Sequence assembly. Jose Blanca COMAV institute bioinf.comav.upv.es
|
|
|
- Clarence Garrison
- 6 years ago
- Views:
Transcription
1 Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es
2
3 Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7


4 Computational requirements Main requirements: Memory. Time. The kmer-based assemblers requirements depend on the genome complexity and not so much on the reads.


5 Read cleaning Some assemblers choke with: bad quality stretches, adapters, low complexity regions, contamination. Bad quality vector Trim Unknown sequence Trim or mask
6 The assembly problem Only one read is usually not capable of producing the complete sequence. Even if the problem sequence is short one read might have sequencing errors. repeats Unknown sequence Reads contig5 (repeat) Contigs / scaffolds Reasons for the gaps contig1 Repeated region contig2 contig3 no coverage contig4
7 Long reads


8 Read size influence
9 Mate pairs and paired-ends Read length is critical, but constrained by sequencing technology, we can sequence molecule ends. Clone (template) reads Useful for dealing with repetitive genomic DNA and with complex transcriptome structure. Paired-ends: Illumina can sequence from both ends of the molecules. ( bp) Mate-pairs: Can be generated from libraries with different lengths (2-20 Kb). BAC-ends: Usually sequenced with Sanger.
10 Mate-pairs
11 Library types Single reads Illumina Pair Ends ( pb) Mate Pairs (2-10 kb) Illumina
12 Illumina mate-pair chimeras

13 Long reads Third-generation sequencing and the future of genomics. Lee et al.
14 Short reads are harder to assemble Taken from Jason Miller
15 The need for paired reads Taken from Jason Miller
16 Algorithm I Overlap Layout - Consensus
17 Overlap Layout - Consensus Algorithm: All-against-all, pair-wise read comparison. Construction of an overlap graph with approximate read layout. Multiple sequence alignment determines the precise layout and the consensus sequence. clean reads Overlaps detection All vs all alignment Contigs Consensus
18 Overlap problems Two reads are similar if they have a good overlap. We assume that overlap implies common origin in the genome. This goodness depends on the overlap quality and similarity. But several things might go wrong repetitive Too many sequencing errors Chimeric clone Short overlap false negative false positive Vector contamination or repetitive
 Overlaps must reach the ends of both reads Ignore high-frequency overlaps (repetitive regions in genomic?) But stringency will induce false negatives.")
19 Overlap problems False positives will be induced by chance and repeats Avoid false positives with stringent criteria: Overlaps must be long enough Sequence similarity must be high (identity threshold) Overlaps must reach the ends of both reads Ignore high-frequency overlaps (repetitive regions in genomic?) But stringency will induce false negatives.
20 Assembly terminology Consensus Scaffold = Contigs + Parings (gap lengths derived from pairings) Contigs = Reads + Overlaps Overlaps Reads & Pairing Taken from Jason Miller ACTGATTAGCTGACGNNNNNNNNNNTCGTATCTAGCATT

21 Unitigs Contigs: High-confidence overlaps Maximal contigs with no contradiction (or almost no contradiction) in the data Ungapped Contigs capture the unique stretches of the genome. Usually where unitigs end, repeats begin Contig example. The ideal Contig is A, B, C. C, D and E have a repetitive sequence.
22 Scaffolds Build with mate pairs Mate pairs help resolve ambiguity in overlap patterns Scaffolds Every contig is a scaffold Every scaffold contains one or more contigs Scaffolds can contain sequence gaps of any size (including negative)
23 Overlap-Consensus-Layout limitation 30 Million 5Kb (long) reads Number of alignments: 30 M reads x 30 M reads = 900 trillion alignments It does not scale
24 Algorithm II kmer-based
25 K-mer based assembly K-mer graphs are de Bruijn graph. Nodes represent all K-mers. Edges represent fixed-length overlaps between consecutive K-mers. Assembly is reduced to a graph reduction problem. Read 1 Read 2 AACCGG CCGGTT Graph: AACC -> ACCG -> CCGG -> CGGT -> GGTT K-mer length = 4

