Sequence File Formats
|
|
|
- Delilah Marsh
- 6 years ago
- Views:
Transcription
1 File Formats
2 Sequence File Formats Different formats for different uses Competing formats developed in parallel Some easy to read, some easy to write programs Don t have to stick to these formats, but parsers already written! All formats are plain text
3 Standard genetic code Symbol Meaning Origin G G Guanine A A Adenine C C Cytosine T T Thymine R G or A purine Y T or C pyrimidine M A or C amino K G or T Keto N G or A or T or C any
4 Standard protein codes One Three Amino acid One Three Amino acid A Ala Alanine M Met Methionine C Cys Cysteine N Asn Asparagine D Asp Aspartic acid P Pro Proline E Glu Glutamate R Arg Arginine F Phe Phenylalanine S Ser Serine G Gly Glycine T Thr Threonine H His Histidine V Val Valine I Ile Isoleucine W Trp Tryptophan K Lys Lysine Y Tyr Tyrosine L Leu Leucine X Xaa Unknown
5 GenBank More complex, includes detailed information on genes, cds, annotation etc Human readable Difficult to parse Use standard parsers (bioperl, biojava, etc)
6 LOCUS NC_ bp ss-dna circular PHG 17-APR-2009 DEFINITION Pseudomonas phage Pf3, complete genome. ACCESSION NC_ VERSION NC_ GI: DBLINK Project:14061 KEYWORDS. SOURCE Pseudomonas phage Pf3 ORGANISM Pseudomonas phage Pf3 Viruses; ssdna viruses; Inoviridae; Inovirus. FEATURES Location/Qualifiers source /organism="pseudomonas phage Pf3" /mol_type="genomic DNA" /host="pseudomonas aeruginosa" /db_xref="taxon:10872" /note="pf3 bacteriophage DNA from P.aeruginosa infected with plasmid RP1." gene join( ,1..106) /locus_tag="pf3_1" /db_xref="geneid: " CDS join( ,1..106) /locus_tag="pf3_1" /note="orf 58, part 2" /codon_start=1 /transl_table=11 /product="hypothetical protein" /protein_id="np_ " /db_xref="gi: " /db_xref="geneid: " /translation="msyyvcvqlvndvchewaersdllslpegsglqiggmllllsat AWGIQQIARLLLNR"
7 3241 aggtcctgtt ggccttaaga tcacccaagg gcatcttgcc agatggtacc gtcattactt 3301 atgagaaaat atcctcaatg ggtaatggct ataccttcga gcttgagtcg cttatatttg 3361 cggctcttgc tcggtcttta tgcgaattac tgggcttacg accgtcagat gttacggtct 3421 atggcgatga cataatattg ccatcagacg cgtgcagtcc tctagttgaa gttttctcct 3481 atgttggttt tcgtaccaac aagaagaaaa cgttttctag tggaccgttc cgagagtcgt 3541 gcggaaagca ctactttttg ggcgttgacg tcacaccttt ctacatacgt cgccgtatag 3601 tgagtccctc cgatctcata ctggttttga accagatgta tcgttgggcc acaattgacg 3661 gcgtatggga tcctagggta tatcctgtat acaccaagta tagacgttac cttccggaaa 3721 ttctccggag gaatgtcgtg cctgatggat acggtgatgg tgccctcgtc ggatctgtct 3781 taatcagtcc tttcgcagaa aatcgcggtt gggttcggcg tgtgccgatg attatagaca 3841 agaggaaaga ccgagttcgt gacgaatatg gttcgtatct ctacgagcta tggtcgttgc 3901 agcaactcga atgtgacagt gagttcccct ttaacgggtc gctggtcgtt ggttccactg 3961 atggcactct cgcttacgca caccgagaac ggttacctac cgttatcagt gatgccgtaa 4021 gtgcgtttga catcatgtgg ataccgtgca gtagtcgtgt cctggctccc tacggggatt 4081 tccggaggca cgaaggctct atcctaaaaa tggggtagcg cctgggaggg gtgcattatg 4141 caccctaggt tagcaatact taaactaacc ttctcaaaag agagagtgaa ggctctgctt 4201 tgccctcact cctccca // LOCUS NC_ bp ds-rna linear PHG 23-AUG-2008 DEFINITION Pseudomonas phage phi8 segment S, complete sequence. ACCESSION NC_ VERSION NC_ GI: DBLINK Project:14731 KEYWORDS. SOURCE Pseudomonas phage phi8 ORGANISM Pseudomonas phage phi8 Viruses; dsrna viruses; Cystoviridae; Cystovirus.
8 GFF3 Tab separated format Easy to parse Attributes are tag/value pairs separated by ; Columns: 1. Contig 2. Source database 3. Feature type 4. Start 5. Stop 6. Score 7. Strand 8. Phase 9. Attributes
9 ASN.1 Developed as computer readable form of GenBank Not widely used
10 ASN.1 seq { id { local id 1 }, descr { title "" }, inst { repr raw, mol aa, length 131, topology linear, { seq-data iupacaa "TSPASIRPPAGPSSRPAMVSSRRTRPSPPGPRRPTGRPCCSAAPRRPQA TGGWKTCSGTCTTSTSTRHRGRSGWSARTTTAACLRASRKSMRAACSRSAGSRPNRFA PTLMSSCITSTTGPPAWAGDRSHE" } }, seq { id { local id 1 }, descr { title "" }, inst { repr raw, mol aa, length 131, topology linear, { seq-data iupacaa "TSPASIRPPAGPSSR RPSPPGPRRPTGRPCCSAAPRRPQAT GGWKTCSGTCTTSTSTRHRGRSGW RASRKSMRAACSRSAGSRPNRFAPTL MSSCITSTTGPPAWAGDRSHE" } }
11 Fasta l Also called Pearson Simplest file format. Easy to parse, easy to use >identifier [optional information] ATGACTAGCATGCATCGATCGATCGACTAGCATG ACTGCACTACGACGACAGCAAC >identifier2 [optional information] ACTAGCTCAGCTAGAGAGCTACGATCAGCACTAC atccgatagcatgacttactacgctagcatcagtcat CAT
12 Qual >identifier [optional information] >identifier2 [optional information]
13 Based on fasta format fastq Contains information about the quality of the sequence Quality comes from sequencing machines! Four lines per sequence: Line = identifier line before the sequence DNA sequence Line starting + = identifier line before the quality scores String = quality scores as ASCII + 33
14 EIXKN4201CFU84 length=93 GGGGGGGGGGGGGGGGCTTTTTTTGTTTGGAACCGA AAGGGTTTTGAATTTCAAACCCTTTTCGGTTTCCAA CCTTCCAAAGCAATGCCAATA +SRR EIXKN4201CFU84 length=93 A1EA1EA5
15 Base calling l l l l l Need to be sure which base you have identified Depends on the technology Each machine includes software Phred is an historical package developed by at U. Washington Phred scores are probability that the base is correct
16 Quality values l l l l Phred 10: 1 x 10 1 chance that the base is wrong Phred 20: 1 x 10 2 chance that the base is wrong Phred 30: 1 x 10 3 chance that the base is wrong Phred 40: 1 x 10 4 chance that the base is wrong l Phred 99: the base is correct! l Fastq scores are the score + 33 then converted to ascii text
17 ASCII character codes ASCII Char ASCII Char ASCII Char ASCII Char ASCII Char 33! F 90 Z 110 n 34 " G 91 [ 111 o 35 # H 92 \ 112 p 36 $ I 93 ] 113 q 37 % J 94 ^ 114 r 38 & K 95 _ 115 s 39 ' L 96 ` 116 t 40 ( M 97 a 117 u 41 ) 58 : 78 N 98 b 118 v 42 * 59 ; 79 O 99 c 119 w < 80 P 100 d 120 x 44, 61 = 81 Q 101 e 121 y > 82 R 102 f 122 z ? 83 S 103 g 123 { 47 / 84 T 104 h A 85 U 105 i 125 } B 86 V 106 j 126 ~
18 EIXKN4201CFU84 length=93 DNA sequence GGGGGGGGGGGGGGGGCTTTTTTTGTTTGGAACCGA AAGGGTTTTGAATTTCAAACCCTTTTCGGTTTCCAA CCTTCCAAAGCAATGCCAATA +SRR EIXKN4201CFU84 length=93 A1EA1EA5 Quality scores Note: Illumina used to have a format of fastq that was not compatible with everyone else s format!


