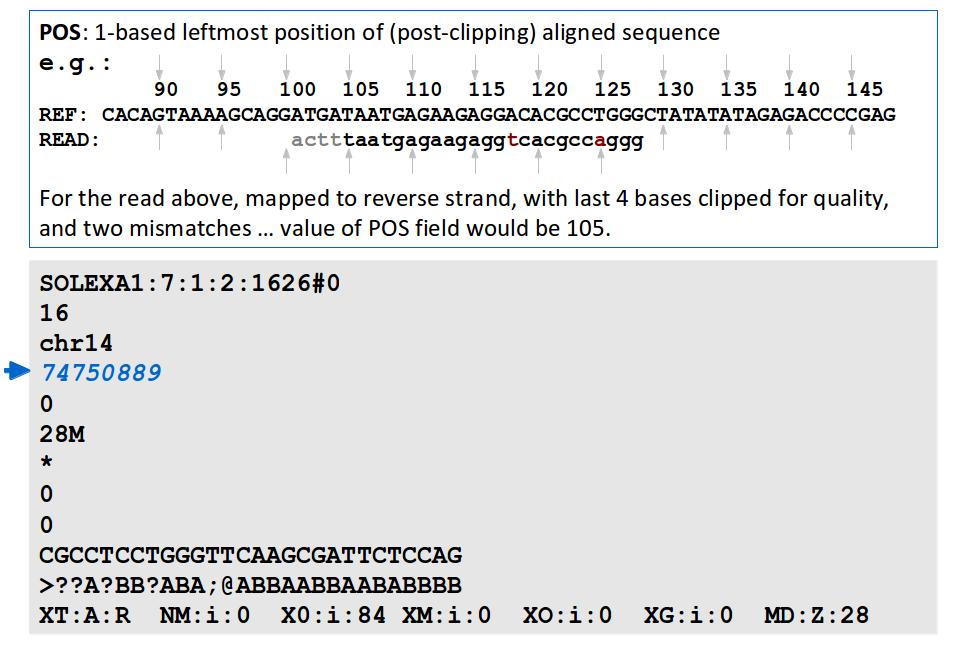
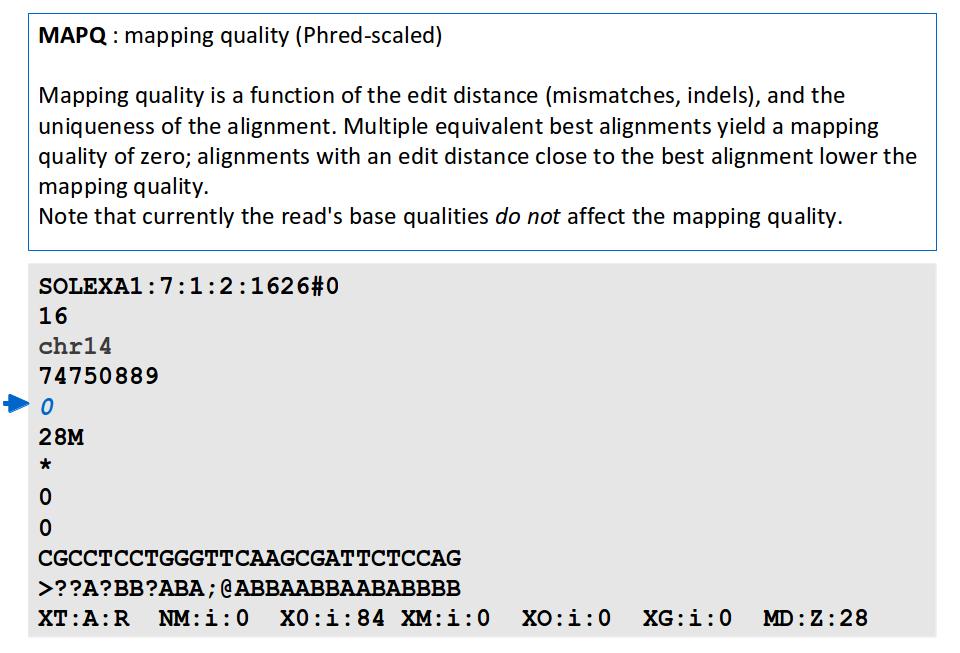
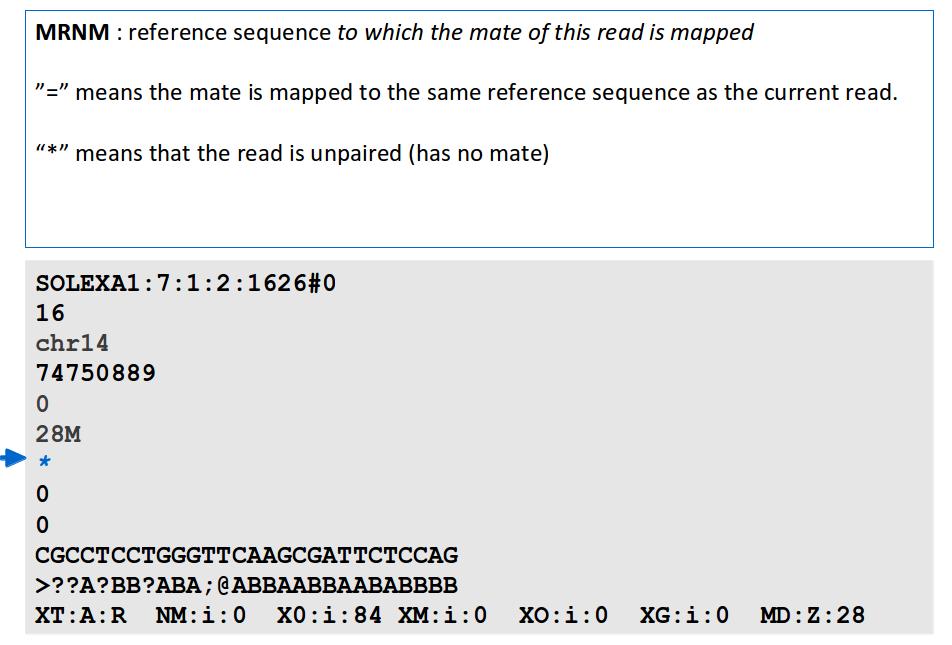
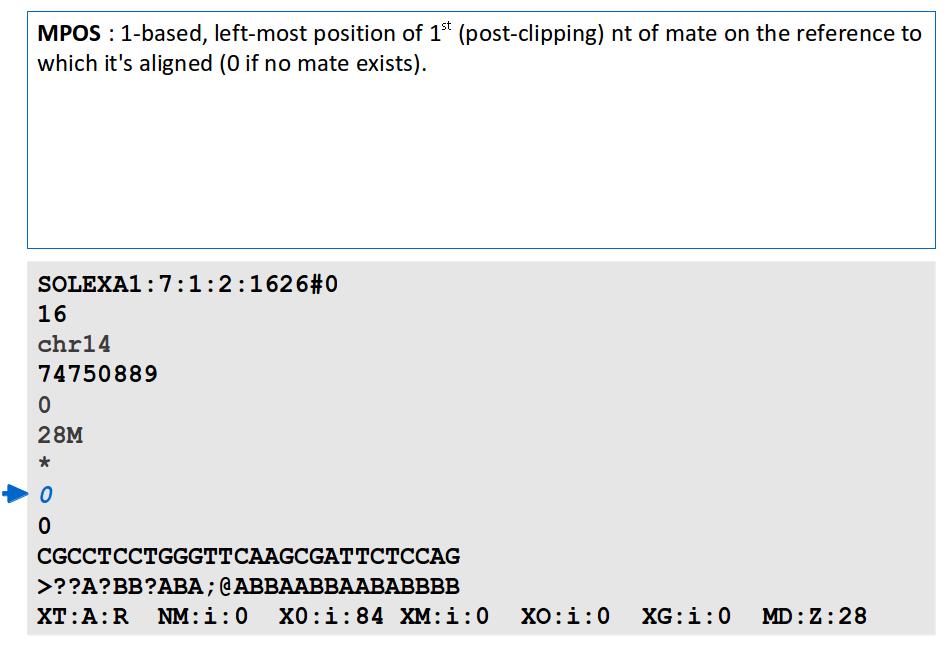
Alignment. J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
|
|
|
- Emory Boyd
- 6 years ago
- Views:
Transcription
1 Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
2 From reads to molecules

3 Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG CAATAAAAGTGCCGTATCATGCTGGTGTTACAATCGCCGCA CGTATCATGCTGGTGTTACAATCGCCGCATGACATGATCAATGG TGTCTGCTCAATAAAAGTGCCGTATCATGCTGGTGTTACAATC ATCGTCGGGTGTCTGCTCAATAAAAGTGCCGTATCATG--GGTGTTATAA CTCAATAAGAGTGCCGTATCATG--GGTGTTATAATCGCCGCA GTTATAATCGCCGCATGACATGATCAATGG To measure variation.