26 K-mer based assembly

27 K-mer bubbles
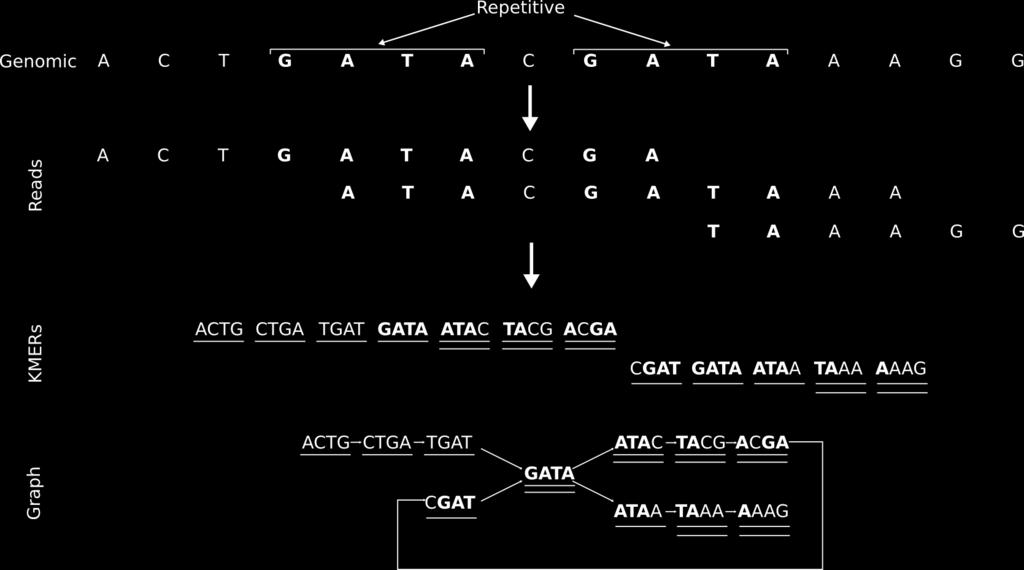
28 K-mer repetitive
29 Graph problems Repeats, sequencing errors and polymorphisms increase graph complexity, leading to tangles difficult to resolve K-mer graphs are more sensitive to repeats and sequencing errors than overlap based methods. Optimal graph reductions algorithms are NP-hard, so assemblers use heuristic algorithms Polymorphism Sequencing error Repeat
30 K-mer analysis 21-mer distribution of sea urchin (Strongylocentrotus purpuratus) genome (900 Mbase long) unique repetitive
31 K-mer analysis Genome K-mer distribution Simulated reads. 50X coverage No errors reads unique Repetitive (duplication)
32 K-mer analysis Genome K-mer distribution real reads. 50X coverage With errors Real reads (with errors) Simulated reads (no error) noise signal
33 Final remarks
34 N50 N50 is defined as the contig length such that using equal or longer contigs produces half the bases of the genome. The N50 statistic is a measure of the average length of a set of sequences, with greater weight given to longer sequences. It is widely used in genome assembly N50: [ [ [ [ [ [ [ [ [ [ [ [ [ [ N95: 20, 7414[, 14728[, 22042[, 29356[, 36670[, 43984[, 51298[, 58612[, 65926[, 73240[, 80554[, 87868[, 95182[, [ Taken from Wikipedia minimum: 100 maximum: 146,370 average: num. seqs.: (131545): ************************************************************************ ( 6247): *** ( 2353): * ( 842): ( 324): ( 118): ( 42): ( 13): ( 10): ( 4): ( 0): ( 1): ( 0): ( 4):
35 Common assembly errors Collapsed repeats Align reads from distinct (polymorphic) repeat copies Fewer repeats in assembly than in genome Fewer tandem repeat copies in assembly than genome Missed joins Missed overlaps due to sequencing error Contradictory evidence from overlaps Contradictory or insufficient evidence from pairs Chimera Enter repeat at copy 1 but exit repeat at copy 2 Assembly joins unrelated sequences
36 Ingredients for Good Assembly Coverage and read length 20X for (corrected) PacBio, 150X for Illumina Paired reads Read lengths long enough to place uniquely Inserts longer than long repeats Pair density sufficient to traverse repeat clusters Tight insert size variance Diversity of insert sizes Read Quality: Sequencing errors No vector contamination Contamination: mitochondrial or chloroplastic The requirements depend greatly on the quality: it is not the same a draft that high quality genome.
37 Common assemblers Genomic SOAPdenovo, Illumina CABOG: Celera assembler, 454 and few Illumina Staden for Sanger reads. Transcriptomic assemblers are specialized software
38 From scafflods to chromosomes

39 Genetic maps

40 Mapping platforms Third-generation sequencing and the future of genomics. Lee et al.
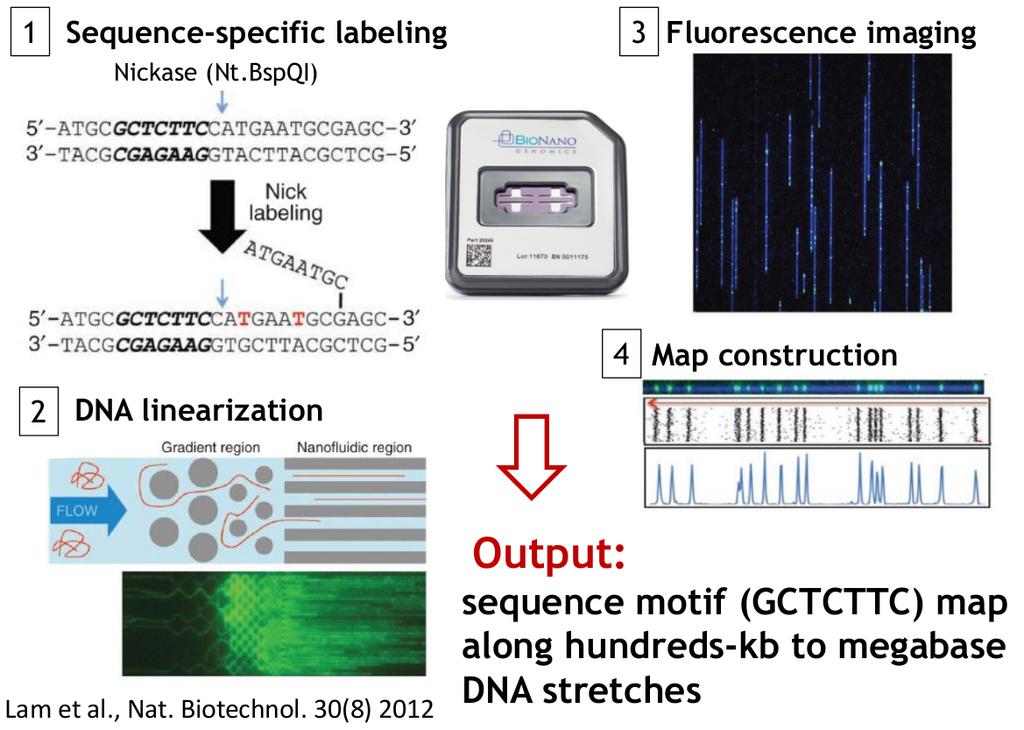
41 Bionano
42 Bionano Bionano helps you solve long range structures (although not chromosome wide assemblies) 1 2
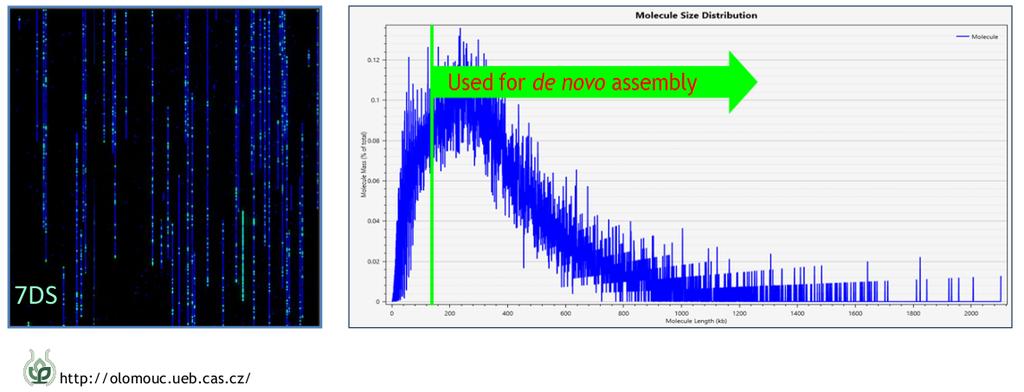
43 Bionano
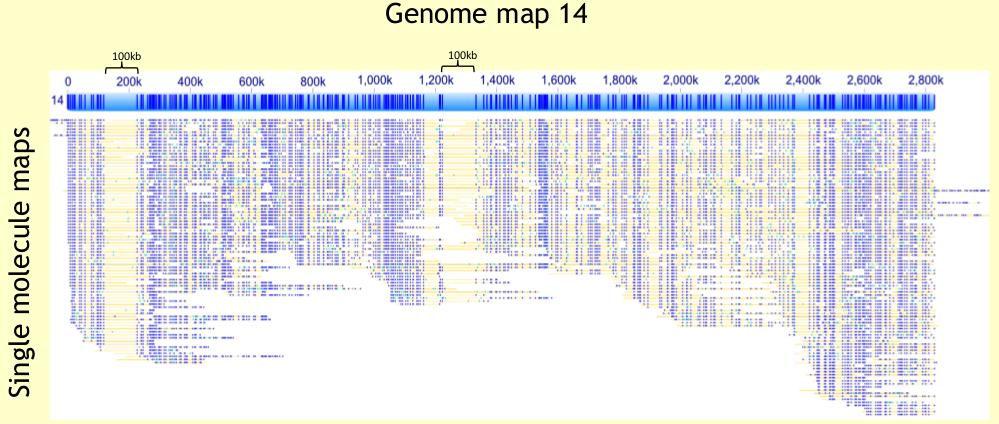
44 Bionano
45 10X linked reads Reads generated from the same long DNA sequence are tagged with the same index and sequenced using Illumina Reads with the same index convey medium range information (Up to 100Kb)

46 10X GemCode
47 10X linked reads assembly
48 10x possible uses De novo genome assembly Haplotype phasing Structural Variants: Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases ( Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficiency. (DOI: /journal.pgen )
49 Tomato assembly example PacBio 10X PacBio-Bionano 10X-Bionano N L N s per 100 Kb Cost $$$ $ $$$$ $$
50 Hi-C Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. (DOI: /science )
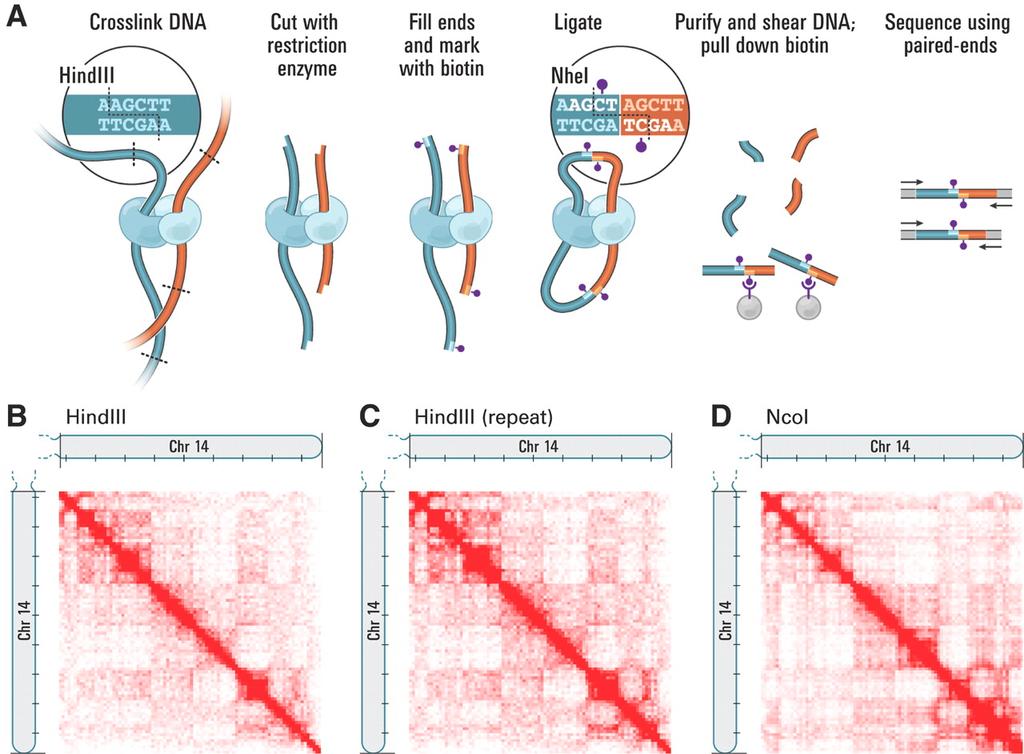
51 Hi-C
52 Hi-C Assembly De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. (DOI: /science.aal3327) Hi-C data provide links across a variety of length scale Unlike paired-end reads Hi-C contact spans an unknown length and may connect loci on different chromosomes Human genome assembly: Pair-end Illumina reads (67X coverage) Hi-C data (6.7 coverage) 23 scaffold that span the 99.5% of the 23 human chromosomes Errors remain in the ordering of short distances than could be fixed by matepairs, long reads or bionano
