RNASEQ WITHOUT A REFERENCE
|
|
|
- Thomasine Bennett
- 6 years ago
- Views:
Transcription
1 RNASEQ WITHOUT A REFERENCE Experimental Design Assembly in Non-Model Organisms And other (hopefully useful) Stuff Meg Staton mstaton1@utk.edu University of Tennessee Knoxville, TN
2 I. Project Design

3 Things you need to know BEFORE you begin Cost Read Count Replicates Pro Tip: Who is your resident statistician? Buy them a coffee and make friends.

4 Replicates What? Biological Replicates independent biological sample, processed separately and barcoded Technical Replicates independent library construction or sequencing of the same biological sample Technical reproducibility is very good for RNASeq Biological variation is much greater! Different genes have different variances and are potentially subject to different errors and biases. Thinking About RNA Seq Experimental Design for Measuring Differential Gene Expression: The Basics Marioni, J.C., et al. (2008) RNA-seq: An assessment of technical reproducibility and
5 Replicates How many? beyond a depth of 10 million reads, replicates provide more statistical power than depth for detecting differential gene expression Many people say at least 3 this enables the t-test What if one fails? (Fishers exact test can utilize no replicates)
6 Replicates Software? Both EdgeR and DeSeq will calculate variance from replicates (but neither do a t-test) From the horse s mouth: to use something like a t test, you need enough replicates to estimate a variance for each gene. With two groups of five samples, you are already entering the regime there this should work well. For comparison, also try a tool that pools information from several genes to get better confidence in variance estimates, such as our DESeq or the Smyth group's edger. Of course, we like to claim that DESeq is better than edger, and for only two or three replicates, I do think so, but for five or more replicates, edger's "moderation" feature really pays off. So, even though I don't like admitting this, for your set-up [of 5 replicates per treatment], edger should work better than DESeq. -Simon Anders on SeqAnswers
7 Replicates And Blocks? Randomized Block Design Randomize - assigning individuals at random to treatments in an experiment Blocking - Experimental units are grouped into homogeneous clusters in an attempt to improve the comparison of treatments Example all organisms from the same location are blocks, multiple locations used Example - each block is a cultivar, with individuals from that cultivar randomly assigned to a treatment
8 Read Count - How to Decide? Standards, Guidelines and Best Practices for RNA-Seq V1.0 (June 2011) The ENCODE Consortium What are you trying to do? Compare two mrna samples for differential expression (30M PE per sample) Discover novel elements, perform more precise quantification, especially of lowly expressed transcripts ( M PE per sample) What resources do you already have? Well assembled and annotated genomes single ends, shorter reads De novo longer reads, paired ends What is being published in your community?

9 Read Count How to Decide? (cont.) Blogosphere disagrees Need half the coverage, double the replicates! Current experiments indicate that we are NOT discovering significantly more transcripts with a hiseq run vs a miseq run. (At least not transcripts that look like genes) A deep biased view
10 Scotty You need more power! Scotty is a web service to plan RNA-Seq experiments that measure differential gene expression. Prototype data required Pilot data -at least two replicates of either control or treatment Pre-loaded data
11 Scotty up to $20k User Inputs Used in the Analysis Control columns in pilot data: 3 Test columns in pilot data: 3 Cost per replicate, control: $200 Cost per replicate, test: $200 Cost per million reads: $23 Alignment Rate: 90% Maximum cost of experiment: $20000 Percentage of genes detected: 50 At p value cutoff: 0.01 For the following true fold change: 2 Maximum percentage of genes with low-powered (biased) measurements: 50 Least expensive: 6 replicates sequenced to a depth of 12 million reads aligned to genes per replicate. $5,712 Most powerful: 20 replicates sequenced to a depth of 34 million reads aligned to genes per replicate. $19,640
12 When is enough enough? Average coverage is not a helpful metric for RNASeq relative expression varies by orders of magnitude Saturation curves the number of transcript references with < 10 read alignments New Discovery Rate How many new genes are being discovered with each additional slice of data?
13 Reality: Variation in # of Sequences green ash blackgum white ash honeylocust sugar maple black walnut sweetgum white oak black cherry redbay - 2,000 4,000 6,000 8,000 10,000 12,000 14,000 16,000 18,000 Thousands

14 Spreading libraries across lanes?
15 Spreading libraries across lanes? Balanced Block Design
16 What s right for your experiment? Cost Read Count? Replicates
17 II. De novo transcriptome sequencing - assembly
18 Model Organism reference genome Trimmomatic quality and adapter trimming Tophat align reads to consensus genome HTSeq count read alignments by library DESeq differential expression

19 Non-model Organism NO reference genome Trimmomatic quality and adapter trimming Trimmomatic quality and adapter trimming Trinity - assembly Tophat align reads to consensus genome Bowtie2 Realign reads to consensus transcripts HTSeq count read alignments by library HTSeq Count read alignments by library DESeq differential expression DESeq differential expression
20 Problems with de novo assemblies plant species have larger and more complex genome sizes and structures than animal species tremendous diversity in both size and structure From a plant perspective Polyploidy Gene family proliferation Heterozygosity Repetitive element proliferation
21 Why plants are difficult (cont) Our best reference, Arabidopsis thaliana has a genome that underwent a 30% reduction in genome size and at least nine rearrangements in the short time since its divergence to Arabidopsis lyrata Maize pan genome - Intraspecific variations of as much as 38.8% from the average of 5.5 pg/2n nucleus driven by LTR retrotransposon expansion Conifer genome sizes Loblolly pine 22Gb (7x bigger than human) largest genome contains roughly 60,000,000,000 more base pairs than the smallest genome Often these difficulties make transcriptome sequencing more attractive than whole genome sequencing!
