Regulatory and ethical requirements in medical devices studies. France
|
|
|
- Avice Hicks
- 6 years ago
- Views:
Transcription

1 Regulatory and ethical in medical devices studies France
2 SECTIONS A.Type of research
3 SECTIONS A.Type of research
4 We have differentiated 8 types of research: Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device Registries
5 SECTIONS A.Type of research
6 H. Data Ptotection Definitions in French law Ecept for «Observational studies with medical device», there is no French definition for studies with medical devices.
7 Conventions/guideline/laws to apply Declaration of Helsinki ICH Guideline of EMA European Directive 2011/20/EC (Only for studies with MD alone and observational studies) Law of 9 aout 2004 (completed by decree du 26 avril 2006) ISO 14155:2011 European Directive 90/385/EC and 93/42/EC
8 Acts to apply Data protection act (Ecept for registries) Genetical engineering act (Ecept for observational studies and registries)
9 SECTIONS A.Type of research
10 For all the type of studies in eception of registries, an insurance must be contracted. It has to cover: - Patients and /or healthy volunteers - Investigators - Sponsor It is not necessary that the insurance covers the manufacturer The compensation sums covered by the insurance depends of the protocol
11 SECTIONS A.Type of research
12 It is mandatory to have a sponsor in all the interventional studies: Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark
13 SECTIONS A.Type of research
14 No French specific /regulations for GCP training of the investigators
15 SECTIONS A.Type of research
16 French Competent : ANSM Agence national de sécurité du médicament et des produits de santé 143/147 Boulevard Anatole France Saint-Denis Cede dedim.dm@ansm.sante.fr or EC.DM- COS@ansm.sante.fr
17 Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device Yes Approval No (if no risky eam)
18 Initial submission When a submission is required, the sponsor is responsible of it The submission to the French Competent is national: you only have to submit one dossier to the national competent authority The submission have to be: - by paper - or by or CD-Rom English documents are accepted, at the eception of the protocol summary, the inform consent form and the information document for patient No submission fee
19 Initial submission In general, 60 days maimum to obtain approval Implicit approval after 60 days without questions No deadlines for submission, you can submit anytime No need to have some kind of representative or a legal entity in France to submit an application to the Competent if you are from the EU
20 F. Competant Initial submission Main documents required for submission: Submission letter Clinical Trial application form Clinical Investigation Plan/Protocol Clinical Investigation Plan/Protocol summary Copies of advertisement materials for research participants Investigator s brochure or CE certificate Instruction for use/technical manual Performance evaluation Pre-clinical evaluation Insurance certificate List of the Competent where protocol was submitted with their decision if available Copy of the required authorization ( authorisation de lieu de recherche )

21 Initial submission Standard application form available on the ANSM s website:
22 - Vigilance SAE definition: Any untoward medical occurrence or effect that at any dose: - Result in death - Is life threatening - Requires hospitalisation or prolongation of eisting hospitalisation - Results in persistent or significant disability or incapacity - Or is a congenital anomaly or birth defect SAE declaration by the sponsor: - If life in danger: 7 days - Other case: 15 days The sponsor has to declare events to the Competent Authorities in the specific countries The sponsor needs to provide to the Competent an annual safety report