53 This work is licensed under the Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit or send a letter to Creative Commons, 171 Second Street, Suite 300, San Francisco, California, 94105, USA. Jose Blanca COMAV institute bioinf.comav.upv.es
Introduction to metagenome assembly. Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014
 Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Alignment and Assembly
 Alignment and Assembly Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which
Alignment and Assembly Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which
De novo genome assembly with next generation sequencing data!! "
 De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
De novo whole genome assembly
 De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore
De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore
TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR)
") tru TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR) Anton Bankevich Center for Algorithmic Biotechnology, SPbSU Sequencing costs 1. Sequencing costs do not follow Moore s law
tru TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR) Anton Bankevich Center for Algorithmic Biotechnology, SPbSU Sequencing costs 1. Sequencing costs do not follow Moore s law
De novo assembly of human genomes with massively parallel short read sequencing. Mikk Eelmets Journal Club
 De novo assembly of human genomes with massively parallel short read sequencing Mikk Eelmets Journal Club 06.04.2010 Problem DNA sequencing technologies: Sanger sequencing (500-1000 bp) Next-generation
De novo assembly of human genomes with massively parallel short read sequencing Mikk Eelmets Journal Club 06.04.2010 Problem DNA sequencing technologies: Sanger sequencing (500-1000 bp) Next-generation
BIOINFORMATICS ORIGINAL PAPER
 BIOINFORMATICS ORIGINAL PAPER Vol. 27 no. 21 2011, pages 2957 2963 doi:10.1093/bioinformatics/btr507 Genome analysis Advance Access publication September 7, 2011 : fast length adjustment of short reads
BIOINFORMATICS ORIGINAL PAPER Vol. 27 no. 21 2011, pages 2957 2963 doi:10.1093/bioinformatics/btr507 Genome analysis Advance Access publication September 7, 2011 : fast length adjustment of short reads
Lecture 14: DNA Sequencing
 Lecture 14: DNA Sequencing Study Chapter 8.9 10/17/2013 COMP 465 Fall 2013 1 Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments (Sanger method) DNA Sequencing
Lecture 14: DNA Sequencing Study Chapter 8.9 10/17/2013 COMP 465 Fall 2013 1 Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments (Sanger method) DNA Sequencing
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
10/20/2009 Comp 590/Comp Fall
 Lecture 14: DNA Sequencing Study Chapter 8.9 10/20/2009 Comp 590/Comp 790-90 Fall 2009 1 DNA Sequencing Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments
Lecture 14: DNA Sequencing Study Chapter 8.9 10/20/2009 Comp 590/Comp 790-90 Fall 2009 1 DNA Sequencing Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments
De novo Genome Assembly
 De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
Genome Assembly. J Fass UCD Genome Center Bioinformatics Core Friday September, 2015
 Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
Outline. The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing
 Illumina Assembly 1 Outline The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing 2 Illumina Sequencing Paired end Illumina
Illumina Assembly 1 Outline The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing 2 Illumina Sequencing Paired end Illumina
Mate-pair library data improves genome assembly
 De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
The Basics of Understanding Whole Genome Next Generation Sequence Data