22 Hornett and Wheat. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC Genomics 2012, 13:361 Problems with de novo assemblies Results Highly fragmented assemblies Chimeras: Paralogs, alleles and alternative splicing variants mushed together or fragmented Metrics based upon contig lengths (e.g. mean, median, N50) do not provide quantitative insights into how much of the target species transcriptome is represented in the de novo TA.
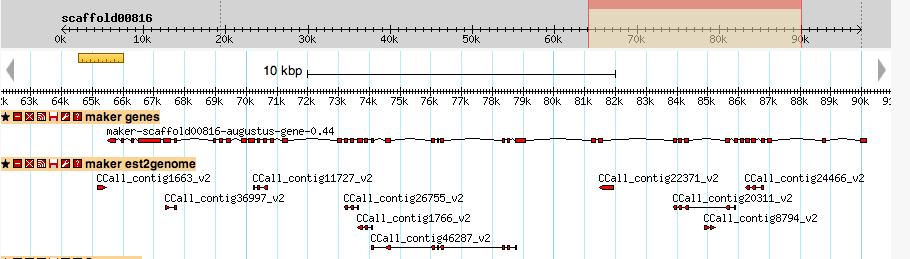
23 Chestnut
24 De novo transcriptome assemblies Is there a close relative with a sequenced genome? How close is close enough? Align then assemble Assemble then align



25 Assemble then align First, assemble Next, align to a close relative Main Problems: Fragmented assemblies gene pieces are scattered in a different consensus pieces More difficult to sort out gene family members Main Advantages: Alignment to a close relative can identify exon/exon boundaries (sort out alternative splicing) Less bias can discover novel gene sequences Weber et al. Strategies for transcriptome analysis in nonmodel plants Am J Bot (2):
 21,362,330 2,926,958,573 Tissues (hiseq) 442,863,286 42,122,511,244 Stress")

26 Green Ash (Fraxinus pennsylvanica) Trimmed reads Trimmed bases ozone project (miseq) 5,151, ,286,276 Tissues (miseq) 21,362,330 2,926,958,573 Tissues (hiseq) 442,863,286 42,122,511,244 Stress (miseq) 27,470,000 3,650,984,673 Stress (hiseq) 350,952,104 35,411,991,796 Data 847,799,220 84,862,732,562 Green Ash transcripts 107,611 peptides 52,899 % ORF discovery 49% 55 libraries Plus 41 technical replicates
27 Ash Genome Richard Buggs' lab Queen Mary, University of London (QMUL) British Ash Tree Genome Project Fraxinus excelsior 89,285 scaffolds, with an N50 of 99 kbp, and total size of 875 Mbp Green Ash 36,944 genes 36,893 proteins transcripts 107,611 peptides 52,899 % ORF discovery 49%
28 Ash Genome From perspective of the genome 36,893 proteins from genome 29,782 have a match to our RNASeq proteome (81%) From perspective of the transcriptome 52,899 proteins from RNASeq 47,657 have a match to the genome proteins (90%) 36,944 genes from genome 35,298 have a match to our RNASeq transcripts (96%) 107,611 transcripts from RNASeq 80,628 have a match to the genome transcripts (75%) BLAST 1e-10



29 Align then assemble First, map reads to (distant) reference Next, do local assemblies for each gene Main Problems Read alignment may be poor due to lack of sequence similarity Gene family expansion/contraction Main Advantage Transcript assembly is less likely to be fragmented Even where it is fragmented, you can identify all the fragments that originate from a single locus Weber et al. Strategies for transcriptome analysis in nonmodel plants Am J Bot (2):
30 Hornett and Wheat. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC Genomics 2012, 13:361 Assemble then align Reasonable Distant References Bad Worse *Likely to vary by phylogenetic neighborhood
31 De novo transcriptome assemblies Completely reference free What are they useful for? Transcriptome characterization Whats there? Resource Building Enabling proteomics experiments Candidate gene discovery Targeted sequencing Marker discovery/development Sequence parents of a cross SNP array (May want to consider genotyping by sequencing/restriction site associated DNA techniques instead)
32 De novo transcriptome assemblies What to do if you want/need differential expression data? Long reads Paired ends, possibly with different insert sizes Analyze gene families for differential expression instead of individual genes Alter the parameters of your assembler Merge at a lower level of heterozygosity 98% or 97% Utilize a closely related relative with a sequenced genome
33 Trinity strategy Inchworm assembles the RNAseq data into the unique sequences of transcripts, often generating full-length transcripts for a dominant isoform, but then reports just the unique portions of alternatively spliced transcripts.
34 Trinity strategy Chrysalis clusters the Inchworm contigs into clusters and constructs complete de Bruijn graphs for each cluster. Each cluster represents the full transcriptonal complexity for a given gene (or sets of genes that share sequences in common). Chrysalis then partitions the full read set among these disjoint graphs.
35 Trinity strategy Butterfly then processes the individual graphs in parallel, tracing the paths that reads and pairs of reads take within the graph, ultimately reporting fulllength transcripts for alternatively spliced isoforms, and teasing apart transcripts that corresponds to paralogous genes.