23 - Vigilance H. Data Ptotection Events mandatory to declare to the Competent : Medical device alone with CE mark use within label Medical device alone with CE mark use outside label AE ADE SADE SAE Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device AE: Adverse Event (Non device related) ADE: Adverse Device Effect (Device or procedure related) SADE: Serious adverse Device Effect (Device or procedure related) SAE: Serious Adverse Event (Non device related)
24 - Vigilance The sponsor is responsible for the declaration to the Competent Special form for the declaration of AE:
25 - Notification It is not mandatory to notify the first patient enrolled to the Competent
26 Substantial amendment There is a specific procedure for submitting a substantial amendment to the Competent (Submission of a dossier and waiting for approval)
27 SECTIONS A.Type of research

 in")
28 40 Comités de Protection des Personnes (CPP) in France:
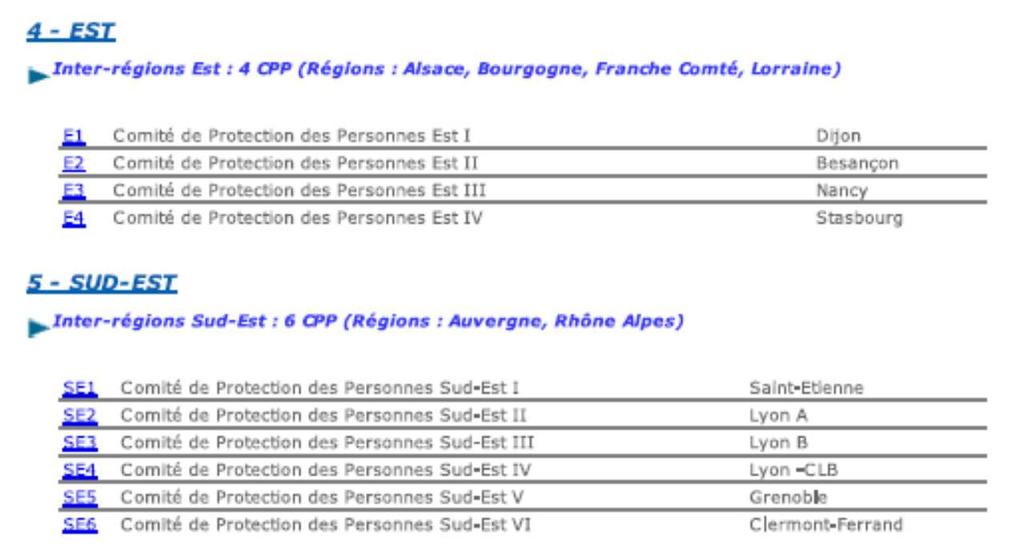
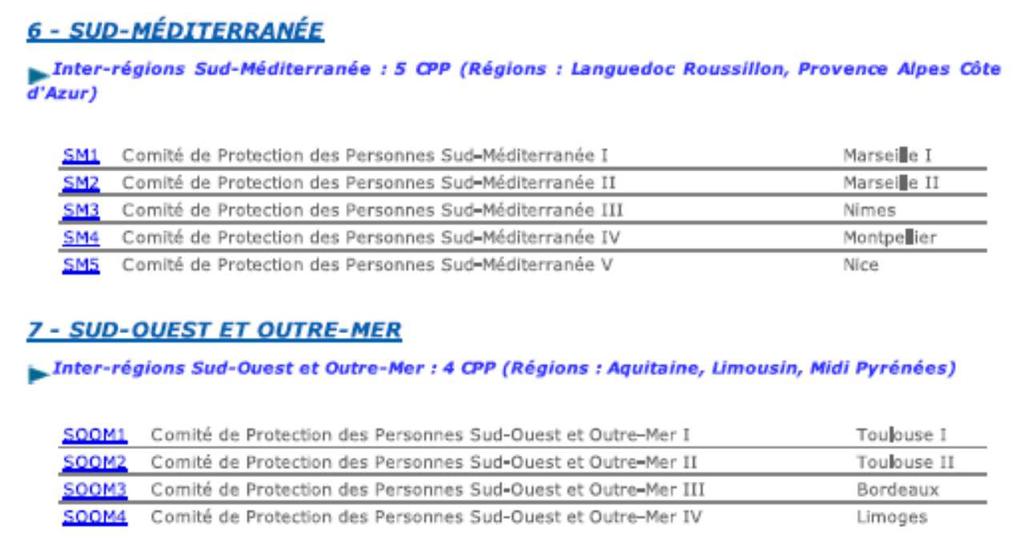
29
30 Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device Positive opinion required Yes No
31 Initial submission When a submission is required, the sponsor is responsible of it The submission to the Ethics Committee is local: you only have to submit one dossier to one of the regional Ethics Committee in the region of the Principal Investigator The submission have to be: - by paper (usually many copies) - and/or by or CD-Rom English documents are accepted, at the eception of the protocol summary, the inform consent form, the information document for patient and additional document No submission fee
32 Initial submission H. Data Ptotection In general, days to obtain approval No implicit approval Deadlines for submissions, once per month in each EC (different dates for each EC) When both Competent approval and Ethics Committee positive opinion are required, it is possible to request the 2 (authorization and approval) in parallel No need to have some kind of representative or a legal entity in France to submit an application to the Competent or Ethics Committee if you are from the EU
33 Initial submission H. Data Ptotection Main documents required for submission: Submission letter Clinical Trial application form Clinical Investigation Plan/Protocol Clinical Investigation Plan/Protocol summary Inform Consent Form and subject Information leaflet CV of PIs Copie of advertisement materials for research participant Investigator s brochure or CE certificate Instruction for use/technical manual Insurance certificate List of the CA where protocol was submitted with their decision if available Copy of the required authorization ( authorisation de lieu de recherche ) Additional document
34 G. Ethics committee Initial submission Use the same standard application form that for the Competent Standard application form available on the ANSM s website:
35 - Vigilance SAE declaration by the sponsor: - If life in danger: 7 days - Other case: 15 days The sponsor also has to declare events to the Ethics Committee in the specific countries The sponsor needs to provide to the Ethics Committee an annual safety report