19 Ion quality scores PRINSEQ Rob Schmieder
20 prinseq
21 Basic data analysis New dataset Assemble data Perform similarity search
22 Bad data analysis
23 Bad data analysis
24 Bad data analysis
25 Bad data analysis
26 Bad data analysis

27 Bad data analysis
28 New dataset Good data analysis
29 Good data analysis New dataset Quality control & Preprocessing
30 Good data analysis New dataset Quality control & Preprocessing Assembly Similarity search
31 Good data analysis New dataset Quality control & Preprocessing Assembly Similarity search
32 3 Tools for metagenomic data
33 Quality control and data preprocessing
34 Number and Length of Sequences

 à Short reads are likely")
35 Number/Length of sequences Bad Good Reads should be approx. same length (same number of cycles) à Short reads are likely lower quality
36 Quality of Sequences

37 Linearly degrading quality across the read àtrim low quality ends
38 Quality filtering Any region with homopolymer will tend to have a lower quality score Huseet al. found that sequences with an average score below 25 had more errors than those with higher averages Huse et al.: Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology (2007)
39 Low quality sequence issue Most assemblers or aligners do not take into account quality scores Errors in reads complicate assembly, might cause misassembly, or make assembly impossible
40 What if quality scores are not available? Alternative: Infer quality from the percent of Ns found in the sequence Removes regions with a high number of Ns Huseet al. found that presence of any ambiguous base calls was a sign for overall poor sequence quality Huse et al.: Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology (2007)
41 Ambiguous bases If you can afford the loss, filter out all reads containing Ns Assemblers (e.g. Velvet) and aligners (SHAHA2, BWA, ) use 2-bit encoding system for nucleotides some replace Ns with random base, some with fixed base (e.g. SHAHA2 & Velvet = A) 2-bit example: 00 A, 01 C, 10 G, 11 - T
42 Tag Sequences

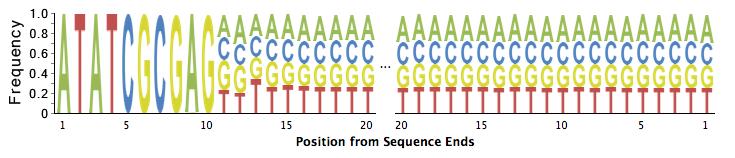
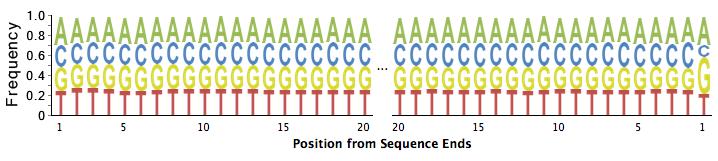
43 o tag ID tag TA tags
44 Detect and remove tag sequences

45 Data upload Tag sequence definition

46 Tag sequence prediction
47 Parameter definition Download results
48 Sequence Contamination
49 Principal component analysis (PCA) of dinucleotide relative abundance Microbial metagenomes Viral metagenomes
50 Identification and removal of sequence contamination
51 Contaminant identification Current methods have critical limitations Dinucleotide relative abundance uses information content in sequences à can not identify single contaminant sequences Sequence similarity seems to be only reliable option to identify single contaminant sequences BLAST against human reference genome is slow and lacks corresponding regions (gaps, variants, ) Novel sequences in every new human genome sequenced* * Li et al.: Building the sequence map of the human pan-genome. Nature Biotechnology (2010)
52 DeconSeq web interface Two types of reference databases Remove Retain
53 DeconSeq web interface (cont.)


54 Human DNA contamination identified in 145 out of 202 metagenomes
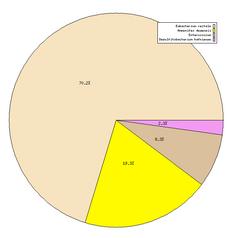
55 Contamination Identification
56
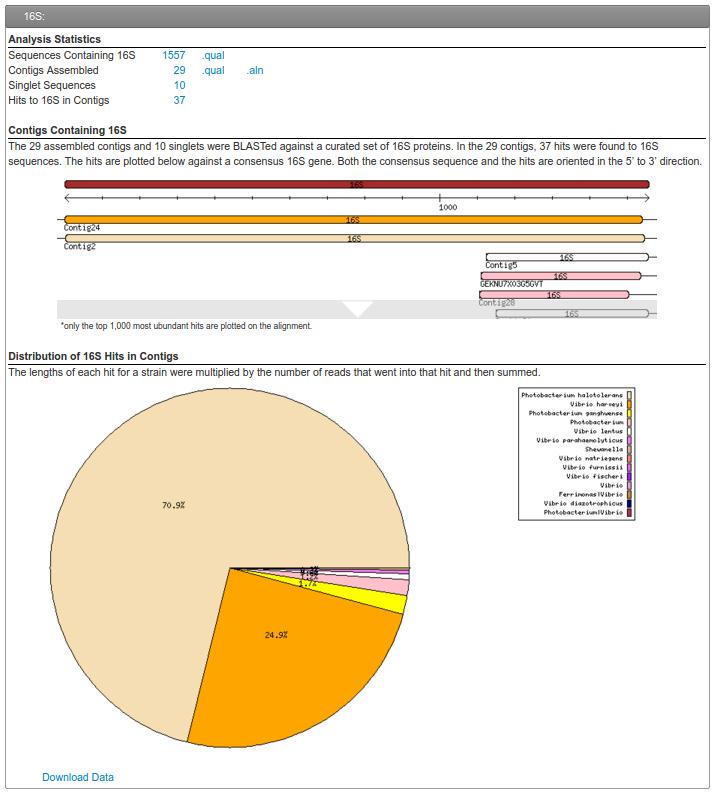
57 16S
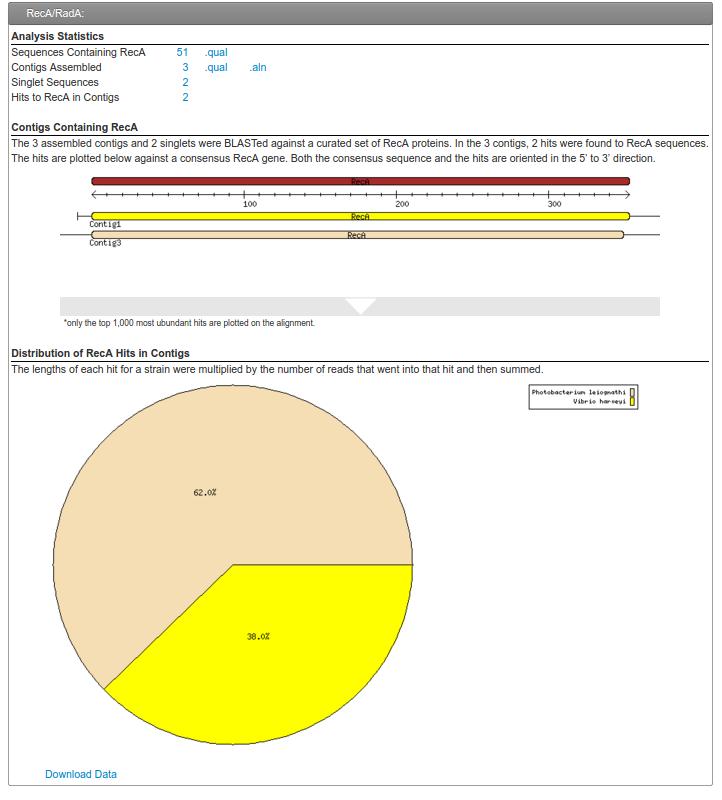
58 reca
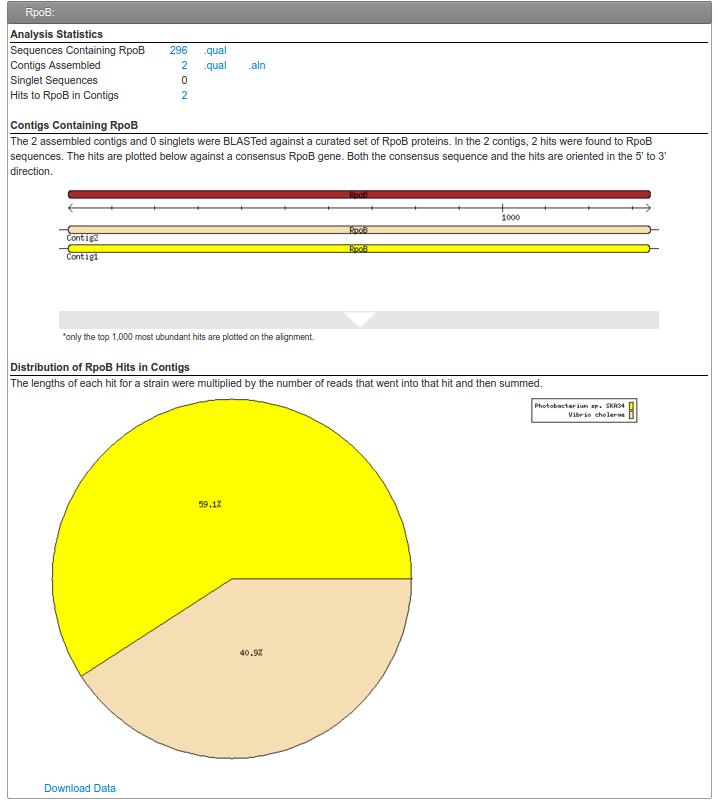
59 rpob
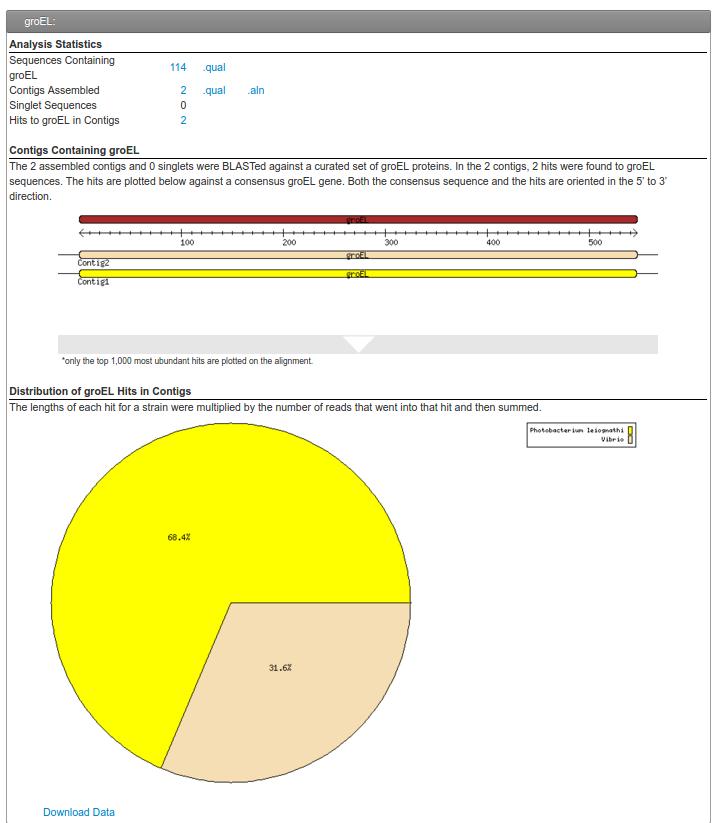
60 groel
61 Using prinseq to check the quality of the data
62 Using prinseq 1. In the folder, go to /home/qiime/documents/ecoli 2. Right click on CIA49E_coli_DH10B_Control_200.zip Choose Extract Here 3. Right click on R_2014_02_04_00_54_34_user_CIA-49-Ion_PGM_E_coli_DH10B_Control_200 Choose open terminal here 4. Run this command: prinseq-lite.pl -fastq Ecoli.fastq -graph_stats ld,gc,qd,ns,ts -graph_data ~/Desktop/DH10B.gd 5. Open firefox: go to Choose Get Report
63 After checking the quality: 1. Trim the sequences to remove low quality (<15) at the right end 2. Filter the sequences to remove sequences less than 200 bp. 3. Create a new fastq file prinseq-lite.pl -h
64 Trimming prinseq-lite.pl -fastq ecoli.fastq -min_len 200 -trim_qual_right 15 -out_good DH10B.trimmed
65 Convert fastq to fasta On the virtual box: l fastq2qual_fasta.pl l fastq_to_fasta l prinseq-lite.pl On the web: l
66 Using prinseq to convert fastq to fasta prinseq-lite.pl -h prinseq-lite.pl -fastq Ecoli_trimmed.fastq -out_format 2 -out_good Ecoli_trimmed
67 Sequence Assembly