36 Trinity Videos!
37 Trinity output deciphering the naming An example Fasta entry for one of the transcripts is formatted like so: >c115_g5_i1 len=247 path=[31015: : ] Component a collection of contigs that are likely to be derived from alternative splice forms or closely related paralogs Gene best guess at an individual locus Isoform alternative splicing events and alleles
38 II. De novo transcriptome sequencing after assembly
39 Trinity TransDecoder and Coverage Maximizes length and likelihood score of ORF Optionally, looks for a putative peptide that has a match to a Pfam domain Full-length transcript analysis for model and nonmodel organisms using BLAST+ Perl script analyze_blastplus_tophit_c overage.pl
 234,546 GO term assignments 29,666 transcripts with go terms (56%) Software:")

40 Functional Annotation InterProScan 329,311 annotations 45,893 transcripts have at least one annotation (87%) 234,546 GO term assignments 29,666 transcripts with go terms (56%) Software: 72,706 PANTHER 51,025 Pfam 41,391 Gene3d 39,965 SUPERFAMILY 27,246 TMHMM 26,189 ProSiteProfiles 20,835 PRINTS 20,078 SMART 8,267 Coils 5,780 TIGR-FAM 2,456 SignalP_EUK 1,224 PIRSF 825 HAMAP
41 Making data public NCBI Short Read Archive stores raw sequence data from "next-generation" sequencing technologies including 454, IonTorrent, Illumina, SOLiD, Helicos and Complete Genomics. In addition to raw sequence data, SRA now stores alignment information in the form of read placements on a reference sequence.
42 NCBI SRA SRA format includes data and metadata Convert using the SRA Toolkit (linux, mac and windows versions available)

43 Upload to SRA

44 NSF Hardwood Genomics Project
Experimental Design. Sequencing. Data Quality Control. Read mapping. Differential Expression analysis
 -Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
-Seq Analysis Quality Control checks Reproducibility Reliability -seq vs Microarray Higher sensitivity and dynamic range Lower technical variation Available for all species Novel transcript identification
Transcriptome Assembly, Functional Annotation (and a few other related thoughts)
") Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
Transcriptome Assembly, Functional Annotation (and a few other related thoughts) Monica Britton, Ph.D. Sr. Bioinformatics Analyst June 23, 2017 Differential Gene Expression Generalized Workflow File Types
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Single Cell Transcriptomics scrnaseq
 Single Cell Transcriptomics scrnaseq Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu Purpose The sequencing of
Single Cell Transcriptomics scrnaseq Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu Purpose The sequencing of
Transcriptome analysis
 Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Statistical Bioinformatics: Transcriptome analysis Stefan Seemann seemann@rth.dk University of Copenhagen April 11th 2018 Outline: a) How to assess the quality of sequencing reads? b) How to normalize
Genomic resources. for non-model systems
 Genomic resources for non-model systems 1 Genomic resources Whole genome sequencing reference genome sequence comparisons across species identify signatures of natural selection population-level resequencing
Genomic resources for non-model systems 1 Genomic resources Whole genome sequencing reference genome sequence comparisons across species identify signatures of natural selection population-level resequencing
Bioinformatics Advice on Experimental Design
 Bioinformatics Advice on Experimental Design Where do I start? Please refer to the following guide to better plan your experiments for good statistical analysis, best suited for your research needs. Statistics
Bioinformatics Advice on Experimental Design Where do I start? Please refer to the following guide to better plan your experiments for good statistical analysis, best suited for your research needs. Statistics
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
NGS part 2: applications. Tobias Österlund
 NGS part 2: applications Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
NGS part 2: applications Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Transcriptomics analysis with RNA seq: an overview Frederik Coppens
 Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
Transcriptomics analysis with RNA seq: an overview Frederik Coppens Platforms Applications Analysis Quantification RNA content Platforms Platforms Short (few hundred bases) Long reads (multiple kilobases)
From reads to results: differential. Alicia Oshlack Head of Bioinformatics
 From reads to results: differential expression analysis with ihrna seq Alicia Oshlack Head of Bioinformatics Murdoch Childrens Research Institute Benefits and opportunities ii of RNA seq All transcripts
From reads to results: differential expression analysis with ihrna seq Alicia Oshlack Head of Bioinformatics Murdoch Childrens Research Institute Benefits and opportunities ii of RNA seq All transcripts
de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ
 de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ de novo transcriptome assembly de novo from the Latin expression meaning from the beginning In bioinformatics, we often use
de novo Transcriptome Assembly Nicole Cloonan 1 st July 2013, Winter School, UQ de novo transcriptome assembly de novo from the Latin expression meaning from the beginning In bioinformatics, we often use
Experimental Design Microbial Sequencing
 Experimental Design Microbial Sequencing Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu General rules for preparing
Experimental Design Microbial Sequencing Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu General rules for preparing
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
ChIP-seq and RNA-seq. Farhat Habib
 ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
ChIP-seq and RNA-seq Farhat Habib fhabib@iiserpune.ac.in Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions
RNA-Seq Analysis. Simon Andrews, Laura v
 RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
RNA-Seq Analysis Simon Andrews, Laura Biggins simon.andrews@babraham.ac.uk @simon_andrews v2018-10 RNA-Seq Libraries rrna depleted mrna Fragment u u u u NNNN Random prime + RT 2 nd strand synthesis (+
Applications of short-read
 Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ Sequencing applications RNA-Seq includes experiments
De novo assembly in RNA-seq analysis.