36 - Vigilance Events mandatory to declare to the competent authority: Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device AE ADE SADE SAE AE: Adverse Event (Non device related) ADE: Adverse Device Effect (Device or procedure related) SADE: Serious adverse Device Effect (Device or procedure related) SAE: Serious Adverse Event (Non device related)
37 - Vigilance The sponsor is responsible for the declaration to the Ethics Committee No special form for the declaration of AE, possible to use the Competent s form:
38 - Notification It is not mandatory to notify the first patient enrolled to the Ethics Committee
39 Substantial amendment There is a specific procedure for submitting a substantial amendment to the Ethics Committee (Submission of a dossier and waiting for approval)
40 SECTIONS A.Type of research
41 H. Data Ptotection French Data Protection: CNIL (Commission Nationale de l Informatique et des Liberté) 8 rue vivienne Paris cede 02 Tel : / Fa :
42 Medical device alone with CE mark use within label Medical device alone with CE mark use outside label Medical device alone without CE mark Medical device combined with medicinal product with CE mark use within label Medical device combined with medicinal product with CE mark use outside label Medical device combined with medicinal product without CE mark Observational studies with medical device Notification required Yes No
43 Submission in French only No submission fee Online submission
44 SECTIONS A.Type of research
45 H. Data Ptotection In France, there are specific /regulations for specific population: - Children - Pregnant women - Lactating women - Adults protected by the law
46 Healthy volunteers participating in clinical research have to be compensated (not mandatory for patients, but possible) National healthy volunteers registry: VRB Obligation to inform the healthy volunteers/patients on the outcomes of the study if she/he asks
47 SECTIONS A.Type of research
48 No specific to publish both positive and negative results of clinical studies in scientific journals Specific /regulations regarding the ICF: needs to be given in writing Specific /regulations regarding archiving of documentation: 15 years Specific regarding blood/tissue samples (circulation and storage) Additional forms to submit the Competent
49 No specific for data management No specific strategies for monitoring Mandatory to register clinical studies which include medicinal product in a registry managed by the Competent Accreditation process for research centres ( Code de santé publique article R to article R )
Regulatory and ethical requirements in medical devices studies. Turkey
 Regulatory and ethical in medical devices studies Turkey SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical in medical devices studies Turkey SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical requirements in medical devices studies. Italy
 Regulatory and ethical in medical devices studies Italy SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical in medical devices studies Italy SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical requirements in medical device studies. Finland
 Regulatory and ethical in medical device studies Finland SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Regulatory and ethical in medical device studies Finland SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Regulatory and ethical requirements in medical devices studies. Portugal
 Regulatory and ethical in medical devices studies Portugal SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical in medical devices studies Portugal SECTIONS A.Type of research H. Data Protection SECTIONS A.Type of research H. Data Protection We have differentiated 8 types of research: Medical