 The Basics of Understanding Whole Genome Next Generation Sequence Data Heather Carleton-Romer, MPH, Ph.D. ASM-CDC Infectious Disease and Public Health Microbiology Postdoctoral Fellow PulseNet USA Next
The Basics of Understanding Whole Genome Next Generation Sequence Data Heather Carleton-Romer, MPH, Ph.D. ASM-CDC Infectious Disease and Public Health Microbiology Postdoctoral Fellow PulseNet USA Next
Genome Projects. Part III. Assembly and sequencing of human genomes
 Genome Projects Part III Assembly and sequencing of human genomes All current genome sequencing strategies are clone-based. 1. ordered clone sequencing e.g., C. elegans well suited for repetitive sequences
Genome Projects Part III Assembly and sequencing of human genomes All current genome sequencing strategies are clone-based. 1. ordered clone sequencing e.g., C. elegans well suited for repetitive sequences
Biol 478/595 Intro to Bioinformatics
 Biol 478/595 Intro to Bioinformatics September M 1 Labor Day 4 W 3 MG Database Searching Ch. 6 5 F 5 MG Database Searching Hw1 6 M 8 MG Scoring Matrices Ch 3 and Ch 4 7 W 10 MG Pairwise Alignment 8 F 12
Biol 478/595 Intro to Bioinformatics September M 1 Labor Day 4 W 3 MG Database Searching Ch. 6 5 F 5 MG Database Searching Hw1 6 M 8 MG Scoring Matrices Ch 3 and Ch 4 7 W 10 MG Pairwise Alignment 8 F 12
NGS developments in tomato genome sequencing
 NGS developments in tomato genome sequencing 16-02-2012, Sandra Smit TATGTTTTGGAAAACATTGCATGCGGAATTGGGTACTAGGTTGGACCTTAGTACC GCGTTCCATCCTCAGACCGATGGTCAGTCTGAGAGAACGATTCAAGTGTTGGAAG ATATGCTTCGTGCATGTGTGATAGAGTTTGGTGGCCATTGGGATAGCTTCTTACC
NGS developments in tomato genome sequencing 16-02-2012, Sandra Smit TATGTTTTGGAAAACATTGCATGCGGAATTGGGTACTAGGTTGGACCTTAGTACC GCGTTCCATCCTCAGACCGATGGTCAGTCTGAGAGAACGATTCAAGTGTTGGAAG ATATGCTTCGTGCATGTGTGATAGAGTTTGGTGGCCATTGGGATAGCTTCTTACC
The Diploid Genome Sequence of an Individual Human
 The Diploid Genome Sequence of an Individual Human Maido Remm Journal Club 12.02.2008 Outline Background (history, assembling strategies) Who was sequenced in previous projects Genome variations in J.
The Diploid Genome Sequence of an Individual Human Maido Remm Journal Club 12.02.2008 Outline Background (history, assembling strategies) Who was sequenced in previous projects Genome variations in J.
Contact us for more information and a quotation
 GenePool Information Sheet #1 Installed Sequencing Technologies in the GenePool The GenePool offers sequencing service on three platforms: Sanger (dideoxy) sequencing on ABI 3730 instruments Illumina SOLEXA
GenePool Information Sheet #1 Installed Sequencing Technologies in the GenePool The GenePool offers sequencing service on three platforms: Sanger (dideoxy) sequencing on ABI 3730 instruments Illumina SOLEXA
DNA Sequencing and Assembly
 DNA Sequencing and Assembly CS 262 Lecture Notes, Winter 2016 February 2nd, 2016 Scribe: Mark Berger Abstract In this lecture, we survey a variety of different sequencing technologies, including their
DNA Sequencing and Assembly CS 262 Lecture Notes, Winter 2016 February 2nd, 2016 Scribe: Mark Berger Abstract In this lecture, we survey a variety of different sequencing technologies, including their
CSE182-L16. LW statistics/assembly
 CSE182-L16 LW statistics/assembly Silly Quiz Who are these people, and what is the occasion? Genome Sequencing and Assembly Sequencing A break at T is shown here. Measuring the lengths using electrophoresis
CSE182-L16 LW statistics/assembly Silly Quiz Who are these people, and what is the occasion? Genome Sequencing and Assembly Sequencing A break at T is shown here. Measuring the lengths using electrophoresis
A shotgun introduction to sequence assembly (with Velvet) MCB Brem, Eisen and Pachter
 MCB Brem, Eisen and Pachter") A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
Consensus Ensemble Approaches Improve De Novo Transcriptome Assemblies
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
Assembly of Ariolimax dolichophallus using SOAPdenovo2
 Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Course summary. Today. PCR Polymerase chain reaction. Obtaining molecular data. Sequencing. DNA sequencing. Genome Projects.