68 Assembling the data l Problem: the longest single sequence possible is 1,000 bp, and most technology is bp. l Microbial genomes are 2,000,000 bp l Therefore how do you sequence a whole genome?
69 Sequencing the genomes l Extract DNA l Shear DNA into small pieces l Ligate adapters on each end l Sequencing using next generation sequencing
70 Sequence assembly l l Before we look at the data Can we make longer pieces
71 The assembly l l l l A hierarchical data structure that maps sequence data to a reconstruction of the target. The assembly groups l l reads into contigs contigs into scaffolds Contigs provide l l multiple sequence alignment of reads consensus sequence. Scaffolds provide l l contig order and orientation sizes of the gaps between contigs.
72 Sequence assembly Reads Contigs Scaffolds
73 Four approaches to assembly Naïve approach Greedy approach Overlap / Layout / Consensus de Bruijn Graphs
74 Naïve approach l l l l Compare every sequence to every other sequence Find stretches that are the same Need to account for phred scores what if a base is wrong? How long of a sequence do you need to be unique?
75 Sequence composition l l 4 bases 4 n chance of finding a sequence if all evenly used (they are not) l 3 bp: 4 3 = 64 l 8 bp: 4 8 = 65,336 l 20 bp: 4 20 = 1,099,511,627,776
76 Problems with this approach l Sequences are not random l Most genomes contain biased information l Repeat sequences in the genome
77 Greedy approaches Start with a sequence Keep extending it while another sequence matches the end When can not be extended further, mark as a contig
78 Improve greedy approachs Only use high quality sequence Use reads that are represented more than n-times in the sample (SSAKE) End to end overlap vs. partial overlap Ignores low coverage regions also incorporate quality scores (SHARCGS) In general, greedy approaches are fast but not very good. Make lots of short contigs
79 Overlap / Layout / Consensus All versus all comparison (done with K-mers for speed). Generate approximate read layout as an overlap graph. Use multiple sequence alignments to resolve layout.
80 Newbler (O/L/C) Makes unitigs Single contigs with no discrepancies Merge unitigs into contigs. May split unitigs and even reads (could be chimeras) Use coverage to compensate for base calls Works in flow space to calculate homopolymeric tracts. More accurate than average of averages
81 Assembly is a graph problem Overlap/Layout/Consensus de Bruijn Graph Greedy graphs A graph is nodes + edges node edge
82 Assemble these two sequences! AACCGGT CCGGTTA Consensus: AACCGGTTA
83 AACCGGT as graphs Node = K-mers; edges = nodes that overlap by K-1 bases. aacc accg ccgg cggt Here K = 4, but in reality K = 19 to 31
84 CCGGTTA as graphs ccgg cggt ggtt gtta
85 Join the two graphs ccgg cggt ggtt gtta aacc accg ccgg cggt
86 Join the two graphs ccgg cggt ggtt gtta aacc accg ccgg cggt
87 Join the two graphs ccgg cggt ggtt gtta aacc accg ccgg cggt AACCGGTTA
88 Differences between an overlap graph and a de Bruijn graph for assembly. Schatz M C et al. Genome Res. 2010;20: by Cold Spring Harbor Laboratory Press
89 Problems with all assemblies l l l Sequences are not random Most genomes contain biased information Repeat sequences in the genome
90 Repeats
91 Repeats
92 Repeats
93 Repeats
94 Repeats have multiple sinks/sources
95 Repeats have multiple sinks/sources 16s Salmonella has 7 rrn operons Salmonella recombines at rrn operons Helm and Maloy
96 Repeat sequences l l l l What happens if the repeat is longer than the read length? Need paired end reads to resolve order Need pairs that span the repeat Need pairs with one end in the repeat
97 Mate pair and paired end sequencing
98 Mate pair Sequencing (Ion Torrent) Add linkers
99 Mate pair sequencing (Ion Torrent) Nick Sequencing migration
100 Paired End Sequencing (Illumina) Left read DNA fragment Right read
101 Paired End Sequencing (Illumina) Number of fragments Fragment length
102 Paired End Sequencing (Illumina)
103 Joining paired end sequences _ \ / \ _ \ _) _ / _ \ _) / / \ _ < _ /_/ \_\_ \_\ PEAR v0.9.6 [January 15, 2015] - [+bzlib +zlib] Citation - PEAR: a fast and accurate Illumina Paired-End read merger Zhang et al (2014) Bioinformatics 30(5): doi: /bioinformatics/btt593 License: Creative Commons Licence Bug-reports and requests to: Tomas.Flouri@h-its.org and Jiajie.Zhang@h-its.org Usage: pear <options> Standard (mandatory): -f, --forward-fastq <str> Forward paired-end FASTQ file. -r, --reverse-fastq <str> Reverse paired-end FASTQ file. -o, --output <str> Output filename.