 De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
De novo assembly in RNA-seq analysis. Joachim Bargsten Wageningen UR/PRI/Plant Breeding October 2012 Motivation Transcriptome sequencing (RNA-seq) Gene expression / differential expression Reconstruct
Experimental Design. Dr. Matthew L. Settles. Genome Center University of California, Davis
 Experimental Design Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu What is Differential Expression Differential expression analysis means taking normalized sequencing
Experimental Design Dr. Matthew L. Settles Genome Center University of California, Davis settles@ucdavis.edu What is Differential Expression Differential expression analysis means taking normalized sequencing
Eucalyptus gene assembly
 Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
Eucalyptus gene assembly ACGT Plant Biotechnology meeting Charles Hefer Bioinformatics and Computational Biology Unit University of Pretoria October 2011 About Eucalyptus Most valuable and widely planted
Analysis of RNA-seq Data. Feb 8, 2017 Peikai CHEN (PHD)
") Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Analysis of RNA-seq Data Feb 8, 2017 Peikai CHEN (PHD) Outline What is RNA-seq? What can RNA-seq do? How is RNA-seq measured? How to process RNA-seq data: the basics How to visualize and diagnose your
Analysis of RNA-seq Data. Bernard Pereira
 Analysis of RNA-seq Data Bernard Pereira The many faces of RNA-seq Applications Discovery Find new transcripts Find transcript boundaries Find splice junctions Comparison Given samples from different experimental
Analysis of RNA-seq Data Bernard Pereira The many faces of RNA-seq Applications Discovery Find new transcripts Find transcript boundaries Find splice junctions Comparison Given samples from different experimental
Sequencing applications. Today's outline. Hands-on exercises. Applications of short-read sequencing: RNA-Seq and ChIP-Seq
 Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
Sequencing applications Applications of short-read sequencing: RNA-Seq and ChIP-Seq BaRC Hot Topics March 2013 George Bell, Ph.D. http://jura.wi.mit.edu/bio/education/hot_topics/ RNA-Seq includes experiments
BST 226 Statistical Methods for Bioinformatics David M. Rocke. March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
BST 226 Statistical Methods for Bioinformatics David M. Rocke March 10, 2014 BST 226 Statistical Methods for Bioinformatics 1 NGS Technologies Illumina Sequencing HiSeq 2500 & MiSeq PacBio Sequencing PacBio
Genomics and Transcriptomics of Spirodela polyrhiza
 Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Post-assembly Data Analysis
 Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
Assembled transcriptome Post-assembly Data Analysis Quantification: the expression level of each gene in each sample DE genes: genes differentially expressed between samples Clustering/network analysis
Long and short/small RNA-seq data analysis
 Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS
 SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
SO YOU WANT TO DO A: RNA-SEQ EXPERIMENT MATT SETTLES, PHD UNIVERSITY OF CALIFORNIA, DAVIS SETTLES@UCDAVIS.EDU Bioinformatics Core Genome Center UC Davis BIOINFORMATICS.UCDAVIS.EDU DISCLAIMER This talk/workshop
Supplementary Materials for De-novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity
 Supplementary Materials for De-novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity Sections: S1. Evaluation of transcriptome assembly completeness S2. Comparison
Supplementary Materials for De-novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity Sections: S1. Evaluation of transcriptome assembly completeness S2. Comparison
ChIP-seq and RNA-seq
 ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
ChIP-seq and RNA-seq Biological Goals Learn how genomes encode the diverse patterns of gene expression that define each cell type and state. Protein-DNA interactions (ChIPchromatin immunoprecipitation)
RNA-SEQUENCING ANALYSIS
 RNA-SEQUENCING ANALYSIS Joseph Powell SISG- 2018 CONTENTS Introduction to RNA sequencing Data structure Analyses Transcript counting Alternative splicing Allele specific expression Discovery APPLICATIONS
RNA-SEQUENCING ANALYSIS Joseph Powell SISG- 2018 CONTENTS Introduction to RNA sequencing Data structure Analyses Transcript counting Alternative splicing Allele specific expression Discovery APPLICATIONS
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ),
,") Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
Analysis of data from high-throughput molecular biology experiments Lecture 6 (F6, RNA-seq ), 2012-01-26 What is a gene What is a transcriptome History of gene expression assessment RNA-seq RNA-seq analysis
The genome of Fraxinus excelsior (European Ash)
") The genome of Fraxinus excelsior (European Ash) Elizabeth Sollars, Laura Kelly, Bernardo Clavijo, David Swarbreck, Jasmin Zohren, David Boshier, Jo Clark, Anika Joecker, Sarah Ayling, Mario Caccamo, Richard
The genome of Fraxinus excelsior (European Ash) Elizabeth Sollars, Laura Kelly, Bernardo Clavijo, David Swarbreck, Jasmin Zohren, David Boshier, Jo Clark, Anika Joecker, Sarah Ayling, Mario Caccamo, Richard
Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro
 Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro Philip Morris International R&D, Philip Morris Products S.A., Neuchatel, Switzerland Introduction Nicotiana sylvestris
Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro Philip Morris International R&D, Philip Morris Products S.A., Neuchatel, Switzerland Introduction Nicotiana sylvestris
Rapid Transcriptome Characterization for a nonmodel organism using 454 pyrosequencing
 Rapid Transcriptome Characterization for a nonmodel organism using 454 pyrosequencing "#$%&'()*+,"(-*."#$%&/.,"*01*0.,(%-*.&0("2*01*3,$,45,"-*4#66&*71** 3"#)(82,"-*2&9:)($*)1*"(03&"2-*#)66(*.(8$6#*;
Rapid Transcriptome Characterization for a nonmodel organism using 454 pyrosequencing "#$%&'()*+,"(-*."#$%&/.,"*01*0.,(%-*.&0("2*01*3,$,45,"-*4#66&*71** 3"#)(82,"-*2&9:)($*)1*"(03&"2-*#)66(*.(8$6#*;
Introduction to RNAseq Analysis. Milena Kraus Apr 18, 2016
 Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Introduction to RNAseq Analysis Milena Kraus Apr 18, 2016 Agenda What is RNA sequencing used for? 1. Biological background 2. From wet lab sample to transcriptome a. Experimental procedure b. Raw data
Transcriptome Assembly and Evaluation, using Sequencing Quality Control (SEQC) Data
 Data") Transcriptome Assembly and Evaluation, using Sequencing Quality Control (SEQC) Data Introduction The US Food and Drug Administration (FDA) has coordinated the Sequencing Quality Control project (SEQC/MAQC-III)
Transcriptome Assembly and Evaluation, using Sequencing Quality Control (SEQC) Data Introduction The US Food and Drug Administration (FDA) has coordinated the Sequencing Quality Control project (SEQC/MAQC-III)
RNA-Seq data analysis course September 7-9, 2015
 RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
RNA-Seq data analysis course September 7-9, 2015 Peter-Bram t Hoen (LUMC) Jan Oosting (LUMC) Celia van Gelder, Jacintha Valk (BioSB) Anita Remmelzwaal (LUMC) Expression profiling DNA mrna protein Comprehensive
How much sequencing do I need? Emily Crisovan Genomics Core
 How much sequencing do I need? Emily Crisovan Genomics Core How much sequencing? Three questions: 1. How much sequence is required for good experimental design? 2. What type of sequencing run is best?