4 Why align?
5 Why align?

6 Short Read Aligners: choices... Fall '12 - Apr '13:... now Gbp / day!* *
7 Burrows-Wheeler Aligners Burrows-Wheeler Transform used in bzip2 file compression tool; FM-index (Ferragina & Manzini) allow efficient finding of substring matches within compressed text algorithm is sub-linear with respect to time and storage space required for a certain set of input data (reference 'ome, essentially). Reduced memory footprint, faster execution.
8 BWA BWA is fast, and can do gapped alignments. When run without seeding, it will find all hits within a given edit distance. Long read aligner is also fast, and can perform well for 454, Ion Torrent, Sanger, and PacBio reads. BWA is actively developed and has a strong user / developer community. bio-bwa.sourceforge.net Short reads under 200 bp Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25: [PMID: ] Long reads over 200 bp chimeric alignments built-in Li H. and Durbin R. (2010) Fast and accurate long read alignment with Burrows-Wheeler Transform. Bioinformatics, 26: [PMID: ] don't forget to join the mailing groups!
9 Bowtie Bowtie (now Bowtie 2) is probably faster than BWA for some types of alignment, but it may not find the best alignments (see discussions on sensitivity, accuracy on SeqAnswers.com). Bowtie is part of a suite of tools (Bowtie, Tophat, Cufflinks, CummeRbund) that address RNAseq experiments. Langmead B., Trapnell C., Pop M., and Salzberg S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Genome Biology 10:R25 [PMID: ] don't forget to join the mailing groups!
10 Alignment concepts / parameters Paired-End reads Mate-Paired reads