104 Repeats A B C Paired end reads or mate pairs
105 Sequence assembly Reads Contigs Scaffolds
106 Current assemblers AMOS Celera WGA Assembler CLC Genomics Workbench DNA Dragon DNAnexus Euler Geneious IDBA (Iterative De Bruijn graph short read Assembler) LIGR Assembler (derived from TIGR Assembler) MIRA (Mimicking Intelligent Read Assembly) Newbler Phrap SSAKE SOAPdenovo SPAdes Velvet
107 Assembly 1. De novo assembly with newbler (runassembly) 2. Map to the genome with newbler (runmapping) 3. De novo assembly with spades (spades.py)
108 De novo assembly with newbler 1. Convert fastq to fasta 2. Use this command: runassembly <fasta file> e.g. (all on one line) runassembly -noinfo -nobig -noace Ecoli.fasta 3. How good is the assembly basic_stats 454AllContigs.fna
109 basic_stats: assembly BASIC FASTA STATISTICS Total number of sequences: 1882 Total number of bases: 4,266,422 bp (4.27 Mb) Average sequence length: Minimum sequence length: Maximum sequence length: N50: 3491 bp 100 bp bp 50% of total sequence length is contained in 369 sequences GC %: %
110 basic_stats: mapping BASIC FASTA STATISTICS Total number of sequences: 654 Total number of bases: 4,339,931 bp (4.34 Mb) Average sequence length: Minimum sequence length: Maximum sequence length: N50: bp 160 bp bp 50% of total sequence length is contained in 109 sequences GC %: %
111 N50
112 N50 Length = N50
113 Mapping to a genome 1. Convert the E. coli genbank file to fasta format GB2Fasta.pl CP000948_DH10B.gbk CP000948_DH10B.fasta 2. Map the reads to the genome runmapping -cpu 2 -noinfo -noace -nobig -gref CP000948_DH10B.fasta DH10B.trimmed.fasta 3. How good is the assembly basic_stats 454AllContigs.fna
114 SPAdes Run this command (all on one line): /opt/spades linux/bin/spades.py --iontorrent -k 21,33,55,77,99,127 --mismatch-correction -t 2 -s ecoli.fastq -o ecoli.spades

115 Hybrid assembly Geni Silva
116 scaffold_builder Silva et al. Source Code for Biology and Medicine 2013, 8:23
117 How do we know if the assembly is good?
118 QUAST 1. Run this command (all on one line): quast -o Ecoli.quast -R ecoli.fasta runmapping/454allcontigs.fna runassembly/454allcontigs.fna python /opt/quast_3.2/quast.py -o Ecoli.quast -R ecoli.fasta runmapping/454allcontigs.fna runassembly/ 454AllContigs.fna 2. When the command finishes: gnome-open DH10B.quast/report.html (also open DH10B.quast/alignment.svg and DH10B.quast/report.pdf)
119 Mauve Locally co-linear blocks Homologous regions shared by any two genomes User specified cutoff to homologous
120 Multi-mums Multiple unique matches Identify k-mers that: Are shared by 2-or more genomes Only appear once per genome Bounded by mismatched base (cannot be extended further)
121 Mauve Find all local multimums Calculate phylogenetic guide tree Select a subset of multimums to create LCBs Anchor regions not covered with multimums Align LCBs
122 How to construct... Construct sorted list of k-mers Find set that match occurrence criteria: Once per genome >1 genome Extend until mismatch

123 Constructing sorted list Hash using hash function: S 1 = start of multimum M in first genome S j = start of multimum M in genome j
124 Join all mums Join all mums together where: M i S j <= M i+1 S j For any genome j, the start of the multimum i is less than or equal to the start of the next multimum.
125 Start with all matches
126 Partition into locally co-linear blocks
127 Remove low scoring lcb's (3 * length of k-mer)

128 Reduce k-mer size, and repeat for unaligned regions
129 Mauve Speed Aligned many prokaryote genomes Human, mouse, and rat genomes
130 Problems - Repeats For a repetitive element appearing r times in G genomes: r G possible combinations r of them are correct!
131 Mauve From the Apps menu on the top left, choose Mauve File Align with ProgressiveMauve
Basic concepts of molecular biology
 Basic concepts of molecular biology Gabriella Trucco Email: gabriella.trucco@unimi.it Life The main actors in the chemistry of life are molecules called proteins nucleic acids Proteins: many different
Basic concepts of molecular biology Gabriella Trucco Email: gabriella.trucco@unimi.it Life The main actors in the chemistry of life are molecules called proteins nucleic acids Proteins: many different
11 questions for a total of 120 points
 Your Name: BYS 201, Final Exam, May 3, 2010 11 questions for a total of 120 points 1. 25 points Take a close look at these tables of amino acids. Some of them are hydrophilic, some hydrophobic, some positive
Your Name: BYS 201, Final Exam, May 3, 2010 11 questions for a total of 120 points 1. 25 points Take a close look at these tables of amino acids. Some of them are hydrophilic, some hydrophobic, some positive
DNA.notebook March 08, DNA Overview
 DNA Overview Deoxyribonucleic Acid, or DNA, must be able to do 2 things: 1) give instructions for building and maintaining cells. 2) be copied each time a cell divides. DNA is made of subunits called nucleotides
DNA Overview Deoxyribonucleic Acid, or DNA, must be able to do 2 things: 1) give instructions for building and maintaining cells. 2) be copied each time a cell divides. DNA is made of subunits called nucleotides
Basic concepts of molecular biology
 Basic concepts of molecular biology Gabriella Trucco Email: gabriella.trucco@unimi.it What is life made of? 1665: Robert Hooke discovered that organisms are composed of individual compartments called cells
Basic concepts of molecular biology Gabriella Trucco Email: gabriella.trucco@unimi.it What is life made of? 1665: Robert Hooke discovered that organisms are composed of individual compartments called cells
Bioinformatics. ONE Introduction to Biology. Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012
 Bioinformatics ONE Introduction to Biology Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012 Biology Review DNA RNA Proteins Central Dogma Transcription Translation
Bioinformatics ONE Introduction to Biology Sami Khuri Department of Computer Science San José State University Biology/CS 123A Fall 2012 Biology Review DNA RNA Proteins Central Dogma Transcription Translation
 Problem: The GC base pairs are more stable than AT base pairs. Why? 5. Triple-stranded DNA was first observed in 1957. Scientists later discovered that the formation of triplestranded DNA involves a type
Problem: The GC base pairs are more stable than AT base pairs. Why? 5. Triple-stranded DNA was first observed in 1957. Scientists later discovered that the formation of triplestranded DNA involves a type
Algorithms in Bioinformatics ONE Transcription Translation
 Algorithms in Bioinformatics ONE Transcription Translation Sami Khuri Department of Computer Science San José State University sami.khuri@sjsu.edu Biology Review DNA RNA Proteins Central Dogma Transcription
Algorithms in Bioinformatics ONE Transcription Translation Sami Khuri Department of Computer Science San José State University sami.khuri@sjsu.edu Biology Review DNA RNA Proteins Central Dogma Transcription
Using DNA sequence, distinguish species in the same genus from one another.