How much sequencing do I need? Emily Crisovan Genomics Core How much sequencing? Three questions: 1. How much sequence is required for good experimental design? 2. What type of sequencing run is best?
Sequence Analysis 2RNA-Seq
 Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
Sequence Analysis 2RNA-Seq Lecture 10 2/21/2018 Instructor : Kritika Karri kkarri@bu.edu Transcriptome Entire set of RNA transcripts in a given cell for a specific developmental stage or physiological
How much sequencing do I need? Emily Crisovan Genomics Core September 26, 2018
 How much sequencing do I need? Emily Crisovan Genomics Core September 26, 2018 How much sequencing? Three questions: 1. How much sequence is required for good experimental design? 2. What type of sequencing
How much sequencing do I need? Emily Crisovan Genomics Core September 26, 2018 How much sequencing? Three questions: 1. How much sequence is required for good experimental design? 2. What type of sequencing
Whole Transcriptome Analysis of Illumina RNA- Seq Data. Ryan Peters Field Application Specialist
 Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
RNA-Seq de novo assembly training
 RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
RNA-Seq de novo assembly training Training session aims Give you some keys elements to look at during read quality check. Transcriptome assembly is not completely a strait forward process : Multiple strategies
RNA-Seq. Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University
 RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
RNA-Seq Joshua Ainsley, PhD Postdoctoral Researcher Lab of Leon Reijmers Neuroscience Department Tufts University joshua.ainsley@tufts.edu Day five Alternative splicing Assembly RNA edits Alternative splicing
Wheat Genome Structural Annotation Using a Modular and Evidence-combined Annotation Pipeline
 Wheat Genome Structural Annotation Using a Modular and Evidence-combined Annotation Pipeline Xi Wang Bioinformatics Scientist Computational Life Science Page 1 Bayer 4:3 Template 2010 March 2016 17/01/2017
Wheat Genome Structural Annotation Using a Modular and Evidence-combined Annotation Pipeline Xi Wang Bioinformatics Scientist Computational Life Science Page 1 Bayer 4:3 Template 2010 March 2016 17/01/2017
measuring gene expression December 11, 2018
 measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and
measuring gene expression December 11, 2018 Intervening Sequences (introns): how does the cell get rid of them? Splicing!!! Highly conserved ribonucleoprotein complex recognizes intron/exon junctions and
RNA-Seq analysis workshop
 RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
Analysis of RNA-seq Data
 Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
Analysis of RNA-seq Data A physicist and an engineer are in a hot-air balloon. Soon, they find themselves lost in a canyon somewhere. They yell out for help: "Helllloooooo! Where are we?" 15 minutes later,
Consensus Ensemble Approaches Improve De Novo Transcriptome Assemblies
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Computer Science and Engineering: Theses, Dissertations, and Student Research Computer Science and Engineering, Department
CBC Data Therapy. Metatranscriptomics Discussion
 CBC Data Therapy Metatranscriptomics Discussion Metatranscriptomics Extract RNA, subtract rrna Sequence cdna QC Gene expression, function Institute for Systems Genomics: Computational Biology Core bioinformatics.uconn.edu
CBC Data Therapy Metatranscriptomics Discussion Metatranscriptomics Extract RNA, subtract rrna Sequence cdna QC Gene expression, function Institute for Systems Genomics: Computational Biology Core bioinformatics.uconn.edu
The Genome Analysis Centre. Building Excellence in Genomics and Computa5onal Bioscience
 Building Excellence in Genomics and Computa5onal Bioscience Resequencing approaches Sarah Ayling Crop Genomics and Diversity sarah.ayling@tgac.ac.uk Why re- sequence plants? To iden
Building Excellence in Genomics and Computa5onal Bioscience Resequencing approaches Sarah Ayling Crop Genomics and Diversity sarah.ayling@tgac.ac.uk Why re- sequence plants? To iden
Next Generation Sequencing. Tobias Österlund
 Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
Next Generation Sequencing Tobias Österlund tobiaso@chalmers.se NGS part of the course Week 4 Friday 13/2 15.15-17.00 NGS lecture 1: Introduction to NGS, alignment, assembly Week 6 Thursday 26/2 08.00-09.45
measuring gene expression December 5, 2017
 measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased
 has increased") Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
Human Genome Sequencing Over the Decades The capacity to sequence all 3.2 billion bases of the human genome (at 30X coverage) has increased exponentially since the 1990s. In 2005, with the introduction
DNA. bioinformatics. genomics. personalized. variation NGS. trio. custom. assembly gene. tumor-normal. de novo. structural variation indel.