Regulatory and ethical requirements in medical device studies DENMARK
 Regulatory and ethical in medical device studies DENMARK SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Regulatory and ethical in medical device studies DENMARK SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Regulatory and ethics bodies involved in approval process for trials. National Ethics Committee; Agency for Data Protection;
 Nutrition/InterventionaI - LUXEMBOURG Competent authority Contact Details Contact Name 1 Ministry of Health Phone +352 247-85592 / 96 Fax +352 24795615 Email General info@ ms.etat.lu Address Allée Marconi
Nutrition/InterventionaI - LUXEMBOURG Competent authority Contact Details Contact Name 1 Ministry of Health Phone +352 247-85592 / 96 Fax +352 24795615 Email General info@ ms.etat.lu Address Allée Marconi
RESEARCH IN HUMAN SUBJECTS OTHER THAN CLINICAL TRIALS OF INVESTIGATIONAL MEDICINAL PRODUCTS
 Page 1 of 5 RESEARCH IN HUMAN SUBJECTS OTHER THAN CLINICAL TRIALS OF INVESTIGATIONAL MEDICINAL PRODUCTS After ethical review guidance for sponsors and investigators This document sets out important guidance
Page 1 of 5 RESEARCH IN HUMAN SUBJECTS OTHER THAN CLINICAL TRIALS OF INVESTIGATIONAL MEDICINAL PRODUCTS After ethical review guidance for sponsors and investigators This document sets out important guidance
Regulatory and ethics bodies involved in approval process. CA - Registration requirements for clinical trials
 Nutrition - LUXEMBOURG Competent authority Contact Details Contact Name 1 Ministry of Health Phone +352 247-85592 / 96 Fax +352 24795615 Email General info@ ms.etat.lu Address Allée Marconi - Villa Louvigny
Nutrition - LUXEMBOURG Competent authority Contact Details Contact Name 1 Ministry of Health Phone +352 247-85592 / 96 Fax +352 24795615 Email General info@ ms.etat.lu Address Allée Marconi - Villa Louvigny
Health Registration and Training Center (HRTC/ENKK) , Mail: 1380 P.O. Box 1188 (Street: Zrinyi u. 3.
 , Mail: 1380 P.O. Box 1188 (Street: Zrinyi u. 3.") Medical Devices - HUNGARY Competent authority Contact Details Contact Name 1 Health Registration and Training Center (HRTC/ENKK) Contact Name 2 Department of Medical Devices Phone +36 1 235-7914, +36 1
Medical Devices - HUNGARY Competent authority Contact Details Contact Name 1 Health Registration and Training Center (HRTC/ENKK) Contact Name 2 Department of Medical Devices Phone +36 1 235-7914, +36 1
ANNEX. CHAPTER I General principles
 ANNEX REGULATIONS on the authorisation by the NMA of clinical trials/notification to the National Medicines Agency of non-interventional studies on medicinal products for human use in Romania CHAPTER I
ANNEX REGULATIONS on the authorisation by the NMA of clinical trials/notification to the National Medicines Agency of non-interventional studies on medicinal products for human use in Romania CHAPTER I
Melbourne Health Guidelines Cover Sheet
 Melbourne Guidelines Cover Sheet NAME OF DEPARTMENT OFFICE FOR RESEARCH NAME OF DOCUMENT Clinical Trials Involving Therapeutic Products and Other Clinical Research NUMBER 18.8 ASSOCIATED MELBOURNE HEALTH
Melbourne Guidelines Cover Sheet NAME OF DEPARTMENT OFFICE FOR RESEARCH NAME OF DOCUMENT Clinical Trials Involving Therapeutic Products and Other Clinical Research NUMBER 18.8 ASSOCIATED MELBOURNE HEALTH
Agence nationale de sécurité du médicament et des produits de santé (ANSM) ansm.sante.fr. 143/147 Boulevard Anatole France