 Species Identification: Penguins 7. It s Not All Black and White! Name: Objective Using DNA sequence, distinguish species in the same genus from one another. Background In this activity, we will observe
Species Identification: Penguins 7. It s Not All Black and White! Name: Objective Using DNA sequence, distinguish species in the same genus from one another. Background In this activity, we will observe
Introduction to metagenome assembly. Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014
 Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Sequence assembly. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
BIOSTAT516 Statistical Methods in Genetic Epidemiology Autumn 2005 Handout1, prepared by Kathleen Kerr and Stephanie Monks
 Rationale of Genetic Studies Some goals of genetic studies include: to identify the genetic causes of phenotypic variation develop genetic tests o benefits to individuals and to society are still uncertain
Rationale of Genetic Studies Some goals of genetic studies include: to identify the genetic causes of phenotypic variation develop genetic tests o benefits to individuals and to society are still uncertain
APPENDIX. Appendix. Table of Contents. Ethics Background. Creating Discussion Ground Rules. Amino Acid Abbreviations and Chemistry Resources
 Appendix Table of Contents A2 A3 A4 A5 A6 A7 A9 Ethics Background Creating Discussion Ground Rules Amino Acid Abbreviations and Chemistry Resources Codons and Amino Acid Chemistry Behind the Scenes with
Appendix Table of Contents A2 A3 A4 A5 A6 A7 A9 Ethics Background Creating Discussion Ground Rules Amino Acid Abbreviations and Chemistry Resources Codons and Amino Acid Chemistry Behind the Scenes with
EE550 Computational Biology
 EE550 Computational Biology Week 1 Course Notes Instructor: Bilge Karaçalı, PhD Syllabus Schedule : Thursday 13:30, 14:30, 15:30 Text : Paul G. Higgs, Teresa K. Attwood, Bioinformatics and Molecular Evolution,
EE550 Computational Biology Week 1 Course Notes Instructor: Bilge Karaçalı, PhD Syllabus Schedule : Thursday 13:30, 14:30, 15:30 Text : Paul G. Higgs, Teresa K. Attwood, Bioinformatics and Molecular Evolution,
Bi Lecture 3 Loss-of-function (Ch. 4A) Monday, April 8, 13
 Monday, April 8, 13") Bi190-2013 Lecture 3 Loss-of-function (Ch. 4A) Infer Gene activity from type of allele Loss-of-Function alleles are Gold Standard If organism deficient in gene A fails to accomplish process B, then gene
Bi190-2013 Lecture 3 Loss-of-function (Ch. 4A) Infer Gene activity from type of allele Loss-of-Function alleles are Gold Standard If organism deficient in gene A fails to accomplish process B, then gene
DNA is normally found in pairs, held together by hydrogen bonds between the bases
 Bioinformatics Biology Review The genetic code is stored in DNA Deoxyribonucleic acid. DNA molecules are chains of four nucleotide bases Guanine, Thymine, Cytosine, Adenine DNA is normally found in pairs,
Bioinformatics Biology Review The genetic code is stored in DNA Deoxyribonucleic acid. DNA molecules are chains of four nucleotide bases Guanine, Thymine, Cytosine, Adenine DNA is normally found in pairs,
Outline. The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing
 Illumina Assembly 1 Outline The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing 2 Illumina Sequencing Paired end Illumina
Illumina Assembly 1 Outline The types of Illumina data Methods of assembly Repeats Selecting k-mer size Assembly Tools Assembly Diagnostics Assembly Polishing 2 Illumina Sequencing Paired end Illumina
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
ENZYMES AND METABOLIC PATHWAYS
 ENZYMES AND METABOLIC PATHWAYS This document is licensed under the Attribution-NonCommercial-ShareAlike 2.5 Italy license, available at http://creativecommons.org/licenses/by-nc-sa/2.5/it/ 1. Enzymes build
ENZYMES AND METABOLIC PATHWAYS This document is licensed under the Attribution-NonCommercial-ShareAlike 2.5 Italy license, available at http://creativecommons.org/licenses/by-nc-sa/2.5/it/ 1. Enzymes build
Unit 1. DNA and the Genome
 Unit 1 DNA and the Genome Gene Expression Key Area 3 Vocabulary 1: Transcription Translation Phenotype RNA (mrna, trna, rrna) Codon Anticodon Ribosome RNA polymerase RNA splicing Introns Extrons Gene Expression
Unit 1 DNA and the Genome Gene Expression Key Area 3 Vocabulary 1: Transcription Translation Phenotype RNA (mrna, trna, rrna) Codon Anticodon Ribosome RNA polymerase RNA splicing Introns Extrons Gene Expression
Materials Protein synthesis kit. This kit consists of 24 amino acids, 24 transfer RNAs, four messenger RNAs and one ribosome (see below).
.") Protein Synthesis Instructions The purpose of today s lab is to: Understand how a cell manufactures proteins from amino acids, using information stored in the genetic code. Assemble models of four very
Protein Synthesis Instructions The purpose of today s lab is to: Understand how a cell manufactures proteins from amino acids, using information stored in the genetic code. Assemble models of four very
Molecular Biology. Biology Review ONE. Protein Factory. Genotype to Phenotype. From DNA to Protein. DNA à RNA à Protein. June 2016
 Molecular Biology ONE Sami Khuri Department of Computer Science San José State University Biology Review DNA RNA Proteins Central Dogma Transcription Translation Genotype to Phenotype Protein Factory DNA
Molecular Biology ONE Sami Khuri Department of Computer Science San José State University Biology Review DNA RNA Proteins Central Dogma Transcription Translation Genotype to Phenotype Protein Factory DNA
Nucleic acid and protein Flow of genetic information
 Nucleic acid and protein Flow of genetic information References: Glick, BR and JJ Pasternak, 2003, Molecular Biotechnology: Principles and Applications of Recombinant DNA, ASM Press, Washington DC, pages.
Nucleic acid and protein Flow of genetic information References: Glick, BR and JJ Pasternak, 2003, Molecular Biotechnology: Principles and Applications of Recombinant DNA, ASM Press, Washington DC, pages.
6-Foot Mini Toober Activity
 Big Idea The interaction between the substrate and enzyme is highly specific. Even a slight change in shape of either the substrate or the enzyme may alter the efficient and selective ability of the enzyme
Big Idea The interaction between the substrate and enzyme is highly specific. Even a slight change in shape of either the substrate or the enzyme may alter the efficient and selective ability of the enzyme
Genomics and Database Mining (HCS 604.3) April 2005
 April 2005") Genomics and Database Mining (HCS 604.3) April 2005 David M. Francis OARDC 1680 Madison Ave Wooster, OH 44691 e-mail: francis.77@osu.edu Introduction: Computers have changed the way biologists go about
Genomics and Database Mining (HCS 604.3) April 2005 David M. Francis OARDC 1680 Madison Ave Wooster, OH 44691 e-mail: francis.77@osu.edu Introduction: Computers have changed the way biologists go about
Granby Transcription and Translation Services plc
 ompany Resources ranby Transcription and Translation Services plc has invested heavily in the Protein Synthesis business. mongst the resources available to new recruits are: the latest cellphones which
ompany Resources ranby Transcription and Translation Services plc has invested heavily in the Protein Synthesis business. mongst the resources available to new recruits are: the latest cellphones which
CECS Introduction to Bioinformatics University of Louisville Spring 2004 Dr. Eric Rouchka
 Dr. Awwad Abdoh Radwan Dept Pharm Organic Chemistry, Faculty of Pharmacy, Assiut University. 29/09/2005 Required Texts Image Source: http://www.amazon.com/ Other Bioinformatics Books Image Source: http://www.amazon.com/
Dr. Awwad Abdoh Radwan Dept Pharm Organic Chemistry, Faculty of Pharmacy, Assiut University. 29/09/2005 Required Texts Image Source: http://www.amazon.com/ Other Bioinformatics Books Image Source: http://www.amazon.com/
Protein Structure Analysis
 BINF 731 Protein Structure Analysis http://binf.gmu.edu/vaisman/binf731/ Secondary Structure: Computational Problems Secondary structure characterization Secondary structure assignment Secondary structure
BINF 731 Protein Structure Analysis http://binf.gmu.edu/vaisman/binf731/ Secondary Structure: Computational Problems Secondary structure characterization Secondary structure assignment Secondary structure
TECHNIQUES FOR STUDYING METAGENOME DATASETS METAGENOMES TO SYSTEMS.
 TECHNIQUES FOR STUDYING METAGENOME DATASETS METAGENOMES TO SYSTEMS. Ian Jeffery I.Jeffery@ucc.ie What is metagenomics Metagenomics is the study of genetic material recovered directly from environmental
TECHNIQUES FOR STUDYING METAGENOME DATASETS METAGENOMES TO SYSTEMS. Ian Jeffery I.Jeffery@ucc.ie What is metagenomics Metagenomics is the study of genetic material recovered directly from environmental
10/20/2009 Comp 590/Comp Fall
 Lecture 14: DNA Sequencing Study Chapter 8.9 10/20/2009 Comp 590/Comp 790-90 Fall 2009 1 DNA Sequencing Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments
Lecture 14: DNA Sequencing Study Chapter 8.9 10/20/2009 Comp 590/Comp 790-90 Fall 2009 1 DNA Sequencing Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments
CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS
 1 CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS * Some contents are adapted from Dr. Jean Gao at UT Arlington Mingon Kang, PhD Computer Science, Kennesaw State University 2 Genetics The discovery of
1 CS 4491/CS 7990 SPECIAL TOPICS IN BIOINFORMATICS * Some contents are adapted from Dr. Jean Gao at UT Arlington Mingon Kang, PhD Computer Science, Kennesaw State University 2 Genetics The discovery of
What is necessary for life?