 DNA Sequencing T TM variation DNA amplicon mendelian trio genomics NGS bioinformatics tumor-normal custom SNP resequencing target validation de novo prediction personalized comparative genomics exome private
DNA Sequencing T TM variation DNA amplicon mendelian trio genomics NGS bioinformatics tumor-normal custom SNP resequencing target validation de novo prediction personalized comparative genomics exome private
High Throughput Sequencing the Multi-Tool of Life Sciences. Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center
 High Throughput Sequencing the Multi-Tool of Life Sciences Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina Still-imaging of clusters (~1000
High Throughput Sequencing the Multi-Tool of Life Sciences Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina Still-imaging of clusters (~1000
Quantifying gene expression
 Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Quantifying gene expression Genome GTF (annotation)? Sequence reads FASTQ FASTQ (+reference transcriptome index) Quality control FASTQ Alignment to Genome: HISAT2, STAR (+reference genome index) (known
Experimental design of RNA-Seq Data
 Experimental design of RNA-Seq Data RNA-seq course: The Power of RNA-seq Thursday June 6 th 2013, Marco Bink Biometris Overview Acknowledgements Introduction Experimental designs Randomization, Replication,
Experimental design of RNA-Seq Data RNA-seq course: The Power of RNA-seq Thursday June 6 th 2013, Marco Bink Biometris Overview Acknowledgements Introduction Experimental designs Randomization, Replication,
RNA-sequencing. Next Generation sequencing analysis Anne-Mette Bjerregaard. Center for biological sequence analysis (CBS)
") RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
RNA-sequencing Next Generation sequencing analysis 2016 Anne-Mette Bjerregaard Center for biological sequence analysis (CBS) Terms and definitions TRANSCRIPTOME The full set of RNA transcripts and their
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Introduction to RNA-Seq Monica Britton, Ph.D. Bioinformatics Analyst September 2014 Workshop Overview of Today s Activities Morning RNA-Seq Concepts, Terminology, and Work Flows Two-Condition Differential
Deep Sequencing technologies
 Deep Sequencing technologies Gabriela Salinas 30 October 2017 Transcriptome and Genome Analysis Laboratory http://www.uni-bc.gwdg.de/index.php?id=709 Microarray and Deep-Sequencing Core Facility University
Deep Sequencing technologies Gabriela Salinas 30 October 2017 Transcriptome and Genome Analysis Laboratory http://www.uni-bc.gwdg.de/index.php?id=709 Microarray and Deep-Sequencing Core Facility University
RNA-Seq with the Tuxedo Suite
 RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
Introduction to RNA-Seq in GeneSpring NGS Software
 Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Introduction to RNA-Seq in GeneSpring NGS Software Dipa Roy Choudhury, Ph.D. Strand Scientific Intelligence and Agilent Technologies Learn more at www.genespring.com Introduction to RNA-Seq In a few years,
Throughput cells cells. Methodology Full transcript or end-counting end-counting. Chemistry SMARTer V SMARTer V. Run time hours.
 PN 101-0984 A1 DATASHEET C1 mrna Sequencing Rapidly characterize heterogeneity, identify critical cell populations. Individual cells are unique they differ by size, protein levels, and expressed mrna transcripts.
PN 101-0984 A1 DATASHEET C1 mrna Sequencing Rapidly characterize heterogeneity, identify critical cell populations. Individual cells are unique they differ by size, protein levels, and expressed mrna transcripts.
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
The Ensembl Database. Dott.ssa Inga Prokopenko. Corso di Genomica
 The Ensembl Database Dott.ssa Inga Prokopenko Corso di Genomica 1 www.ensembl.org Lecture 7.1 2 What is Ensembl? Public annotation of mammalian and other genomes Open source software Relational database
The Ensembl Database Dott.ssa Inga Prokopenko Corso di Genomica 1 www.ensembl.org Lecture 7.1 2 What is Ensembl? Public annotation of mammalian and other genomes Open source software Relational database
RNA
 RNA sequencing Michael Inouye Baker Heart and Diabetes Institute Univ of Melbourne / Monash Univ Summer Institute in Statistical Genetics 2017 Integrative Genomics Module Seattle @minouye271 www.inouyelab.org
RNA sequencing Michael Inouye Baker Heart and Diabetes Institute Univ of Melbourne / Monash Univ Summer Institute in Statistical Genetics 2017 Integrative Genomics Module Seattle @minouye271 www.inouyelab.org
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
Introduction to metagenome assembly. Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014
 Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
Introduction to metagenome assembly Bas E. Dutilh Metagenomic Methods for Microbial Ecologists, NIOO September 18 th 2014 Sequencing specs* Method Read length Accuracy Million reads Time Cost per M 454
1. Introduction Gene regulation Genomics and genome analyses
 1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
1. Introduction Gene regulation Genomics and genome analyses 2. Gene regulation tools and methods Regulatory sequences and motif discovery TF binding sites Databases 3. Technologies Microarrays Deep sequencing
Shuji Shigenobu. April 3, 2013 Illumina Webinar Series
 Shuji Shigenobu April 3, 2013 Illumina Webinar Series RNA-seq RNA-seq is a revolutionary tool for transcriptomics using deepsequencing technologies. genome HiSeq2000@NIBB (Wang 2009 with modifications)
Shuji Shigenobu April 3, 2013 Illumina Webinar Series RNA-seq RNA-seq is a revolutionary tool for transcriptomics using deepsequencing technologies. genome HiSeq2000@NIBB (Wang 2009 with modifications)
Integrative Genomics 1a. Introduction
 2016 Course Outline Integrative Genomics 1a. Introduction ggibson.gt@gmail.com http://www.cig.gatech.edu 1a. Experimental Design and Hypothesis Testing (GG) 1b. Normalization (GG) 2a. RNASeq (MI) 2b. Clustering
2016 Course Outline Integrative Genomics 1a. Introduction ggibson.gt@gmail.com http://www.cig.gatech.edu 1a. Experimental Design and Hypothesis Testing (GG) 1b. Normalization (GG) 2a. RNASeq (MI) 2b. Clustering
An introduction to RNA-seq. Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy
 An introduction to RNA-seq Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy The central dogma Genome = all DNA in an organism (genotype) Transcriptome = all RNA (molecular
An introduction to RNA-seq Nicole Cloonan - 4 th July 2018 #UQWinterSchool #Bioinformatics #GroupTherapy The central dogma Genome = all DNA in an organism (genotype) Transcriptome = all RNA (molecular
GREG GIBSON SPENCER V. MUSE
 A Primer of Genome Science ience THIRD EDITION TAGCACCTAGAATCATGGAGAGATAATTCGGTGAGAATTAAATGGAGAGTTGCATAGAGAACTGCGAACTG GREG GIBSON SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc.