11 Alignment concepts / parameters 454 "Paired-End" reads Single End Construct

12 Alignment concepts / parameters

13 Alignment concepts / parameters

14 Alignment concepts / parameters
15 Alignment concepts / parameters
16 File Format: SAM / BAM / CRAM! NEW - deprecated! - SAMtools 1.0 and up Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, [PMID: ] SAM specification (currently v1, renumbered 1 is after old v1.4) samtools man page example workflow(s) mailing list!

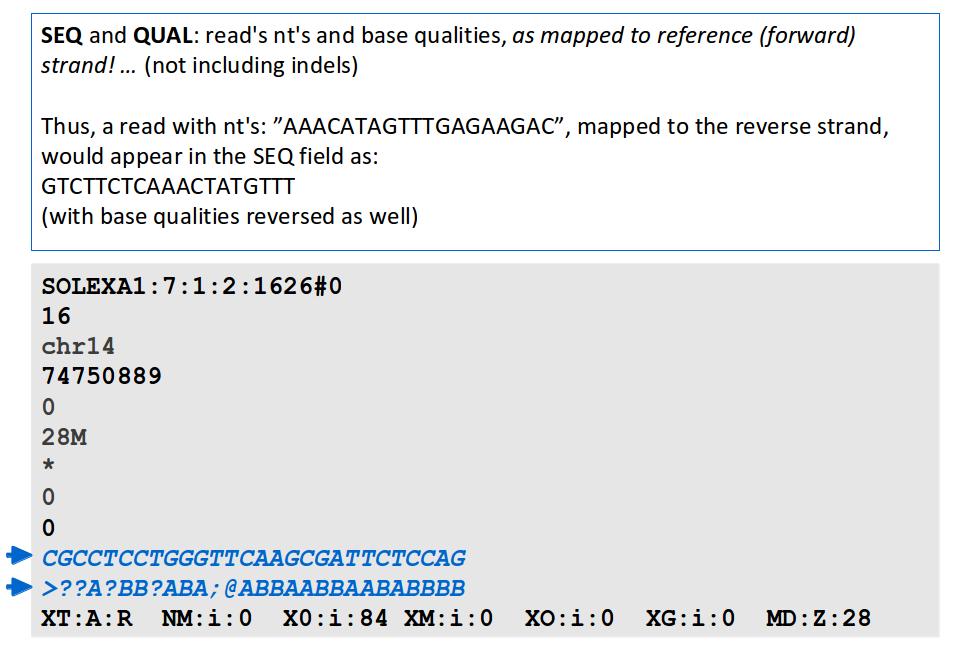
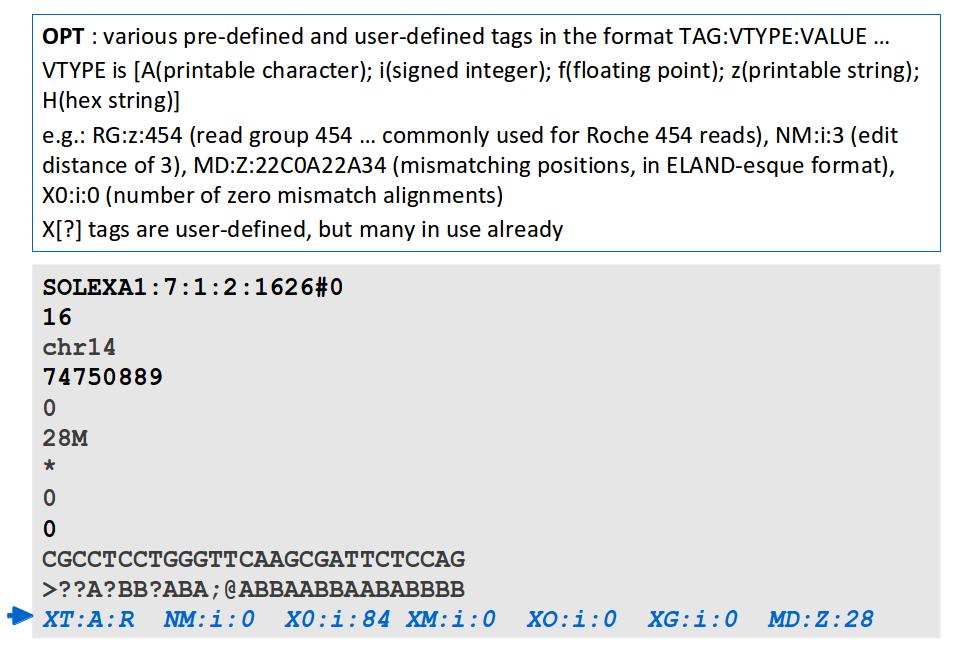
17 File Format: SAM
18 File Format: SAM SAM Format Specification v1.4-r985 7,8 - formerly MRNM, MPOS (mate reference name, mate position) 9 - formerly ISIZE ("insert" size)

19
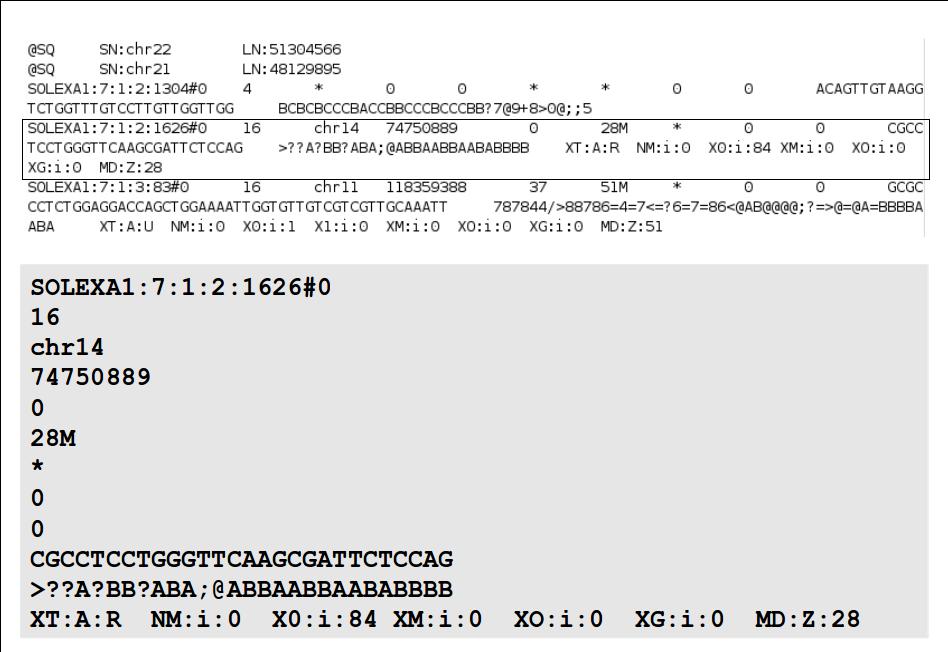
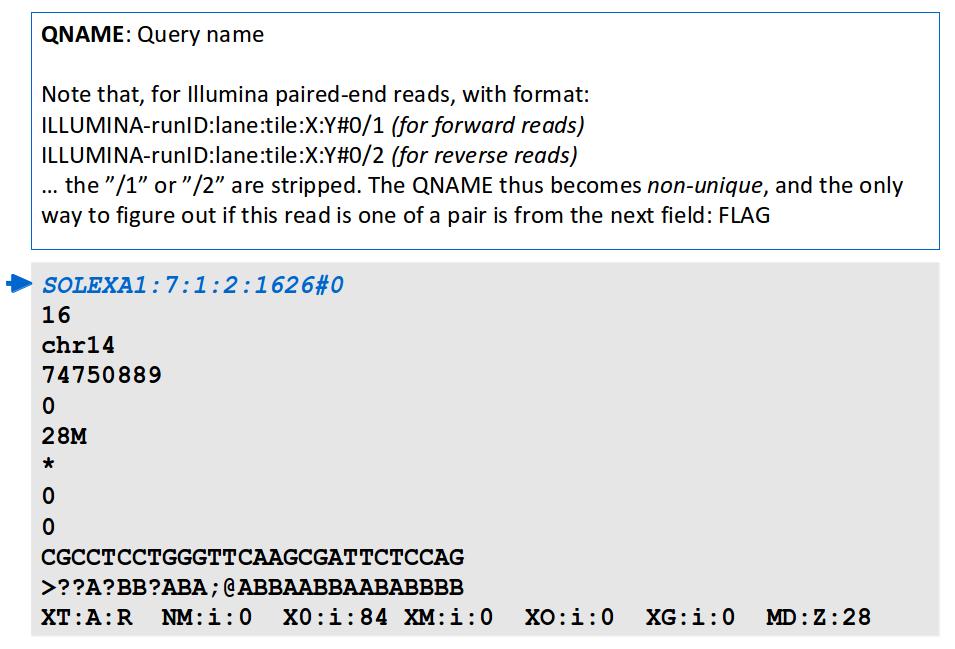
20
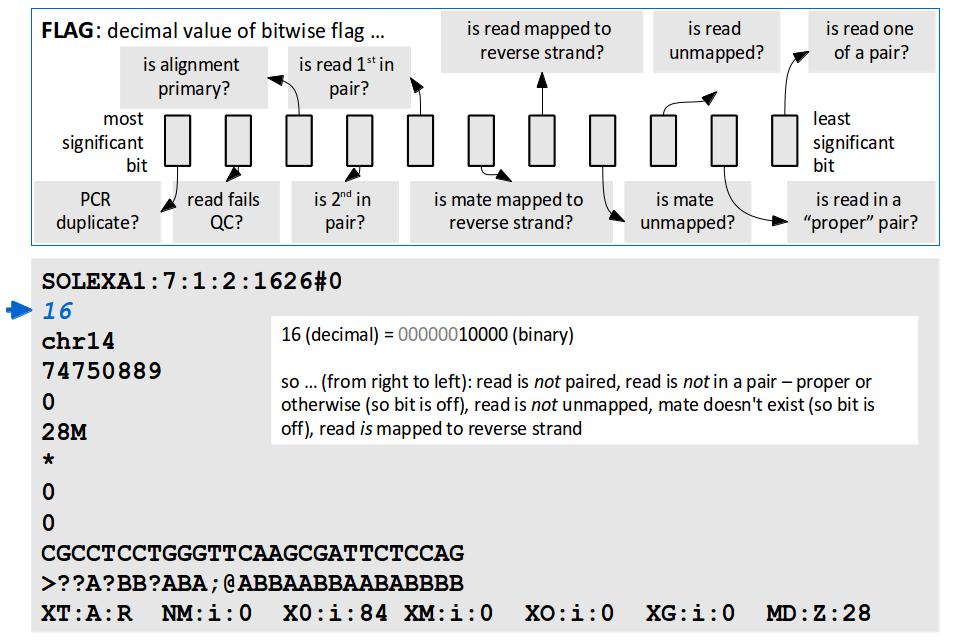
21
22
23

24
25
26
27

28
29
30
31
32
33
34 File Format: SAM google "Heng Li slides" - Challenges and Solutions in the Analysis of Next Generation Sequencing Data (2010)
35 File Format: BAM BAMs are compressed SAMs (so, binary, not human-readable text don't look directly at them!). They can be indexed to allow rapid extraction of information, so alignment viewers do not need to uncompress the whole BAM file in order to look at information for a particular read or coordinate range, somewhere in the file. Indexing your BAM file, mycoolbamfile.bam, will create an index file, mycoolbamfile.bam.bai, which is needed (in addition to the BAM file) by viewers and other downstream tools. An occasional downstream tool will require an index called mycoolbamfile.bai (notice that the.bai replaces the.bam, instead of being appended after it).