 Life What is necessary for life? Most life familiar to us: Eukaryotes FREE LIVING Or Parasites First appeared ~ 1.5-2 10 9 years ago Requirements: DNA, proteins, lipids, carbohydrates, complex structure,
Life What is necessary for life? Most life familiar to us: Eukaryotes FREE LIVING Or Parasites First appeared ~ 1.5-2 10 9 years ago Requirements: DNA, proteins, lipids, carbohydrates, complex structure,
Protein Synthesis. Application Based Questions
 Protein Synthesis Application Based Questions MRNA Triplet Codons Note: Logic behind the single letter abbreviations can be found at: http://www.biology.arizona.edu/biochemistry/problem_sets/aa/dayhoff.html
Protein Synthesis Application Based Questions MRNA Triplet Codons Note: Logic behind the single letter abbreviations can be found at: http://www.biology.arizona.edu/biochemistry/problem_sets/aa/dayhoff.html
BME205: Lecture 2 Bio systems. David Bernick
 BME205: Lecture 2 Bio systems David Bernick Bioinforma;cs Infer pa>erns from life biological sequences structures molecular pathways. Suggest hypotheses from inferred pa>erns Structure and Func;on Novel
BME205: Lecture 2 Bio systems David Bernick Bioinforma;cs Infer pa>erns from life biological sequences structures molecular pathways. Suggest hypotheses from inferred pa>erns Structure and Func;on Novel
Lecture 19A. DNA computing
 Lecture 19A. DNA computing What exactly is DNA (deoxyribonucleic acid)? DNA is the material that contains codes for the many physical characteristics of every living creature. Your cells use different
Lecture 19A. DNA computing What exactly is DNA (deoxyribonucleic acid)? DNA is the material that contains codes for the many physical characteristics of every living creature. Your cells use different
Key Concept Translation converts an mrna message into a polypeptide, or protein.
 8.5 Translation VOBLRY translation codon stop codon start codon anticodon Key oncept Translation converts an mrn message into a polypeptide, or protein. MIN IDES mino acids are coded by mrn base sequences.
8.5 Translation VOBLRY translation codon stop codon start codon anticodon Key oncept Translation converts an mrn message into a polypeptide, or protein. MIN IDES mino acids are coded by mrn base sequences.
A Zero-Knowledge Based Introduction to Biology
 A Zero-Knowledge Based Introduction to Biology Konstantinos (Gus) Katsiapis 25 Sep 2009 Thanks to Cory McLean and George Asimenos Cells: Building Blocks of Life cell, membrane, cytoplasm, nucleus, mitochondrion
A Zero-Knowledge Based Introduction to Biology Konstantinos (Gus) Katsiapis 25 Sep 2009 Thanks to Cory McLean and George Asimenos Cells: Building Blocks of Life cell, membrane, cytoplasm, nucleus, mitochondrion
NEXT GENERATION SEQUENCING. Farhat Habib
 NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
NEXT GENERATION SEQUENCING HISTORY HISTORY Sanger Dominant for last ~30 years 1000bp longest read Based on primers so not good for repetitive or SNPs sites HISTORY Sanger Dominant for last ~30 years 1000bp
36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L-
 36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L- 37. The essential fatty acids are A. palmitic acid B. linoleic acid C. linolenic
36. The double bonds in naturally-occuring fatty acids are usually isomers. A. cis B. trans C. both cis and trans D. D- E. L- 37. The essential fatty acids are A. palmitic acid B. linoleic acid C. linolenic
De novo meta-assembly of ultra-deep sequencing data
 De novo meta-assembly of ultra-deep sequencing data Hamid Mirebrahim 1, Timothy J. Close 2 and Stefano Lonardi 1 1 Department of Computer Science and Engineering 2 Department of Botany and Plant Sciences
De novo meta-assembly of ultra-deep sequencing data Hamid Mirebrahim 1, Timothy J. Close 2 and Stefano Lonardi 1 1 Department of Computer Science and Engineering 2 Department of Botany and Plant Sciences
BIOLOGY. Monday 14 Mar 2016
 BIOLOGY Monday 14 Mar 2016 Entry Task List the terms that were mentioned last week in the video. Translation, Transcription, Messenger RNA (mrna), codon, Ribosomal RNA (rrna), Polypeptide, etc. Agenda
BIOLOGY Monday 14 Mar 2016 Entry Task List the terms that were mentioned last week in the video. Translation, Transcription, Messenger RNA (mrna), codon, Ribosomal RNA (rrna), Polypeptide, etc. Agenda
Introduction to Next Generation Sequencing
 The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
The Sequencing Revolution Introduction to Next Generation Sequencing Dena Leshkowitz,WIS 1 st BIOmics Workshop High throughput Short Read Sequencing Technologies Highly parallel reactions (millions to
Data Basics. Josef K Vogt Slides by: Simon Rasmussen Next Generation Sequencing Analysis
 Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
De novo assembly of human genomes with massively parallel short read sequencing. Mikk Eelmets Journal Club
 De novo assembly of human genomes with massively parallel short read sequencing Mikk Eelmets Journal Club 06.04.2010 Problem DNA sequencing technologies: Sanger sequencing (500-1000 bp) Next-generation
De novo assembly of human genomes with massively parallel short read sequencing Mikk Eelmets Journal Club 06.04.2010 Problem DNA sequencing technologies: Sanger sequencing (500-1000 bp) Next-generation
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Lecture 14: DNA Sequencing
 Lecture 14: DNA Sequencing Study Chapter 8.9 10/17/2013 COMP 465 Fall 2013 1 Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments (Sanger method) DNA Sequencing
Lecture 14: DNA Sequencing Study Chapter 8.9 10/17/2013 COMP 465 Fall 2013 1 Shear DNA into millions of small fragments Read 500 700 nucleotides at a time from the small fragments (Sanger method) DNA Sequencing
2018 Protein Modeling Exam Key
 2018 Protein Modeling Exam Key Multiple Choice: 1. Which of the following amino acids has a negative charge at ph 7? a. Gln b. Glu c. Ser d. Cys 2. Which of the following is an example of secondary structure?
2018 Protein Modeling Exam Key Multiple Choice: 1. Which of the following amino acids has a negative charge at ph 7? a. Gln b. Glu c. Ser d. Cys 2. Which of the following is an example of secondary structure?
NORWEGIAN UNIVERSITY OF SCIENCE AND TECHNOLOGY DEPARTMENT OF BIOTECHNOLOGY Professor Bjørn E. Christensen, Department of Biotechnology
 Page 1 NRWEGIAN UNIVERSITY F SCIENCE AND TECNLGY DEPARTMENT F BITECNLGY Professor Bjørn E. Christensen, Department of Biotechnology Contact during the exam: phone: 73593327, 92634016 EXAM TBT4135 BIPLYMERS
Page 1 NRWEGIAN UNIVERSITY F SCIENCE AND TECNLGY DEPARTMENT F BITECNLGY Professor Bjørn E. Christensen, Department of Biotechnology Contact during the exam: phone: 73593327, 92634016 EXAM TBT4135 BIPLYMERS
De novo genome assembly with next generation sequencing data!! "
 De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools
 CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
Introduction to Bioinformatics
 Introduction to Bioinformatics Contents Cell biology Organisms and cells Building blocks of cells How genes encode proteins? Bioinformatics What is bioinformatics? Practical applications Tools and databases
Introduction to Bioinformatics Contents Cell biology Organisms and cells Building blocks of cells How genes encode proteins? Bioinformatics What is bioinformatics? Practical applications Tools and databases
Alignment and Assembly
 Alignment and Assembly Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which
Alignment and Assembly Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which
De Novo Assembly (Pseudomonas aeruginosa MAPO1 ) Sample to Insight
 Sample to Insight") De Novo Assembly (Pseudomonas aeruginosa MAPO1 ) Sample to Insight 1 Workflow Import NGS raw data QC on reads De novo assembly Trim reads Finding Genes BLAST Sample to Insight Case Study Pseudomonas aeruginosa
De Novo Assembly (Pseudomonas aeruginosa MAPO1 ) Sample to Insight 1 Workflow Import NGS raw data QC on reads De novo assembly Trim reads Finding Genes BLAST Sample to Insight Case Study Pseudomonas aeruginosa
What is necessary for life?
 Life What is necessary for life? Most life familiar to us: Eukaryotes FREE LIVING Or Parasites First appeared ~ 1.5-2 10 9 years ago Requirements: DNA, proteins, lipids, carbohydrates, complex structure,
Life What is necessary for life? Most life familiar to us: Eukaryotes FREE LIVING Or Parasites First appeared ~ 1.5-2 10 9 years ago Requirements: DNA, proteins, lipids, carbohydrates, complex structure,
Mate-pair library data improves genome assembly
 De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
Basic Bioinformatics: Homology, Sequence Alignment,
 Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
CBC Data Therapy. Metagenomics Discussion
 CBC Data Therapy Metagenomics Discussion General Workflow Microbial sample Generate Metaomic data Process data (QC, etc.) Analysis Marker Genes Extract DNA Amplify with targeted primers Filter errors,
CBC Data Therapy Metagenomics Discussion General Workflow Microbial sample Generate Metaomic data Process data (QC, etc.) Analysis Marker Genes Extract DNA Amplify with targeted primers Filter errors,
A Guide to Consed Michelle Itano, Carolyn Cain, Tien Chusak, Justin Richner, and SCR Elgin.