 Goals Organization Labs Project Reading Course summary DNA sequencing. Genome Projects. Today New DNA sequencing technologies. Obtaining molecular data PCR Typically used in empirical molecular evolution
Goals Organization Labs Project Reading Course summary DNA sequencing. Genome Projects. Today New DNA sequencing technologies. Obtaining molecular data PCR Typically used in empirical molecular evolution
Workflow of de novo assembly
 Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Genome Assembly Software for Different Technology Platforms. PacBio Canu Falcon. Illumina Soap Denovo Discovar Platinus MaSuRCA.
 Genome Assembly Software for Different Technology Platforms PacBio Canu Falcon 10x SuperNova Illumina Soap Denovo Discovar Platinus MaSuRCA Experimental design using Illumina Platform Estimate genome size:
Genome Assembly Software for Different Technology Platforms PacBio Canu Falcon 10x SuperNova Illumina Soap Denovo Discovar Platinus MaSuRCA Experimental design using Illumina Platform Estimate genome size:
De Novo and Hybrid Assembly
 On the PacBio RS Introduction The PacBio RS utilizes SMRT technology to generate both Continuous Long Read ( CLR ) and Circular Consensus Read ( CCS ) data. In this document, we describe sequencing the
On the PacBio RS Introduction The PacBio RS utilizes SMRT technology to generate both Continuous Long Read ( CLR ) and Circular Consensus Read ( CCS ) data. In this document, we describe sequencing the
Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I
 Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I Dr David Studholme. 18 th February 2014. BIO1033 theme lecture. 1 28 February 2014 @davidjstudholme 28 February 2014 @davidjstudholme
Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I Dr David Studholme. 18 th February 2014. BIO1033 theme lecture. 1 28 February 2014 @davidjstudholme 28 February 2014 @davidjstudholme
Finishing of Fosmid 1042D14. Project 1042D14 is a roughly 40 kb segment of Drosophila ananassae
 Schefkind 1 Adam Schefkind Bio 434W 03/08/2014 Finishing of Fosmid 1042D14 Abstract Project 1042D14 is a roughly 40 kb segment of Drosophila ananassae genomic DNA. Through a comprehensive analysis of forward-
Schefkind 1 Adam Schefkind Bio 434W 03/08/2014 Finishing of Fosmid 1042D14 Abstract Project 1042D14 is a roughly 40 kb segment of Drosophila ananassae genomic DNA. Through a comprehensive analysis of forward-
Assembly and Validation of Large Genomes from Short Reads Michael Schatz. March 16, 2011 Genome Assembly Workshop / Genome 10k
 Assembly and Validation of Large Genomes from Short Reads Michael Schatz March 16, 2011 Genome Assembly Workshop / Genome 10k A Brief Aside 4.7GB / disc ~20 discs / 1G Genome X 10,000 Genomes = 1PB Data
Assembly and Validation of Large Genomes from Short Reads Michael Schatz March 16, 2011 Genome Assembly Workshop / Genome 10k A Brief Aside 4.7GB / disc ~20 discs / 1G Genome X 10,000 Genomes = 1PB Data
State of the art de novo assembly of human genomes from massively parallel sequencing data
 State of the art de novo assembly of human genomes from massively parallel sequencing data Yingrui Li, 1 Yujie Hu, 1,2 Lars Bolund 1,3 and Jun Wang 1,2* 1 BGI-Shenzhen, Shenzhen, Guangdong 518083, China
State of the art de novo assembly of human genomes from massively parallel sequencing data Yingrui Li, 1 Yujie Hu, 1,2 Lars Bolund 1,3 and Jun Wang 1,2* 1 BGI-Shenzhen, Shenzhen, Guangdong 518083, China
Genomic resources. for non-model systems
 Genomic resources for non-model systems 1 Genomic resources Whole genome sequencing reference genome sequence comparisons across species identify signatures of natural selection population-level resequencing
Genomic resources for non-model systems 1 Genomic resources Whole genome sequencing reference genome sequence comparisons across species identify signatures of natural selection population-level resequencing
ChIP-seq and RNA-seq. Farhat Habib
 ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
Next Generation Sequences & Chloroplast Assembly. 8 June, 2012 Jongsun Park
 Next Generation Sequences & Chloroplast Assembly 8 June, 2012 Jongsun Park Table of Contents 1 History of Sequencing Technologies 2 Genome Assembly Processes With NGS Sequences 3 How to Assembly Chloroplast
Next Generation Sequences & Chloroplast Assembly 8 June, 2012 Jongsun Park Table of Contents 1 History of Sequencing Technologies 2 Genome Assembly Processes With NGS Sequences 3 How to Assembly Chloroplast
short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014
 1 short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014 2 Genomathica Assembler Mathematica notebook for genome assembly simulation Assembler can be found at:
1 short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014 2 Genomathica Assembler Mathematica notebook for genome assembly simulation Assembler can be found at:
NEXT GENERATION SEQUENCING. Farhat Habib
 NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
Data Basics. Josef K Vogt Slides by: Simon Rasmussen Next Generation Sequencing Analysis
 Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
AMOS Assembly Validation and Visualization
 AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland August 13, 2006 University of Hawaii Outline AMOS Validation Pipeline
AMOS Assembly Validation and Visualization Michael Schatz Center for Bioinformatics and Computational Biology University of Maryland August 13, 2006 University of Hawaii Outline AMOS Validation Pipeline
We begin with a high-level overview of sequencing. There are three stages in this process.
