA Office of the Commissioner NORD Rare Diseases & Orphan Products Breakthrough