 1 A Guide to Consed Michelle Itano, Carolyn Cain, Tien Chusak, Justin Richner, and SCR Elgin. Main Window Figure 1. The Main Window is the starting point when Consed is opened. From here, you can access
1 A Guide to Consed Michelle Itano, Carolyn Cain, Tien Chusak, Justin Richner, and SCR Elgin. Main Window Figure 1. The Main Window is the starting point when Consed is opened. From here, you can access
De Novo and Hybrid Assembly
 On the PacBio RS Introduction The PacBio RS utilizes SMRT technology to generate both Continuous Long Read ( CLR ) and Circular Consensus Read ( CCS ) data. In this document, we describe sequencing the
On the PacBio RS Introduction The PacBio RS utilizes SMRT technology to generate both Continuous Long Read ( CLR ) and Circular Consensus Read ( CCS ) data. In this document, we describe sequencing the
BIOLOGY LTF DIAGNOSTIC TEST DNA to PROTEIN & BIOTECHNOLOGY
 Biology Multiple Choice 016074 BIOLOGY LTF DIAGNOSTIC TEST DNA to PROTEIN & BIOTECHNOLOGY Test Code: 016074 Directions: Each of the questions or incomplete statements below is followed by five suggested
Biology Multiple Choice 016074 BIOLOGY LTF DIAGNOSTIC TEST DNA to PROTEIN & BIOTECHNOLOGY Test Code: 016074 Directions: Each of the questions or incomplete statements below is followed by five suggested
Station 1: DNA Structure Use the figure above to answer each of the following questions. 1.This is the subunit that DNA is composed of. 2.
 1. Station 1: DNA Structure Use the figure above to answer each of the following questions. 1.This is the subunit that DNA is composed of. 2.This subunit is composed of what 3 parts? 3.What molecules make
1. Station 1: DNA Structure Use the figure above to answer each of the following questions. 1.This is the subunit that DNA is composed of. 2.This subunit is composed of what 3 parts? 3.What molecules make
Assembling a Cassava Transcriptome using Galaxy on a High Performance Computing Cluster
 Assembling a Cassava Transcriptome using Galaxy on a High Performance Computing Cluster Aobakwe Matshidiso Supervisor: Prof Chrissie Rey Co-Supervisor: Prof Scott Hazelhurst Next Generation Sequencing
Assembling a Cassava Transcriptome using Galaxy on a High Performance Computing Cluster Aobakwe Matshidiso Supervisor: Prof Chrissie Rey Co-Supervisor: Prof Scott Hazelhurst Next Generation Sequencing
Concepts and methods in genome assembly and annotation
 BCM-2002 Concepts and methods in genome assembly and annotation B. Franz LANG, Département de Biochimie Bureau: H307-15 Courrier électronique: Franz.Lang@Umontreal.ca Outline 1. What is genome assembly?
BCM-2002 Concepts and methods in genome assembly and annotation B. Franz LANG, Département de Biochimie Bureau: H307-15 Courrier électronique: Franz.Lang@Umontreal.ca Outline 1. What is genome assembly?
Homework. A bit about the nature of the atoms of interest. Project. The role of electronega<vity
 Homework Why cited articles are especially useful. citeulike science citation index When cutting and pasting less is more. Project Your protein: I will mail these out this weekend If you haven t gotten
Homework Why cited articles are especially useful. citeulike science citation index When cutting and pasting less is more. Project Your protein: I will mail these out this weekend If you haven t gotten
CHAPTER 1. DNA: The Hereditary Molecule SECTION D. What Does DNA Do? Chapter 1 Modern Genetics for All Students S 33
 HPER 1 DN: he Hereditary Molecule SEION D What Does DN Do? hapter 1 Modern enetics for ll Students S 33 D.1 DN odes For Proteins PROEINS DO HE nitty-gritty jobs of every living cell. Proteins are the molecules
HPER 1 DN: he Hereditary Molecule SEION D What Does DN Do? hapter 1 Modern enetics for ll Students S 33 D.1 DN odes For Proteins PROEINS DO HE nitty-gritty jobs of every living cell. Proteins are the molecules
Forensic Science: DNA Evidence Unit
 Day 2 : Cooperative Lesson Topic: Protein Synthesis Duration: 55 minutes Grade Level: 10 th Grade Forensic Science: DNA Evidence Unit Purpose: The purpose of this lesson is to review and build upon prior
Day 2 : Cooperative Lesson Topic: Protein Synthesis Duration: 55 minutes Grade Level: 10 th Grade Forensic Science: DNA Evidence Unit Purpose: The purpose of this lesson is to review and build upon prior
How life. constructs itself.
 How life constructs itself Life constructs itself using few simple rules of information processing. On the one hand, there is a set of rules determining how such basic chemical reactions as transcription,
How life constructs itself Life constructs itself using few simple rules of information processing. On the one hand, there is a set of rules determining how such basic chemical reactions as transcription,
Next Generation Sequencing. Tobias Österlund
 Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
GENOME ASSEMBLY FINAL PIPELINE AND RESULTS
 GENOME ASSEMBLY FINAL PIPELINE AND RESULTS Faction 1 Yanxi Chen Carl Dyson Sean Lucking Chris Monaco Shashwat Deepali Nagar Jessica Rowell Ankit Srivastava Camila Medrano Trochez Venna Wang Seyed Alireza
GENOME ASSEMBLY FINAL PIPELINE AND RESULTS Faction 1 Yanxi Chen Carl Dyson Sean Lucking Chris Monaco Shashwat Deepali Nagar Jessica Rowell Ankit Srivastava Camila Medrano Trochez Venna Wang Seyed Alireza
CFSSP: Chou and Fasman Secondary Structure Prediction server
 Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Lecture 7. Next-generation sequencing technologies
 Lecture 7 Next-generation sequencing technologies Next-generation sequencing technologies General principles of short-read NGS Construct a library of fragments Generate clonal template populations Massively
Lecture 7 Next-generation sequencing technologies Next-generation sequencing technologies General principles of short-read NGS Construct a library of fragments Generate clonal template populations Massively
Genome Assembly Background and Strategy
 Genome Assembly Background and Strategy February 6th, 2017 BIOL 7210 - Faction I (Outbreak) - Genome Assembly Group Yanxi Chen Carl Dyson Zhiqiang Lin Sean Lucking Chris Monaco Shashwat Deepali Nagar Jessica
Genome Assembly Background and Strategy February 6th, 2017 BIOL 7210 - Faction I (Outbreak) - Genome Assembly Group Yanxi Chen Carl Dyson Zhiqiang Lin Sean Lucking Chris Monaco Shashwat Deepali Nagar Jessica
THE GENETIC CODE Figure 1: The genetic code showing the codons and their respective amino acids
 THE GENETIC CODE As DNA is a genetic material, it carries genetic information from cell to cell and from generation to generation. There are only four bases in DNA and twenty amino acids in protein, so
THE GENETIC CODE As DNA is a genetic material, it carries genetic information from cell to cell and from generation to generation. There are only four bases in DNA and twenty amino acids in protein, so
The combination of a phosphate, sugar and a base forms a compound called a nucleotide.
 History Rosalin Franklin: Female scientist (x-ray crystallographer) who took the picture of DNA James Watson and Francis Crick: Solved the structure of DNA from information obtained by other scientist.
History Rosalin Franklin: Female scientist (x-ray crystallographer) who took the picture of DNA James Watson and Francis Crick: Solved the structure of DNA from information obtained by other scientist.
DNA stands for deoxyribose nucleic acid
 1 DNA 2 DNA stands for deoxyribose nucleic acid This chemical substance is present in the nucleus of all cells in all living organisms DNA controls all the chemical changes which take place in cells The
1 DNA 2 DNA stands for deoxyribose nucleic acid This chemical substance is present in the nucleus of all cells in all living organisms DNA controls all the chemical changes which take place in cells The
DE NOVO GENOME ASSEMBLY OF THE AFRICAN CATFISH (CLARIAS GARIEPINUS)
") DE NOVO GENOME ASSEMBLY OF THE AFRICAN CATFISH (CLARIAS GARIEPINUS) Kovács B. a,, Barta E. c, Pongor S. L. b, Uri Cs. a, Patócs A. b, Orbán L. d, Müller T. a, Urbányi B. a a Department of Aquaculture,
DE NOVO GENOME ASSEMBLY OF THE AFRICAN CATFISH (CLARIAS GARIEPINUS) Kovács B. a,, Barta E. c, Pongor S. L. b, Uri Cs. a, Patócs A. b, Orbán L. d, Müller T. a, Urbányi B. a a Department of Aquaculture,
Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I
 Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I Dr David Studholme. 18 th February 2014. BIO1033 theme lecture. 1 28 February 2014 @davidjstudholme 28 February 2014 @davidjstudholme
Bioinformatic analysis of Illumina sequencing data for comparative genomics Part I Dr David Studholme. 18 th February 2014. BIO1033 theme lecture. 1 28 February 2014 @davidjstudholme 28 February 2014 @davidjstudholme
Daily Agenda. Warm Up: Review. Translation Notes Protein Synthesis Practice. Redos
 Daily Agenda Warm Up: Review Translation Notes Protein Synthesis Practice Redos 1. What is DNA Replication? 2. Where does DNA Replication take place? 3. Replicate this strand of DNA into complimentary
Daily Agenda Warm Up: Review Translation Notes Protein Synthesis Practice Redos 1. What is DNA Replication? 2. Where does DNA Replication take place? 3. Replicate this strand of DNA into complimentary
Dynamic Programming Algorithms
 Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Chemistry 121 Winter 17
 Chemistry 121 Winter 17 Introduction to Organic Chemistry and Biochemistry Instructor Dr. Upali Siriwardane (Ph.D. Ohio State) E-mail: upali@latech.edu Office: 311 Carson Taylor Hall ; Phone: 318-257-4941;
Chemistry 121 Winter 17 Introduction to Organic Chemistry and Biochemistry Instructor Dr. Upali Siriwardane (Ph.D. Ohio State) E-mail: upali@latech.edu Office: 311 Carson Taylor Hall ; Phone: 318-257-4941;
Create a model to simulate the process by which a protein is produced, and how a mutation can impact a protein s function.