A Primer of Genome Science ience THIRD EDITION TAGCACCTAGAATCATGGAGAGATAATTCGGTGAGAATTAAATGGAGAGTTGCATAGAGAACTGCGAACTG GREG GIBSON SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc.
DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN. (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN
 JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN") DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN ... 2014 2015 2016 2017 ... 2014 2015 2016 2017 Synthetic
DE NOVO WHOLE GENOME ASSEMBLY AND SEQUENCING OF THE SUPERB FAIRYWREN (Malurus cyaneus) JOSHUA PEÑALBA LEO JOSEPH CRAIG MORITZ ANDREW COCKBURN ... 2014 2015 2016 2017 ... 2014 2015 2016 2017 Synthetic
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq. David Wood Winter School in Mathematics and Computational Biology July 1, 2013
 Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
Introduction to RNA-Seq David Wood Winter School in Mathematics and Computational Biology July 1, 2013 Abundance RNA is... Diverse Dynamic Central DNA rrna Epigenetics trna RNA mrna Time Protein Abundance
resequencing storage SNP ncrna metagenomics private trio de novo exome ncrna RNA DNA bioinformatics RNA-seq comparative genomics
 RNA Sequencing T TM variation genetics validation SNP ncrna metagenomics private trio de novo exome mendelian ChIP-seq RNA DNA bioinformatics custom target high-throughput resequencing storage ncrna comparative
RNA Sequencing T TM variation genetics validation SNP ncrna metagenomics private trio de novo exome mendelian ChIP-seq RNA DNA bioinformatics custom target high-throughput resequencing storage ncrna comparative
Statistical Genomics and Bioinformatics Workshop. Genetic Association and RNA-Seq Studies
 Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
Statistical Genomics and Bioinformatics Workshop: Genetic Association and RNA-Seq Studies RNA Seq and Differential Expression Analysis Brooke L. Fridley, PhD University of Kansas Medical Center 1 Next-generation
High performance sequencing and gene expression quantification
 High performance sequencing and gene expression quantification Ana Conesa Genomics of Gene Expression Lab Centro de Investigaciones Príncipe Felipe Valencia aconesa@cipf.es Next Generation Sequencing NGS
High performance sequencing and gene expression quantification Ana Conesa Genomics of Gene Expression Lab Centro de Investigaciones Príncipe Felipe Valencia aconesa@cipf.es Next Generation Sequencing NGS
Introduction to Microbial Sequencing
 Introduction to Microbial Sequencing Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu General rules for preparing
Introduction to Microbial Sequencing Matthew L. Settles Genome Center Bioinformatics Core University of California, Davis settles@ucdavis.edu; bioinformatics.core@ucdavis.edu General rules for preparing
Next Generation Sequencing: An Overview
 Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
RADSeq Data Analysis. Through STACKS on Galaxy. Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé
 RADSeq Data Analysis Through STACKS on Galaxy Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé RAD sequencing: next-generation tools for an old problem INTRODUCTION source: Karim Gharbi
RADSeq Data Analysis Through STACKS on Galaxy Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé RAD sequencing: next-generation tools for an old problem INTRODUCTION source: Karim Gharbi
Exploring and Understanding ChIP-Seq data. Simon v
 Exploring and Understanding ChIP-Seq data Simon Andrews simon.andrews@babraham.ac.uk @simon_andrews v2018-03-02 Data Creation and Processing Starting DNA Fragmented DNA ChIPped DNA Mapped BAM File FastQ
Exploring and Understanding ChIP-Seq data Simon Andrews simon.andrews@babraham.ac.uk @simon_andrews v2018-03-02 Data Creation and Processing Starting DNA Fragmented DNA ChIPped DNA Mapped BAM File FastQ
Workflows and Pipelines for NGS analysis: Lessons from proteomics
 Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
From assembled genome to annotated genome
 From assembled genome to annotated genome Procaryotic genomes Eucaryotic genomes Genome annotation servers (web based) 1. RAST 2. NCBI Gene prediction pipeline: Maker Function annotation pipeline: Blast2GO
From assembled genome to annotated genome Procaryotic genomes Eucaryotic genomes Genome annotation servers (web based) 1. RAST 2. NCBI Gene prediction pipeline: Maker Function annotation pipeline: Blast2GO
Session 8. Differential gene expression analysis using RNAseq data
 Functional and Comparative Genomics 2018 Session 8. Differential gene expression analysis using RNAseq data Tutors: Hrant Hovhannisyan, PhD student, email: grant.hovhannisyan@gmail.com Uciel Chorostecki,
Functional and Comparative Genomics 2018 Session 8. Differential gene expression analysis using RNAseq data Tutors: Hrant Hovhannisyan, PhD student, email: grant.hovhannisyan@gmail.com Uciel Chorostecki,
Post-assembly Data Analysis