36 File Format: CRAM Available as of SAMtools 1.0, and is a binary format like BAM. Uses data-specific compression tools (i.e. compressing letters is different than compressing numbers), specifically reference-based compression (e.g. for aligned reads, only mis-matching bases need to be stored). Also can employ lossy compression of base qualities, which appears to have a negligible effect on, say, variant calling (see Illumina white paper). Indexing your CRAM file, mycoolbamfile.cram, will create an index file, mycoolbamfile.cram.crai, which is needed (in addition to the CRAM file) by viewers and other downstream tools. This is a very recent development, so it may be a while before tools are CRAM-capable.
, IGB, GenomeView, SAMscope.")
37 Alignment Viewers IGV (Integrated Genomics Viewer) BAMview, tview (in SAMtools), IGB, GenomeView, SAMscope... UCSC Genome Browser, GBrowse

38 IGV red box indicates region of reference in view below coverage track: read coverage depth plot read alignments: (various view styles - squished shown here) read positions, orientations, pairing, sequence that disagrees with reference highlighted, improper pairs highlighted, etc. annotation tracks (GTF, BED, etc.)
 improper pairs (mate aligns far away, in wrong orientation, or on another chromosome) reference")
39 IGV colored bases where they disagree with reference (substitution, indel, etc.) improper pairs (mate aligns far away, in wrong orientation, or on another chromosome) reference sequence, reading frames, etc.
40
 is now the standard format for variant reporting. http://vcftools.sourceforge.net/specs.html.")
41 Variant Calling - VCF format One main application of read alignment. A.k.a. "resequencing", SNP / indel discovery. VCF (variant call format) is now the standard format for variant reporting. VCF poster
42 Variant Call Format ##fileformat=vcfv4.1 ##filedate= ##source=freebayes v gfbf46fc-dirty ##reference=../results/8/8.fa ##phasing=none ##commandline="../tools/freebayes/bin/freebayes -f../results/8/8.fa --min-alternate-fraction minmapping-quality 20 --min-base-quality 20 --ploidy 1 --pooled-continuous --use-best-n-alleles 4 --usemapping-quality --min-alternate-fraction min-alternate-count 1../results/8/8.bam" ##INFO=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description="Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description="Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description="Sum of quality of the alternate observations"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB1 26. TGTTACGCG GCTTTTGC,TGTTTCTAC AO=1,2;RO=0;TYPE=complex, complex GT:DP:RO:QR:AO:QA:GL 2:3:0:0:1,2:31,70:-4.46,-1.65,0 8_PB1 38. TCA ACG,TA,AGA AO=1,1,1;RO=3;TYPE=complex,del,mnp GT:DP:RO:QR:AO:QA:GL 2:6:3:101:1,1,1:31,37,34:0,-4.556,-4.004, _PB1 42. G A e-14. AO=8;RO=128;TYPE=snp GT:DP:RO:QR:AO:QA: GL
43 Variant Call Format ##fileformat=vcfv4.1 ##filedate= ##source=freebayes v gfbf46fc-dirty ##reference=../results/8/8.fa ##phasing=none ##commandline="../tools/freebayes/bin/freebayes -f../results/8/8.fa --min-alternate-fraction minmapping-quality 20 --min-base-quality 20 --ploidy 1 --pooled-continuous --use-best-n-alleles 4 --usemapping-quality --min-alternate-fraction min-alternate-count 1../results/8/8.bam" ##INFO=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description="Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description="Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description="Sum of quality of the alternate observations"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB1 26. TGTTACGCG GCTTTTGC,TGTTTCTAC AO=1,2;RO=0;TYPE=complex, complex GT:DP:RO:QR:AO:QA:GL 2:3:0:0:1,2:31,70:-4.46,-1.65,0 8_PB1 38. TCA ACG,TA,AGA AO=1,1,1;RO=3;TYPE=complex,del,mnp GT:DP:RO:QR:AO:QA:GL 2:6:3:101:1,1,1:31,37,34:0,-4.556,-4.004, _PB1 42. G A e-14. AO=8;RO=128;TYPE=snp GT:DP:RO:QR:AO:QA: GL
44 Variant Call Format #CHROM POS ID REF 8_PB A 170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = ALT G QUAL FILTER INFO FORMAT 8 AO=149;RO=21;TYPE=snp GT:DP:RO:QR:AO:QA:GL 1: FILTER =. INFO = AO=149;RO=21;TYPE=snp FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0

45 Variant Call Format

46 Variant Call Format

47 Variant Call Format
48 Variant Call Format #CHROM POS ID REF 8_PB A 170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = ALT G QUAL FILTER INFO FORMAT 8 AO=149;RO=21;TYPE=snp GT:DP:RO:QR:AO:QA:GL 1: ##FORMAT=<ID=DP,Number=1,Type=Integer, Description="Read Depth"> FILTER =. INFO = AO=149;RO=21;TYPE=snp FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0
49 Variant Call Format ##INFO=<ID=RO,Number=1,Type=Integer,Description=" Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description=" Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex.">
50 Variant Call Format ##FORMAT=<ID=GT,Number=1,Type=String,Description=" Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description=" Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description=" Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description=" Read Depth">
51 Variant Call Format ##FORMAT=<ID=RO,Number=1,Type=Integer,Description=" Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description=" Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description=" Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description=" Sum of quality of the alternate observations">
52 Variant Effect Prediction snpeff Variant Effect Predictor (EMBL) SIFT
53 VCF after Effect Prediction #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB A G AO=149;RO=21;TYPE=snp;EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1) GT:DP:RO:QR:AO:QA:GL 1:170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = FILTER =. INFO = AO=149;RO=21;TYPE=snp;EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1) FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0
54 VCF after Effect Prediction ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##INFO=<ID=EFF,Number=.,Type=String,Description="Predicted effects for this variant.format: 'Effect ( Effect_Impact Functional_Class Codon_Change Amino_Acid_change Amino_Acid_length Gene_Name Transcript_BioType Gene_Coding Transcript_ID Exon GenotypeNum [ ERRORS WARNINGS ] )' "> INFO = AO=149;RO=21;TYPE=snp; EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1)
Alignment & Variant Discovery. J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014
 Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Short Read Alignment. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 Introduction to Short Read Alignment UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to Short Read Alignment UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Variant Finding. UCD Genome Center Bioinformatics Core Wednesday 30 August 2016
 Variant Finding UCD Genome Center Bioinformatics Core Wednesday 30 August 2016 Types of Variants Adapted from Alkan et al, Nature Reviews Genetics 2011 Why Look For Variants? Genotyping Correlation with
Variant Finding UCD Genome Center Bioinformatics Core Wednesday 30 August 2016 Types of Variants Adapted from Alkan et al, Nature Reviews Genetics 2011 Why Look For Variants? Genotyping Correlation with
C3BI. VARIANTS CALLING November Pierre Lechat Stéphane Descorps-Declère
 C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
C3BI VARIANTS CALLING November 2016 Pierre Lechat Stéphane Descorps-Declère General Workflow (GATK) software websites software bwa picard samtools GATK IGV tablet vcftools website http://bio-bwa.sourceforge.net/
RNAseq and Variant discovery
 RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
RNAseq and Variant discovery RNAseq Gene discovery Gene valida5on training gene predic5on programs Gene expression studies Paris japonica Gene discovery Understanding physiological processes Dissec5ng
Variant Analysis. CB2-201 Computational Biology and Bioinformatics! February 27, Emidio Capriotti!