 Lecture 11 Sequence Assembly February 10, 1998 Lecturer: Phil Green Notes: Kavita Garg 11.1. Introduction This is the first of two lectures by Phil Green on Sequence Assembly. Yeast and some of the bacterial
Lecture 11 Sequence Assembly February 10, 1998 Lecturer: Phil Green Notes: Kavita Garg 11.1. Introduction This is the first of two lectures by Phil Green on Sequence Assembly. Yeast and some of the bacterial
Gap Filling for a Human MHC Haplotype Sequence
 American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
ChIP-seq and RNA-seq
 ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
PERGA: A Paired-End Read Guided De Novo Assembler for Extending Contigs Using SVM and Look Ahead Approach
 Title for Extending Contigs Using SVM and Look Ahead Approach Author(s) Zhu, X; Leung, HCM; Chin, FYL; Yiu, SM; Quan, G; Liu, B; Wang, Y Citation PLoS ONE, 2014, v. 9 n. 12, article no. e114253 Issued
Title for Extending Contigs Using SVM and Look Ahead Approach Author(s) Zhu, X; Leung, HCM; Chin, FYL; Yiu, SM; Quan, G; Liu, B; Wang, Y Citation PLoS ONE, 2014, v. 9 n. 12, article no. e114253 Issued
Greene 1. Finishing of DEUG The entire genome of Drosophila eugracilis has recently been sequenced using Roche
 Greene 1 Harley Greene Bio434W Elgin Finishing of DEUG4927002 Abstract The entire genome of Drosophila eugracilis has recently been sequenced using Roche 454 pyrosequencing and Illumina paired-end reads
Greene 1 Harley Greene Bio434W Elgin Finishing of DEUG4927002 Abstract The entire genome of Drosophila eugracilis has recently been sequenced using Roche 454 pyrosequencing and Illumina paired-end reads
De novo meta-assembly of ultra-deep sequencing data
 De novo meta-assembly of ultra-deep sequencing data Hamid Mirebrahim 1, Timothy J. Close 2 and Stefano Lonardi 1 1 Department of Computer Science and Engineering 2 Department of Botany and Plant Sciences
De novo meta-assembly of ultra-deep sequencing data Hamid Mirebrahim 1, Timothy J. Close 2 and Stefano Lonardi 1 1 Department of Computer Science and Engineering 2 Department of Botany and Plant Sciences
Copyright (c) 2008 Daniel Huson. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation
 2008 Daniel Huson. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation") Assembly of the Human Genome Daniel Huson Informatics Research Copyright (c) 2008 Daniel Huson. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation
Assembly of the Human Genome Daniel Huson Informatics Research Copyright (c) 2008 Daniel Huson. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation
DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN. (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN
 JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN") DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN ... 2014 2015 2016 2017 ... 2014 2015 2016 2017 Synthetic
DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN ... 2014 2015 2016 2017 ... 2014 2015 2016 2017 Synthetic
Analysis of structural variation. Alistair Ward USTAR Center for Genetic Discovery University of Utah
 Analysis of structural variation Alistair Ward USTAR Center for Genetic Discovery University of Utah What is structural variation? What differentiates SV from short variants? What are the major SV types?
Analysis of structural variation Alistair Ward USTAR Center for Genetic Discovery University of Utah What is structural variation? What differentiates SV from short variants? What are the major SV types?
COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly
 Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Bionano Access : Assembly Report Guidelines
 Bionano Access : Assembly Report Guidelines Document Number: 30255 Document Revision: A For Research Use Only. Not for use in diagnostic procedures. Copyright 2018 Bionano Genomics Inc. All Rights Reserved
Bionano Access : Assembly Report Guidelines Document Number: 30255 Document Revision: A For Research Use Only. Not for use in diagnostic procedures. Copyright 2018 Bionano Genomics Inc. All Rights Reserved
Lecture 10 : Whole genome sequencing and analysis. Introduction to Computational Biology Teresa Przytycka, PhD
 Lecture 10 : Whole genome sequencing and analysis Introduction to Computational Biology Teresa Przytycka, PhD Sequencing DNA Goal obtain the string of bases that make a given DNA strand. Problem Typically
Lecture 10 : Whole genome sequencing and analysis Introduction to Computational Biology Teresa Przytycka, PhD Sequencing DNA Goal obtain the string of bases that make a given DNA strand. Problem Typically
Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Supplemental Materials
 Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads Supplemental Materials 1. Supplemental Methods... 3 1.1 Algorithm Detail... 3 1.1.1 k-mer coverage distribution
Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads Supplemental Materials 1. Supplemental Methods... 3 1.1 Algorithm Detail... 3 1.1.1 k-mer coverage distribution
Genome Sequencing-- Strategies
 Genome Sequencing-- Strategies Bio 4342 Spring 04 What is a genome? A genome can be defined as the entire DNA content of each nucleated cell in an organism Each organism has one or more chromosomes that
Genome Sequencing-- Strategies Bio 4342 Spring 04 What is a genome? A genome can be defined as the entire DNA content of each nucleated cell in an organism Each organism has one or more chromosomes that
Genomics and Transcriptomics of Spirodela polyrhiza
 Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
De novo assembly in RNA-seq analysis.
 De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
ABSTRACT. Genome Assembly:
 ABSTRACT Title of dissertation: Genome Assembly: Novel Applications by Harnessing Emerging Sequencing Technologies and Graph Algorithms Sergey Koren, Doctor of Philosophy, 2012 Dissertation directed by:
ABSTRACT Title of dissertation: Genome Assembly: Novel Applications by Harnessing Emerging Sequencing Technologies and Graph Algorithms Sergey Koren, Doctor of Philosophy, 2012 Dissertation directed by:
Analysis of structural variation. Alistair Ward - Boston College
 Analysis of structural variation Alistair Ward - Boston College What is structural variation? What differentiates SV from short variants? What are the major SV types? Summary of MEI detection What is an
Analysis of structural variation Alistair Ward - Boston College What is structural variation? What differentiates SV from short variants? What are the major SV types? Summary of MEI detection What is an
Why are we here? Introduction
 Why are we here? Introduction Genome assembly Original DNA Fragments Sequenced ends Fragments Contigs Scaffold A correct assembly The right motifs, the correct number of times, in correct order and position.
Why are we here? Introduction Genome assembly Original DNA Fragments Sequenced ends Fragments Contigs Scaffold A correct assembly The right motifs, the correct number of times, in correct order and position.