 Assembled transcriptome Post-assembly Data Analysis Quantification: get expression for each gene in each sample Genes differentially expressed between samples Clustering/network analysis Identifying over-represented
Assembled transcriptome Post-assembly Data Analysis Quantification: get expression for each gene in each sample Genes differentially expressed between samples Clustering/network analysis Identifying over-represented
Purpose of sequence assembly
 Sequence Assembly Purpose of sequence assembly Reconstruct long DNA/RNA sequences from short sequence reads Genome sequencing RNA sequencing for gene discovery But not for transcript quantification Variant
Sequence Assembly Purpose of sequence assembly Reconstruct long DNA/RNA sequences from short sequence reads Genome sequencing RNA sequencing for gene discovery But not for transcript quantification Variant
Basics of RNA-Seq. (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility
 Michael Kelly, PhD Team Lead, NCI Single Cell Analysis Facility") 2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
2018 ABRF Meeting Satellite Workshop 4 Bridging the Gap: Isolation to Translation (Single Cell RNA-Seq) Sunday, April 22 Basics of RNA-Seq (With a Focus on Application to Single Cell RNA-Seq) Michael Kelly,
TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR)
") tru TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR) Anton Bankevich Center for Algorithmic Biotechnology, SPbSU Sequencing costs 1. Sequencing costs do not follow Moore s law
tru TruSPAdes: analysis of variations using TruSeq Synthetic Long Reads (TSLR) Anton Bankevich Center for Algorithmic Biotechnology, SPbSU Sequencing costs 1. Sequencing costs do not follow Moore s law
From reads to results: differen1al expression analysis with RNA seq. Alicia Oshlack Bioinforma1cs Division Walter and Eliza Hall Ins1tute
 From reads to results: differen1al expression analysis with RNA seq Alicia Oshlack Bioinforma1cs Division Walter and Eliza Hall Ins1tute Purported benefits and opportuni1es of RNA seq All transcripts are
From reads to results: differen1al expression analysis with RNA seq Alicia Oshlack Bioinforma1cs Division Walter and Eliza Hall Ins1tute Purported benefits and opportuni1es of RNA seq All transcripts are
Introduction of RNA-Seq Analysis
 Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Introduction of RNA-Seq Analysis Jiang Li, MS Bioinformatics System Engineer I Center for Quantitative Sciences(CQS) Vanderbilt University September 21, 2012 Goal of this talk 1. Act as a practical resource
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Introduction to bioinformatics (NGS data analysis)
") Introduction to bioinformatics (NGS data analysis) Alexander Jueterbock 2015-06-02 1 / 45 Got your sequencing data - now, what to do with it? File size: several Gb Number of lines: >1,000,000 @M02443:17:000000000-ABPBW:1:1101:12675:1533
Introduction to bioinformatics (NGS data analysis) Alexander Jueterbock 2015-06-02 1 / 45 Got your sequencing data - now, what to do with it? File size: several Gb Number of lines: >1,000,000 @M02443:17:000000000-ABPBW:1:1101:12675:1533
Data Basics. Josef K Vogt Slides by: Simon Rasmussen Next Generation Sequencing Analysis
 Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Data Basics Josef K Vogt Slides by: Simon Rasmussen 2017 Generalized NGS analysis Sample prep & Sequencing Data size Main data reductive steps SNPs, genes, regions Application Assembly: Compare Raw Pre-
Finding Genes with Genomics Technologies
 PLNT2530 Plant Biotechnology (2018) Unit 7 Finding Genes with Genomics Technologies Unless otherwise cited or referenced, all content of this presenataion is licensed under the Creative Commons License
PLNT2530 Plant Biotechnology (2018) Unit 7 Finding Genes with Genomics Technologies Unless otherwise cited or referenced, all content of this presenataion is licensed under the Creative Commons License
Wet-lab Considerations for Illumina data analysis
 Wet-lab Considerations for Illumina data analysis Based on a presentation by Henriette O Geen Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina
Wet-lab Considerations for Illumina data analysis Based on a presentation by Henriette O Geen Lutz Froenicke DNA Technologies and Expression Analysis Cores UCD Genome Center Complementary Approaches Illumina
Parts of a standard FastQC report
 FastQC FastQC, written by Simon Andrews of Babraham Bioinformatics, is a very popular tool used to provide an overview of basic quality control metrics for raw next generation sequencing data. There are
FastQC FastQC, written by Simon Andrews of Babraham Bioinformatics, is a very popular tool used to provide an overview of basic quality control metrics for raw next generation sequencing data. There are
De novo whole genome assembly
 De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore
De novo whole genome assembly Qi Sun Bioinformatics Facility Cornell University Sequencing platforms Short reads: o Illumina (150 bp, up to 300 bp) Long reads (>10kb): o PacBio SMRT; o Oxford Nanopore