 Variant Analysis CB2-201 Computational Biology and Bioinformatics February 27, 2015 Emidio Capriotti http://biofold.org/emidio Division of Informatics Department of Pathology Variant Call Format The final
Variant Analysis CB2-201 Computational Biology and Bioinformatics February 27, 2015 Emidio Capriotti http://biofold.org/emidio Division of Informatics Department of Pathology Variant Call Format The final
Bioinformatics in next generation sequencing projects
 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013 Standard sequence library generation Illumina
Bioinformatics small variants Data Analysis. Guidelines. genomescan.nl
 Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Mapping Next Generation Sequence Reads. Bingbing Yuan Dec. 2, 2010
 Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Mapping Next Generation Sequence Reads Bingbing Yuan Dec. 2, 2010 1 What happen if reads are not mapped properly? Some data won t be used, thus fewer reads would be aligned. Reads are mapped to the wrong
Genome 373: Mapping Short Sequence Reads II. Doug Fowler
 Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Genome 373: Mapping Short Sequence Reads II Doug Fowler The final Will be in this room on June 6 th at 8:30a Will be focused on the second half of the course, but will include material from the first half
Introduc)on to Genomics
on to Genomics") Introduc)on to Genomics Libor Mořkovský, Václav Janoušek, Anastassiya Zidkova, Anna Přistoupilová, Filip Sedlák h1p://ngs-course.readthedocs.org/en/praha-january-2017/ Genome The genome is the gene,c material
Introduc)on to Genomics Libor Mořkovský, Václav Janoušek, Anastassiya Zidkova, Anna Přistoupilová, Filip Sedlák h1p://ngs-course.readthedocs.org/en/praha-january-2017/ Genome The genome is the gene,c material
Exome Sequencing and Disease Gene Search
 Exome Sequencing and Disease Gene Search Erzurumluoglu AM, Rodriguez S, Shihab HA, Baird D, Richardson TG, Day IN, Gaunt TR. Identifying Highly Penetrant Disease Causal Mutations Using Next Generation
Exome Sequencing and Disease Gene Search Erzurumluoglu AM, Rodriguez S, Shihab HA, Baird D, Richardson TG, Day IN, Gaunt TR. Identifying Highly Penetrant Disease Causal Mutations Using Next Generation
Prioritization: from vcf to finding the causative gene
 Prioritization: from vcf to finding the causative gene vcf file making sense A vcf file from an exome sequencing project may easily contain 40-50 thousand variants. In order to optimize the search for
Prioritization: from vcf to finding the causative gene vcf file making sense A vcf file from an exome sequencing project may easily contain 40-50 thousand variants. In order to optimize the search for
Read Mapping and Variant Calling. Johannes Starlinger
 Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
About Strand NGS. Strand Genomics, Inc All rights reserved.
 About Strand NGS Strand NGS-formerly known as Avadis NGS, is an integrated platform that provides analysis, management and visualization tools for next-generation sequencing data. It supports extensive
About Strand NGS Strand NGS-formerly known as Avadis NGS, is an integrated platform that provides analysis, management and visualization tools for next-generation sequencing data. It supports extensive
Alignment methods. Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics
 Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Variant Detection in Next Generation Sequencing Data. John Osborne Sept 14, 2012
 + Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
+ Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
Analysis Datasheet Exosome RNA-seq Analysis
 Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
Analysis Datasheet Exosome RNA-seq Analysis Overview RNA-seq is a high-throughput sequencing technology that provides a genome-wide assessment of the RNA content of an organism, tissue, or cell. Small
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Variant calling in NGS experiments
 Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
Data Basics. Josef K Vogt Slides by: Simon Rasmussen Next Generation Sequencing Analysis
 Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Illumina (Solexa) Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme
 Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme") Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
From raw reads to variants
 From raw reads to variants Sebastian DiLorenzo Sebastian.DiLorenzo@NBIS.se Talk Overview Concepts Reference genome Variants Paired-end data NGS Workflow Quality control & Trimming Alignment Local realignment
From raw reads to variants Sebastian DiLorenzo Sebastian.DiLorenzo@NBIS.se Talk Overview Concepts Reference genome Variants Paired-end data NGS Workflow Quality control & Trimming Alignment Local realignment
NGS in Pathology Webinar
 NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
NGS in Pathology Webinar NGS Data Analysis March 10 2016 1 Topics for today s presentation 2 Introduction Next Generation Sequencing (NGS) is becoming a common and versatile tool for biological and medical
NEXT GENERATION SEQUENCING. Farhat Habib
 NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
Genomic DNA ASSEMBLY BY REMAPPING. Course overview
 ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
Introduc)on to Bioinforma)cs of next- genera)on sequencing. Sequence acquisi)on and processing; genome mapping and alignment manipula)on
on to Bioinforma)cs of next- genera)on sequencing. Sequence acquisi)on and processing; genome mapping and alignment manipula)on") Introduc)on to Bioinforma)cs of next- genera)on sequencing Sequence acquisi)on and processing; genome mapping and alignment manipula)on Ruslan Sadreyev Director of Bioinformatics Department of Molecular
Introduc)on to Bioinforma)cs of next- genera)on sequencing Sequence acquisi)on and processing; genome mapping and alignment manipula)on Ruslan Sadreyev Director of Bioinformatics Department of Molecular
SNP calling. Jose Blanca COMAV institute bioinf.comav.upv.es
 SNP calling Jose Blanca COMAV institute bioinf.comav.upv.es SNP calling Genotype matrix Genotype matrix: Samples x SNPs SNPs and errors A change in a read may due to: Sample contamination Cloning or PCR
SNP calling Jose Blanca COMAV institute bioinf.comav.upv.es SNP calling Genotype matrix Genotype matrix: Samples x SNPs SNPs and errors A change in a read may due to: Sample contamination Cloning or PCR
Next Generation Sequencing: Data analysis for genetic profiling
 Next Generation Sequencing: Data analysis for genetic profiling Raed Samara, Ph.D. Global Product Manager Raed.Samara@QIAGEN.com Welcome to the NGS webinar series - 2015 NGS Technology Webinar 1 NGS: Introduction
Next Generation Sequencing: Data analysis for genetic profiling Raed Samara, Ph.D. Global Product Manager Raed.Samara@QIAGEN.com Welcome to the NGS webinar series - 2015 NGS Technology Webinar 1 NGS: Introduction