Supplementary Data for Hybrid error correction and de novo assembly of single-molecule sequencing reads
 Supplementary Data for Hybrid error correction and de novo assembly of single-molecule sequencing reads Online Resources Pre&compiledsourcecodeanddatasetsusedforthispublication: http://www.cbcb.umd.edu/software/pbcr
Supplementary Data for Hybrid error correction and de novo assembly of single-molecule sequencing reads Online Resources Pre&compiledsourcecodeanddatasetsusedforthispublication: http://www.cbcb.umd.edu/software/pbcr
Each cell of a living organism contains chromosomes
 COVER FEATURE Genome Sequence Assembly: Algorithms and Issues Algorithms that can assemble millions of small DNA fragments into gene sequences underlie the current revolution in biotechnology, helping
COVER FEATURE Genome Sequence Assembly: Algorithms and Issues Algorithms that can assemble millions of small DNA fragments into gene sequences underlie the current revolution in biotechnology, helping
De novo genome assembly. Dr Torsten Seemann
 De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
Genome Assembly Using de Bruijn Graphs. Biostatistics 666
 Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Genome Assembly Using de Bruijn Graphs Biostatistics 666 Previously: Reference Based Analyses Individual short reads are aligned to reference Genotypes generated by examining reads overlapping each position
Next Genera*on Sequencing II: Personal Genomics. Jim Noonan Department of Gene*cs
 Next Genera*on Sequencing II: Personal Genomics Jim Noonan Department of Gene*cs Personal genome sequencing Iden*fying the gene*c basis of phenotypic diversity among humans Gene*c risk factors for disease
Next Genera*on Sequencing II: Personal Genomics Jim Noonan Department of Gene*cs Personal genome sequencing Iden*fying the gene*c basis of phenotypic diversity among humans Gene*c risk factors for disease
A Short Sequence Splicing Method for Genome Assembly Using a Three- Dimensional Mixing-Pool of BAC Clones and High-throughput Technology
 Send Orders for Reprints to reprints@benthamscience.ae 210 The Open Biotechnology Journal, 2015, 9, 210-215 Open Access A Short Sequence Splicing Method for Genome Assembly Using a Three- Dimensional Mixing-Pool
Send Orders for Reprints to reprints@benthamscience.ae 210 The Open Biotechnology Journal, 2015, 9, 210-215 Open Access A Short Sequence Splicing Method for Genome Assembly Using a Three- Dimensional Mixing-Pool
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Supplementary Material
 Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory
 Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title Glomus intraradices: Initial Whole-Genome Shotgun Sequencing and Assembly Results Permalink https://escholarship.org/uc/item/4c13k1dh
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title Glomus intraradices: Initial Whole-Genome Shotgun Sequencing and Assembly Results Permalink https://escholarship.org/uc/item/4c13k1dh
A Roadmap to the De-novo Assembly of the Banana Slug Genome
 A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased
 has increased") Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
Finishing Drosophila ananassae Fosmid 2410F24
 Nick Spies Research Explorations in Genomics Finishing Report Elgin, Shaffer and Leung 23 February 2013 Abstract: Finishing Drosophila ananassae Fosmid 2410F24 Finishing Drosophila ananassae fosmid clone
Nick Spies Research Explorations in Genomics Finishing Report Elgin, Shaffer and Leung 23 February 2013 Abstract: Finishing Drosophila ananassae Fosmid 2410F24 Finishing Drosophila ananassae fosmid clone
High-Throughput Bioinformatics: Re-sequencing and de novo assembly. Elena Czeizler
 High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
Finishing Drosophila Ananassae Fosmid 2728G16
 Finishing Drosophila Ananassae Fosmid 2728G16 Kyle Jung March 8, 2013 Bio434W Professor Elgin Page 1 Abstract For my finishing project, I chose to finish fosmid 2728G16. This fosmid carries a segment of
Finishing Drosophila Ananassae Fosmid 2728G16 Kyle Jung March 8, 2013 Bio434W Professor Elgin Page 1 Abstract For my finishing project, I chose to finish fosmid 2728G16. This fosmid carries a segment of
Direct determination of diploid genome sequences. Supplemental material: contents
 Direct determination of diploid genome sequences Neil I. Weisenfeld, Vijay Kumar, Preyas Shah, Deanna M. Church, David B. Jaffe Supplemental material: contents Supplemental Note 1. Comparison of performance
Direct determination of diploid genome sequences Neil I. Weisenfeld, Vijay Kumar, Preyas Shah, Deanna M. Church, David B. Jaffe Supplemental material: contents Supplemental Note 1. Comparison of performance
SUPPLEMENTARY MATERIAL FOR THE PAPER: RASCAF: IMPROVING GENOME ASSEMBLY WITH RNA-SEQ DATA
 SUPPLEMENTARY MATERIAL FOR THE PAPER: RASCAF: IMPROVING GENOME ASSEMBLY WITH RNA-SEQ DATA Authors: Li Song, Dhruv S. Shankar, Liliana Florea Table of contents: Figure S1. Methods finding contig connections
SUPPLEMENTARY MATERIAL FOR THE PAPER: RASCAF: IMPROVING GENOME ASSEMBLY WITH RNA-SEQ DATA Authors: Li Song, Dhruv S. Shankar, Liliana Florea Table of contents: Figure S1. Methods finding contig connections
Genome Sequencing and Assembly
 Genome Sequencing and Assembly History of Sequencing What was the first fully sequenced nucleic acid? Yeast trna (alanine trna) Robert Holley 1965 Image: Wikipedia History of Sequencing Sequencing began
Genome Sequencing and Assembly History of Sequencing What was the first fully sequenced nucleic acid? Yeast trna (alanine trna) Robert Holley 1965 Image: Wikipedia History of Sequencing Sequencing began
Genome Assembly. Microbial Single Cell Genomics Workshop 2010 Sergey Koren JCVI, CBCB at UMD
 Genome Assembly Microbial Single Cell Genomics Workshop 2010 Sergey Koren JCVI, CBCB at UMD Introduction Platform: 3730 Model length reads bases ABI 3730 800 96 80K Applied Biosystems 3730xl DNA amplification
Genome Assembly Microbial Single Cell Genomics Workshop 2010 Sergey Koren JCVI, CBCB at UMD Introduction Platform: 3730 Model length reads bases ABI 3730 800 96 80K Applied Biosystems 3730xl DNA amplification
de novo paired-end short reads assembly
 1/54 de novo paired-end short reads assembly Rayan Chikhi ENS Cachan Brittany Symbiose, Irisa, France 2/54 THESIS FOCUS Graph theory for assembly models Indexing large sequencing datasets Practical implementation