 HASPI Medical Biology Lab 0 Purpose Create a model to simulate the process by which a protein is produced, and how a mutation can impact a protein s function. Background http://mssdbio.weebly.com/uploads/1//7/6/17618/970_orig.jpg
HASPI Medical Biology Lab 0 Purpose Create a model to simulate the process by which a protein is produced, and how a mutation can impact a protein s function. Background http://mssdbio.weebly.com/uploads/1//7/6/17618/970_orig.jpg
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Tutorial for Stop codon reassignment in the wild
 Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Biochemistry and Cell Biology
 Biochemistry and Cell Biology Monomersare simple molecules that can be linked into chains. (Nucleotides, Amino acids, monosaccharides) Polymersare the long chains of monomers. (DNA, RNA, Cellulose, Protein,
Biochemistry and Cell Biology Monomersare simple molecules that can be linked into chains. (Nucleotides, Amino acids, monosaccharides) Polymersare the long chains of monomers. (DNA, RNA, Cellulose, Protein,
short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014
 1 short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014 2 Genomathica Assembler Mathematica notebook for genome assembly simulation Assembler can be found at:
1 short read genome assembly Sorin Istrail CSCI1820 Short-read genome assembly algorithms 3/6/2014 2 Genomathica Assembler Mathematica notebook for genome assembly simulation Assembler can be found at:
De novo whole genome assembly
 De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore
De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore
de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ
 de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ de novo transcriptome assembly de novo from the Latin expression meaning from the beginning In bioinformatics, we often use
de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ de novo transcriptome assembly de novo from the Latin expression meaning from the beginning In bioinformatics, we often use
Aipotu II: Biochemistry
 Aipotu II: Biochemistry Introduction: The Biological Phenomenon Under Study In this lab, you will continue to explore the biological mechanisms behind the expression of flower color in a hypothetical plant.
Aipotu II: Biochemistry Introduction: The Biological Phenomenon Under Study In this lab, you will continue to explore the biological mechanisms behind the expression of flower color in a hypothetical plant.
First&year&tutorial&in&Chemical&Biology&(amino&acids,&peptide&and&proteins)&! 1.&!
&! 1.&!") First&year&tutorial&in&Chemical&Biology&(amino&acids,&peptide&and&proteins& 1.& a. b. c. d. e. 2.& a. b. c. d. e. f. & UsingtheCahn Ingold Prelogsystem,assignstereochemicaldescriptorstothe threeaminoacidsshownbelow.
First&year&tutorial&in&Chemical&Biology&(amino&acids,&peptide&and&proteins& 1.& a. b. c. d. e. 2.& a. b. c. d. e. f. & UsingtheCahn Ingold Prelogsystem,assignstereochemicaldescriptorstothe threeaminoacidsshownbelow.
Bioinformatics for Genomics
 Bioinformatics for Genomics It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material. When I was young my Father
Bioinformatics for Genomics It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material. When I was young my Father
The Effect of Using Different Neural Networks Architectures on the Protein Secondary Structure Prediction
 The Effect of Using Different Neural Networks Architectures on the Protein Secondary Structure Prediction Hanan Hendy, Wael Khalifa, Mohamed Roushdy, Abdel Badeeh Salem Computer Science Department, Faculty
The Effect of Using Different Neural Networks Architectures on the Protein Secondary Structure Prediction Hanan Hendy, Wael Khalifa, Mohamed Roushdy, Abdel Badeeh Salem Computer Science Department, Faculty
High-Throughput Bioinformatics: Re-sequencing and de novo assembly. Elena Czeizler
 High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
High-Throughput Bioinformatics: Re-sequencing and de novo assembly Elena Czeizler 13.11.2015 Sequencing data Current sequencing technologies produce large amounts of data: short reads The outputted sequences
Big Idea 3C Basic Review
 Big Idea 3C Basic Review 1. A gene is a. A sequence of DNA that codes for a protein. b. A sequence of amino acids that codes for a protein. c. A sequence of codons that code for nucleic acids. d. The end
Big Idea 3C Basic Review 1. A gene is a. A sequence of DNA that codes for a protein. b. A sequence of amino acids that codes for a protein. c. A sequence of codons that code for nucleic acids. d. The end
Fundamentals of Protein Structure
 Outline Fundamentals of Protein Structure Yu (Julie) Chen and Thomas Funkhouser Princeton University CS597A, Fall 2005 Protein structure Primary Secondary Tertiary Quaternary Forces and factors Levels
Outline Fundamentals of Protein Structure Yu (Julie) Chen and Thomas Funkhouser Princeton University CS597A, Fall 2005 Protein structure Primary Secondary Tertiary Quaternary Forces and factors Levels
Supplementary Figure 1. Design of the control microarray. a, Genomic DNA from the
 Supplementary Information Supplementary Figures Supplementary Figure 1. Design of the control microarray. a, Genomic DNA from the strain M8 of S. ruber and a fosmid containing the S. ruber M8 virus M8CR4
Supplementary Information Supplementary Figures Supplementary Figure 1. Design of the control microarray. a, Genomic DNA from the strain M8 of S. ruber and a fosmid containing the S. ruber M8 virus M8CR4
From Infection to Genbank
 From Infection to Genbank How a pathogenic bacterium gets its genome to NCBI Torsten Seemann VLSCI - Life Sciences Computation Centre - Genomics Theme - Lab Meeting - Friday 27 April 2012 The steps 1.
From Infection to Genbank How a pathogenic bacterium gets its genome to NCBI Torsten Seemann VLSCI - Life Sciences Computation Centre - Genomics Theme - Lab Meeting - Friday 27 April 2012 The steps 1.
Translating the Genetic Code. DANILO V. ROGAYAN JR. Faculty, Department of Natural Sciences
 Translating the Genetic Code DANILO V. ROGAYAN JR. Faculty, Department of Natural Sciences An overview of gene expression Figure 13.2 The Idea of A Code 20 amino acids 4 nucleotides How do nucleic acids
Translating the Genetic Code DANILO V. ROGAYAN JR. Faculty, Department of Natural Sciences An overview of gene expression Figure 13.2 The Idea of A Code 20 amino acids 4 nucleotides How do nucleic acids
Biology: The substrate of bioinformatics
 Bi01_1 Unit 01: Biology: The substrate of bioinformatics What is Bioinformatics? Bi01_2 handling of information related to living organisms understood on the basis of molecular biology Nature does it.
Bi01_1 Unit 01: Biology: The substrate of bioinformatics What is Bioinformatics? Bi01_2 handling of information related to living organisms understood on the basis of molecular biology Nature does it.
Contact us for more information and a quotation
 GenePool Information Sheet #1 Installed Sequencing Technologies in the GenePool The GenePool offers sequencing service on three platforms: Sanger (dideoxy) sequencing on ABI 3730 instruments Illumina SOLEXA
GenePool Information Sheet #1 Installed Sequencing Technologies in the GenePool The GenePool offers sequencing service on three platforms: Sanger (dideoxy) sequencing on ABI 3730 instruments Illumina SOLEXA
Additional Case Study: Amino Acids and Evolution
 Student Worksheet Additional Case Study: Amino Acids and Evolution Objectives To use biochemical data to determine evolutionary relationships. To test the hypothesis that living things that are morphologically
Student Worksheet Additional Case Study: Amino Acids and Evolution Objectives To use biochemical data to determine evolutionary relationships. To test the hypothesis that living things that are morphologically