Gene Expression analysis with RNA-Seq data
 Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
Gene Expression analysis with RNA-Seq data C3BI Hands-on NGS course November 24th 2016 Frédéric Lemoine Plan 1. 2. Quality Control 3. Read Mapping 4. Gene Expression Analysis 5. Splicing/Transcript Analysis
UAB DNA-Seq Analysis Workshop. John Osborne Research Associate Centers for Clinical and Translational Science
 + UAB DNA-Seq Analysis Workshop John Osborne Research Associate Centers for Clinical and Translational Science ozborn@uab.,edu + Thanks in advance You are the Guinea pigs for this workshop! At this point
+ UAB DNA-Seq Analysis Workshop John Osborne Research Associate Centers for Clinical and Translational Science ozborn@uab.,edu + Thanks in advance You are the Guinea pigs for this workshop! At this point
Short Read Alignment to a Reference Genome
 Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Summer School in Bioinformatics Cambridge, September 2018 Aligning to a reference genome BWA Bowtie2 STAR GEM Pseudo Aligners for RNA-seq
Exploring structural variation in the tomato genome with JBrowse
 Exploring structural variation in the tomato genome with JBrowse Richard Finkers, Wageningen UR Plant Breeding Richard.Finkers@wur.nl; @rfinkers Version 1.0, December 2013 This work is licensed under the
Exploring structural variation in the tomato genome with JBrowse Richard Finkers, Wageningen UR Plant Breeding Richard.Finkers@wur.nl; @rfinkers Version 1.0, December 2013 This work is licensed under the
SNP detection in allopolyploid crops
 SNP detection in allopolyploid crops using NGS data Abstract Homologous SNP detection in polyploid organisms is complicated due to the presence of subgenome polymorphisms, i.e. homeologous SNPs. Several
SNP detection in allopolyploid crops using NGS data Abstract Homologous SNP detection in polyploid organisms is complicated due to the presence of subgenome polymorphisms, i.e. homeologous SNPs. Several
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
Genomic Technologies. Michael Schatz. Feb 1, 2018 Lecture 2: Applied Comparative Genomics
 Genomic Technologies Michael Schatz Feb 1, 2018 Lecture 2: Applied Comparative Genomics Welcome! The primary goal of the course is for students to be grounded in theory and leave the course empowered to
Genomic Technologies Michael Schatz Feb 1, 2018 Lecture 2: Applied Comparative Genomics Welcome! The primary goal of the course is for students to be grounded in theory and leave the course empowered to
High-Throughput Bioinformatics: Re-sequencing and de novo assembly. Elena Czeizler
 High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017
 Andrew Parrish Exeter, 2 nd November 2017") Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017 Topics to cover today What is Next Generation Sequencing (NGS)? Why do we need NGS? Common approaches to NGS NGS
Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017 Topics to cover today What is Next Generation Sequencing (NGS)? Why do we need NGS? Common approaches to NGS NGS
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Variation detection based on second generation sequencing data. Xin LIU Department of Science and Technology, BGI
 Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Introduction to RNA-Seq in GeneSpring NGS Software
 Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Identifying copy number alterations and genotype with Control-FREEC
 Identifying copy number alterations and genotype with Control-FREEC Valentina Boeva contact: freec@curie.fr Most approaches for predicting copy number alterations (CNAs) require you to have whole exomesequencing
Identifying copy number alterations and genotype with Control-FREEC Valentina Boeva contact: freec@curie.fr Most approaches for predicting copy number alterations (CNAs) require you to have whole exomesequencing
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12
 Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Galaxy for Next Generation Sequencing 初探次世代序列分析平台 蘇聖堯 2013/9/12 What s Galaxy? Bringing Developers And Biologists Together. Reproducible Science Is Our Goal An open, web-based platform for data intensive
Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4
 WHITE PAPER Oncomine Comprehensive Assay Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4 Contents Scope and purpose of document...2 Content...2 How Torrent
WHITE PAPER Oncomine Comprehensive Assay Variant calling workflow for the Oncomine Comprehensive Assay using Ion Reporter Software v4.4 Contents Scope and purpose of document...2 Content...2 How Torrent
Reads to Discovery. Visualize Annotate Discover. Small DNA-Seq ChIP-Seq Methyl-Seq. MeDIP-Seq. RNA-Seq. RNA-Seq.
 Reads to Discovery RNA-Seq Small DNA-Seq ChIP-Seq Methyl-Seq RNA-Seq MeDIP-Seq www.strand-ngs.com Analyze Visualize Annotate Discover Data Import Alignment Vendor Platforms: Illumina Ion Torrent Roche
Reads to Discovery RNA-Seq Small DNA-Seq ChIP-Seq Methyl-Seq RNA-Seq MeDIP-Seq www.strand-ngs.com Analyze Visualize Annotate Discover Data Import Alignment Vendor Platforms: Illumina Ion Torrent Roche
10/06/2014. RNA-Seq analysis. With reference assembly. Cormier Alexandre, PhD student UMR8227, Algal Genetics Group
 RNA-Seq analysis With reference assembly Cormier Alexandre, PhD student UMR8227, Algal Genetics Group Summary 2 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference
RNA-Seq analysis With reference assembly Cormier Alexandre, PhD student UMR8227, Algal Genetics Group Summary 2 Typical RNA-seq workflow Introduction Reference genome Reference transcriptome Reference
Variant Callers. J Fass 24 August 2017
 Variant Callers J Fass 24 August 2017 Variant Types Caller Consistency Pabinger (2014) Briefings Bioinformatics 15:256 Freebayes Bayesian haplotype caller that can call SNPs, short CNVs / duplications,
Variant Callers J Fass 24 August 2017 Variant Types Caller Consistency Pabinger (2014) Briefings Bioinformatics 15:256 Freebayes Bayesian haplotype caller that can call SNPs, short CNVs / duplications,
Next Generation Sequencing: An Overview
 Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Variant Discovery. Jie (Jessie) Li PhD Bioinformatics Analyst Bioinformatics Core, UCD
 Li PhD Bioinformatics Analyst Bioinformatics Core, UCD") Variant Discovery Jie (Jessie) Li PhD Bioinformatics Analyst Bioinformatics Core, UCD Variant Type Alkan et al, Nature Reviews Genetics 2011 doi:10.1038/nrg2958 Variant Type http://www.broadinstitute.org/education/glossary/snp
Variant Discovery Jie (Jessie) Li PhD Bioinformatics Analyst Bioinformatics Core, UCD Variant Type Alkan et al, Nature Reviews Genetics 2011 doi:10.1038/nrg2958 Variant Type http://www.broadinstitute.org/education/glossary/snp
RNA-Seq Module 2 From QC to differential gene expression.
 RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
RNA-Seq Module 2 From QC to differential gene expression. Ying Zhang Ph.D, Informatics Analyst Research Informatics Support System (RISS) MSI Apr. 24, 2012 RNA-Seq Tutorials Tutorial 1: Introductory (Mar.
Read Quality Assessment & Improvement. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
BST 226 Statistical Methods for Bioinformatics David M. Rocke. March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
QIAseq Targeted Panel Analysis Plugin USER MANUAL
 QIAseq Targeted Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted Panel Analysis 1.1 Windows, macos and Linux June 18, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej
QIAseq Targeted Panel Analysis Plugin USER MANUAL User manual for QIAseq Targeted Panel Analysis 1.1 Windows, macos and Linux June 18, 2018 This software is for research purposes only. QIAGEN Aarhus Silkeborgvej
MPG NGS workshop I: SNP calling
 MPG NGS workshop I: SNP calling Mark DePristo Manager, Medical and Popula
MPG NGS workshop I: SNP calling Mark DePristo Manager, Medical and Popula
Reference genomes and common file formats
 Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
Reference genomes and common file formats Dóra Bihary MRC Cancer Unit, University of Cambridge CRUK Functional Genomics Workshop September 2017 Overview Reference genomes and GRC Fasta and FastQ (unaligned
NGS Data Analysis and Galaxy
 NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
NGS Data Analysis and Galaxy University of Pretoria Pretoria, South Africa 14-18 October 2013 Dave Clements, Emory University http://galaxyproject.org/ Fourie Joubert, Burger van Jaarsveld Bioinformatics
IDENTIFYING A DISEASE CAUSING MUTATION
 IDENTIFYING A DISEASE CAUSING MUTATION Targeted resequencing MARCELA DAVILA 3/MZO/2016 Core Facilities at Sahlgrenska Academy Statistics Software bioinformatics@gu.se www.cf.gu.se/english// Increasing
IDENTIFYING A DISEASE CAUSING MUTATION Targeted resequencing MARCELA DAVILA 3/MZO/2016 Core Facilities at Sahlgrenska Academy Statistics Software bioinformatics@gu.se www.cf.gu.se/english// Increasing
Introduction to RNAseq Analysis. Milena Kraus Apr 18, 2016
 Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Analysis of RNA-seq Data. Feb 8, 2017 Peikai CHEN (PHD)
") Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Quantifying gene expression
 Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Introduction to Next Generation Sequencing
 The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
Introduction to NGS analyses
 Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Introduction to NGS analyses Giorgio L Papadopoulos Institute of Molecular Biology and Biotechnology Bioinformatics Support Group 04/12/2015 Papadopoulos GL (IMBB, FORTH) IMBB NGS Seminar 04/12/2015 1
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
Comparing a few SNP calling algorithms using low-coverage sequencing data
 Yu and Sun BMC Bioinformatics 2013, 14:274 RESEARCH ARTICLE Open Access Comparing a few SNP calling algorithms using low-coverage sequencing data Xiaoqing Yu 1 and Shuying Sun 1,2* Abstract Background:
Yu and Sun BMC Bioinformatics 2013, 14:274 RESEARCH ARTICLE Open Access Comparing a few SNP calling algorithms using low-coverage sequencing data Xiaoqing Yu 1 and Shuying Sun 1,2* Abstract Background:
RNAseq Applications in Genome Studies. Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford
 RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
RNAseq Applications in Genome Studies Alexander Kanapin, PhD Wellcome Trust Centre for Human Genetics, University of Oxford RNAseq Protocols Next generation sequencing protocol cdna, not RNA sequencing
CNV and variant detection for human genome resequencing data - for biomedical researchers (II)
") CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
BIOINFORMATICS. Lacking alignments? The next-generation sequencing mapper segemehl revisited
 BIOINFORMATICS Vol. 00 no. 00 2014 Pages 1 7 Lacking alignments? The next-generation sequencing mapper segemehl revisited Christian Otto 1,2, Peter F. Stadler 2 8, Steve Hoffmann 1,2 1 Transcriptome Bioinformatics
BIOINFORMATICS Vol. 00 no. 00 2014 Pages 1 7 Lacking alignments? The next-generation sequencing mapper segemehl revisited Christian Otto 1,2, Peter F. Stadler 2 8, Steve Hoffmann 1,2 1 Transcriptome Bioinformatics
Analytics Behind Genomic Testing
 A Quick Guide to the Analytics Behind Genomic Testing Elaine Gee, PhD Director, Bioinformatics ARUP Laboratories 1 Learning Objectives Catalogue various types of bioinformatics analyses that support clinical
A Quick Guide to the Analytics Behind Genomic Testing Elaine Gee, PhD Director, Bioinformatics ARUP Laboratories 1 Learning Objectives Catalogue various types of bioinformatics analyses that support clinical
BIGGIE: A Distributed Pipeline for Genomic Variant Calling
 BIGGIE: A Distributed Pipeline for Genomic Variant Calling Richard Xia, Sara Sheehan, Yuchen Zhang, Ameet Talwalkar, Matei Zaharia Jonathan Terhorst, Michael Jordan, Yun S. Song, Armando Fox, David Patterson
BIGGIE: A Distributed Pipeline for Genomic Variant Calling Richard Xia, Sara Sheehan, Yuchen Zhang, Ameet Talwalkar, Matei Zaharia Jonathan Terhorst, Michael Jordan, Yun S. Song, Armando Fox, David Patterson
Genome STRiP ASHG Workshop demo materials. Bob Handsaker October 19, 2014
 Genome STRiP ASHG Workshop demo materials Bob Handsaker October 19, 2014 Running Genome STRiP directly on AWS Genome STRiP Structure in Populations Popula'on)aware-discovery-andgenotyping-of-structural-varia'onfrom-whole)genome-sequencing-
Genome STRiP ASHG Workshop demo materials Bob Handsaker October 19, 2014 Running Genome STRiP directly on AWS Genome STRiP Structure in Populations Popula'on)aware-discovery-andgenotyping-of-structural-varia'onfrom-whole)genome-sequencing-
Reference genomes and common file formats
 Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John DiCarlo Yexun Wang
 Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Variant detection analysis in the BRCA1/2 genes from Ion torrent PGM data
 Variant detection analysis in the BRCA1/2 genes from Ion torrent PGM data Bruno Zeitouni Bionformatics department of the Institut Curie Inserm U900 Mines ParisTech Ion Torrent User Meeting 2012, October
Variant detection analysis in the BRCA1/2 genes from Ion torrent PGM data Bruno Zeitouni Bionformatics department of the Institut Curie Inserm U900 Mines ParisTech Ion Torrent User Meeting 2012, October
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow
 From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
HiSeq Whole Exome Sequencing Report. BGI Co., Ltd.