1/54 de novo paired-end short reads assembly Rayan Chikhi ENS Cachan Brittany Symbiose, Irisa, France 2/54 THESIS FOCUS Graph theory for assembly models Indexing large sequencing datasets Practical implementation
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Purpose of sequence assembly
 Sequence Assembly Purpose of sequence assembly Reconstruct long DNA/RNA sequences from short sequence reads Genome sequencing RNA sequencing for gene discovery But not for transcript quantification Variant
Sequence Assembly Purpose of sequence assembly Reconstruct long DNA/RNA sequences from short sequence reads Genome sequencing RNA sequencing for gene discovery But not for transcript quantification Variant
Finished (Almost) Sequence of Drosophila littoralis Chromosome 4 Fosmid Clone XAAA73. Seth Bloom Biology 4342 March 7, 2004
 Sequence of Drosophila littoralis Chromosome 4 Fosmid Clone XAAA73. Seth Bloom Biology 4342 March 7, 2004") Finished (Almost) Sequence of Drosophila littoralis Chromosome 4 Fosmid Clone XAAA73 Seth Bloom Biology 4342 March 7, 2004 Summary: I successfully sequenced Drosophila littoralis fosmid clone XAAA73. The
Finished (Almost) Sequence of Drosophila littoralis Chromosome 4 Fosmid Clone XAAA73 Seth Bloom Biology 4342 March 7, 2004 Summary: I successfully sequenced Drosophila littoralis fosmid clone XAAA73. The
The Irys System. Rapid Genome Wide Mapping for de novo Assembly and Structural Variation Analysis. Jack Peart, Ph.D. Director of Sales EMEA
 The Irys System Rapid Genome Wide Mapping for de novo Assembly and Structural Variation Analysis Jack Peart, Ph.D. Director of Sales EMEA BioNano Snapshot Developed & Commercialized the Irys System for
The Irys System Rapid Genome Wide Mapping for de novo Assembly and Structural Variation Analysis Jack Peart, Ph.D. Director of Sales EMEA BioNano Snapshot Developed & Commercialized the Irys System for
Bionano Solve Theory of Operation: Variant Annotation Pipeline
 Bionano Solve Theory of Operation: Variant Annotation Pipeline Document Number: 30190 Document Revision: B For Research Use Only. Not for use in diagnostic procedures. Copyright 2018 Bionano Genomics,
Bionano Solve Theory of Operation: Variant Annotation Pipeline Document Number: 30190 Document Revision: B For Research Use Only. Not for use in diagnostic procedures. Copyright 2018 Bionano Genomics,
The Human Genome and its upcoming Dynamics
 The Human Genome and its upcoming Dynamics Matthias Platzer Genome Analysis Leibniz Institute for Age Research - Fritz-Lipmann Institute (FLI) Sequencing of the Human Genome Publications 2004 2001 2001
The Human Genome and its upcoming Dynamics Matthias Platzer Genome Analysis Leibniz Institute for Age Research - Fritz-Lipmann Institute (FLI) Sequencing of the Human Genome Publications 2004 2001 2001
ARACHNE: A Whole-Genome Shotgun Assembler
 Methods Serafim Batzoglou, 1,2,3 David B. Jaffe, 2,3,4 Ken Stanley, 2 Jonathan Butler, 2 Sante Gnerre, 2 Evan Mauceli, 2 Bonnie Berger, 1,5 Jill P. Mesirov, 2 and Eric S. Lander 2,6,7 1 Laboratory for
Methods Serafim Batzoglou, 1,2,3 David B. Jaffe, 2,3,4 Ken Stanley, 2 Jonathan Butler, 2 Sante Gnerre, 2 Evan Mauceli, 2 Bonnie Berger, 1,5 Jill P. Mesirov, 2 and Eric S. Lander 2,6,7 1 Laboratory for
Understanding Accuracy in SMRT Sequencing
 Understanding Accuracy in SMRT Sequencing Jonas Korlach, Chief Scientific Officer, Pacific Biosciences Introduction Single Molecule, Real-Time (SMRT ) DNA sequencing achieves highly accurate sequencing
Understanding Accuracy in SMRT Sequencing Jonas Korlach, Chief Scientific Officer, Pacific Biosciences Introduction Single Molecule, Real-Time (SMRT ) DNA sequencing achieves highly accurate sequencing
Conditional Random Fields, DNA Sequencing. Armin Pourshafeie. February 10, 2015
 Conditional Random Fields, DNA Sequencing Armin Pourshafeie February 10, 2015 CRF Continued HMMs represent a distribution for an observed sequence x and a parse P(x, ). However, usually we are interested
Conditional Random Fields, DNA Sequencing Armin Pourshafeie February 10, 2015 CRF Continued HMMs represent a distribution for an observed sequence x and a parse P(x, ). However, usually we are interested
3) This diagram represents: (Indicate all correct answers)
 This diagram represents: (Indicate all correct answers)") Functional Genomics Midterm II (self-questions) 2/4/05 1) One of the obstacles in whole genome assembly is dealing with the repeated portions of DNA within the genome. How do repeats cause complications
Functional Genomics Midterm II (self-questions) 2/4/05 1) One of the obstacles in whole genome assembly is dealing with the repeated portions of DNA within the genome. How do repeats cause complications
GENETICS - CLUTCH CH.15 GENOMES AND GENOMICS.
 !! www.clutchprep.com CONCEPT: OVERVIEW OF GENOMICS Genomics is the study of genomes in their entirety Bioinformatics is the analysis of the information content of genomes - Genes, regulatory sequences,
!! www.clutchprep.com CONCEPT: OVERVIEW OF GENOMICS Genomics is the study of genomes in their entirety Bioinformatics is the analysis of the information content of genomes - Genes, regulatory sequences,
BroadE Workshop: Genome Assembly. March 20 th, 2013
 BroadE Workshop: Genome Assembly March 20 th, 2013 Introduc@on & Logis@cs De- Bruijn Graph Interac@ve Problem (45 minutes) Assembly Theory Lecture (45 minutes) Break (10-15 minutes) Assembly in Prac@ce
BroadE Workshop: Genome Assembly March 20 th, 2013 Introduc@on & Logis@cs De- Bruijn Graph Interac@ve Problem (45 minutes) Assembly Theory Lecture (45 minutes) Break (10-15 minutes) Assembly in Prac@ce
SUPPLEMENTARY INFORMATION
 Contents De novo assembly... 2 Assembly statistics for all 150 individuals... 2 HHV6b integration... 2 Comparison of assemblers... 4 Variant calling and genotyping... 4 Protein truncating variants (PTV)...
Contents De novo assembly... 2 Assembly statistics for all 150 individuals... 2 HHV6b integration... 2 Comparison of assemblers... 4 Variant calling and genotyping... 4 Protein truncating variants (PTV)...
Genome annotation & EST
 Genome annotation & EST What is genome annotation? The process of taking the raw DNA sequence produced by the genome sequence projects and adding the layers of analysis and interpretation necessary
Genome annotation & EST What is genome annotation? The process of taking the raw DNA sequence produced by the genome sequence projects and adding the layers of analysis and interpretation necessary