 HiSeq Whole Exome Sequencing Report BGI Co., Ltd. Friday, 11th Nov., 2016 Table of Contents Results 1 Data Production 2 Summary Statistics of Alignment on Target Regions 3 Data Quality Control 4 SNP Results
HiSeq Whole Exome Sequencing Report BGI Co., Ltd. Friday, 11th Nov., 2016 Table of Contents Results 1 Data Production 2 Summary Statistics of Alignment on Target Regions 3 Data Quality Control 4 SNP Results
RNA Seq: Methods and Applica6ons. Prat Thiru
 RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Nature Biotechnology: doi: /nbt Supplementary Figure 1. Read Complexity
 Supplementary Figure 1 Read Complexity A) Density plot showing the percentage of read length masked by the dust program, which identifies low-complexity sequence (simple repeats). Scrappie outputs a significantly
Supplementary Figure 1 Read Complexity A) Density plot showing the percentage of read length masked by the dust program, which identifies low-complexity sequence (simple repeats). Scrappie outputs a significantly
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
BST227 Introduction to Statistical Genetics. Lecture 8: Variant calling from high-throughput sequencing data
 BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
Distributed Pipeline for Genomic Variant Calling
 Distributed Pipeline for Genomic Variant Calling Richard Xia, Sara Sheehan, Yuchen Zhang, Ameet Talwalkar, Matei Zaharia Jonathan Terhorst, Michael Jordan, Yun S. Song, Armando Fox, David Patterson Division
Distributed Pipeline for Genomic Variant Calling Richard Xia, Sara Sheehan, Yuchen Zhang, Ameet Talwalkar, Matei Zaharia Jonathan Terhorst, Michael Jordan, Yun S. Song, Armando Fox, David Patterson Division
Next Generation Sequencing. Tobias Österlund
 Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Normal-Tumor Comparison using Next-Generation Sequencing Data
 Normal-Tumor Comparison using Next-Generation Sequencing Data Chun Li Vanderbilt University Taichung, March 16, 2011 Next-Generation Sequencing First-generation (Sanger sequencing): 115 kb per day per
Normal-Tumor Comparison using Next-Generation Sequencing Data Chun Li Vanderbilt University Taichung, March 16, 2011 Next-Generation Sequencing First-generation (Sanger sequencing): 115 kb per day per
Data Analysis Report: Variant Analysis v1.2
 GATC Biotech AG, Jakob-Stadler-Platz 7, 78467 Konstanz Data Analysis Report: Variant Analysis v1.2 Project / Study: GATC-Demo Date: February 28, 2018 Table of Contents 1 Analysis workflow 1 2 Samples Analysed
GATC Biotech AG, Jakob-Stadler-Platz 7, 78467 Konstanz Data Analysis Report: Variant Analysis v1.2 Project / Study: GATC-Demo Date: February 28, 2018 Table of Contents 1 Analysis workflow 1 2 Samples Analysed
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
VM origin. Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE
 X2go: LXDE") VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
VM origin Okeanos: Image Trinity_U16 (upgrade to Ubuntu16.04, thanks to Alexandros Dimopoulos) X2go: LXDE NGS intro + Genome-Based Transcript Reconstruction and Analysis Using RNA-Seq Data Based on material
Bulked Segregant Analysis For Fine Mapping Of Genes. Cheng Zou, Qi Sun Bioinformatics Facility Cornell University
 Bulked Segregant Analysis For Fine Mapping Of enes heng Zou, Qi Sun Bioinformatics Facility ornell University Outline What is BSA? Keys for a successful BSA study Pipeline of BSA extended reading ompare
Bulked Segregant Analysis For Fine Mapping Of enes heng Zou, Qi Sun Bioinformatics Facility ornell University Outline What is BSA? Keys for a successful BSA study Pipeline of BSA extended reading ompare
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
L3: Short Read Alignment to a Reference Genome
 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA Expression Time Course Analysis
 RNA Expression Time Course Analysis April 23, 2014 Introduction The increased efficiency and reduced per-read cost of next generation sequencing (NGS) has opened new and exciting opportunities for data
RNA Expression Time Course Analysis April 23, 2014 Introduction The increased efficiency and reduced per-read cost of next generation sequencing (NGS) has opened new and exciting opportunities for data
BICF Variant Analysis Tools. Using the BioHPC Workflow Launching Tool Astrocyte
 BICF Variant Analysis Tools Using the BioHPC Workflow Launching Tool Astrocyte Prioritization of Variants SNP INDEL SV Astrocyte BioHPC Workflow Platform Allows groups to give easy-access to their analysis
BICF Variant Analysis Tools Using the BioHPC Workflow Launching Tool Astrocyte Prioritization of Variants SNP INDEL SV Astrocyte BioHPC Workflow Platform Allows groups to give easy-access to their analysis
Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer. Project XX1001. Customer Detail
 Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer Project XX Customer Detail Table of Contents. Bioinformatics analysis pipeline...3.. Read quality check. 3.2. Read alignment...3.3.
Analysis of neo-antigens to identify T-cell neo-epitopes in human Head & Neck cancer Project XX Customer Detail Table of Contents. Bioinformatics analysis pipeline...3.. Read quality check. 3.2. Read alignment...3.3.
Genomic Dark Matter: The limitations of short read mapping illustrated by the Genome Mappability Score (GMS)
") Genomic Dark Matter: The limitations of short read mapping illustrated by the Genome Mappability Score (GMS) Hayan Lee Advised by Prof. Michael Schatz Sep. 28, 2011 Quantitative Biology Seminar 1 Outline
Genomic Dark Matter: The limitations of short read mapping illustrated by the Genome Mappability Score (GMS) Hayan Lee Advised by Prof. Michael Schatz Sep. 28, 2011 Quantitative Biology Seminar 1 Outline
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Transcriptomics analysis with RNA seq: an overview Frederik Coppens
 Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Illumina Read QC. UCD Genome Center Bioinformatics Core Monday 29 August 2016
 Illumina Read QC UCD Genome Center Bioinformatics Core Monday 29 August 2016 QC should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical processes involved
Illumina Read QC UCD Genome Center Bioinformatics Core Monday 29 August 2016 QC should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical processes involved
Mining GWAS Catalog & 1000 Genomes Dataset. Segun Fatumo
 Mining GWAS Catalog & 1000 Genomes Dataset Segun Fatumo What is GWAS Catalog NHGRI GWA Catalog www.genome.gov/gwastudies Citation How to cite the NHGRI GWAS Catalog: Hindorff LA, MacArthur J (European
Mining GWAS Catalog & 1000 Genomes Dataset Segun Fatumo What is GWAS Catalog NHGRI GWA Catalog www.genome.gov/gwastudies Citation How to cite the NHGRI GWAS Catalog: Hindorff LA, MacArthur J (European
Virus-Clip: a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability
 Title Virus-Clip: a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability Author(s) Ho, DWH; Sze, MF; Ng, IOL Citation, 2015, v. 6, n. 25,
Title Virus-Clip: a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability Author(s) Ho, DWH; Sze, MF; Ng, IOL Citation, 2015, v. 6, n. 25,