 ansm.sante.fr. 143/147 Boulevard Anatole France") Medicinal Products for Human Use - FRANCE Competent authority Contact Details Contact Name 1 Agence nationale de sécurité du médicament et des produits de santé (ANSM) Phone +33 (0)1 55 87 36 58 Fax +33
Medicinal Products for Human Use - FRANCE Competent authority Contact Details Contact Name 1 Agence nationale de sécurité du médicament et des produits de santé (ANSM) Phone +33 (0)1 55 87 36 58 Fax +33
Belgian Center for Pharamcovigilance for medicines for Human use (BCPH): faggafmps.be
: faggafmps.be") Medicinal Products for Human Use - BELGIUM Competent authority Contact Details Contact Name 1 Federal Agency for Medicines and Health Products (FAMHP) / Agence Fédérale des Médicaments et des Produits
Medicinal Products for Human Use - BELGIUM Competent authority Contact Details Contact Name 1 Federal Agency for Medicines and Health Products (FAMHP) / Agence Fédérale des Médicaments et des Produits
Impact of Clinical EU Directive on the implementation of Early development studies in Europe
 Impact of Clinical EU Directive on the implementation of Early development studies in Europe Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Syposium Strasbourg
Impact of Clinical EU Directive on the implementation of Early development studies in Europe Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Syposium Strasbourg
Pharmacovigilance and safety reporting for sponsored ATIMPs/CTIMPs
 Joint Research Management Office (JRMO) Standard Operating Procedure (SOP) for: Pharmacovigilance and safety reporting for sponsored ATIMPs/CTIMPs SOP Number: 26a Version Number: 13.0 Effective Date: 31
Joint Research Management Office (JRMO) Standard Operating Procedure (SOP) for: Pharmacovigilance and safety reporting for sponsored ATIMPs/CTIMPs SOP Number: 26a Version Number: 13.0 Effective Date: 31
State Institute for Control of Drugs-/ Státní ústav pro control léčiv (SÚKL) Regulatory and ethics bodies involved in approval process
 Regulatory and ethics bodies involved in approval process") Medical Devices - CZECH REPUBLIC Competent authority Contact Details Contact Name 1 State Institute for Control of Drugs-/ Státní ústav pro control léčiv (SÚKL) Phone +420 272 185 111 Fax +420 271 732
Medical Devices - CZECH REPUBLIC Competent authority Contact Details Contact Name 1 State Institute for Control of Drugs-/ Státní ústav pro control léčiv (SÚKL) Phone +420 272 185 111 Fax +420 271 732
Investigator Site File Index (Medical Devices)
") Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol (Clinical Investigation Plan) Current version plus all previous versions (or provide file note
Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol (Clinical Investigation Plan) Current version plus all previous versions (or provide file note
1 PROCEDURE S OBJECT 2 PROCEDURE S SCOPE 3 RESPONSIBILITIES AND AUTHORITIES SOP FOR THE PHARMACOVIGILANCE OF THE CLINICAL TRIALS
 SOP FOR THE PHARMACOVIGILANCE OF THE CLINICAL TRIALS N : AAHRPP-SOP-015 / REV 002 N ENGLISH VERSION : 104 "Please do take into account that this is a translation of the original French version validated
SOP FOR THE PHARMACOVIGILANCE OF THE CLINICAL TRIALS N : AAHRPP-SOP-015 / REV 002 N ENGLISH VERSION : 104 "Please do take into account that this is a translation of the original French version validated
Regulatory Newsletter January - March 2014
 Regulatory Newsletter January - March 2014 Introduction CROMSOURCE is committed to sharing our expertise with our clients and future clients. This reflects the first part of our Advise Agree Deliver motto!
Regulatory Newsletter January - March 2014 Introduction CROMSOURCE is committed to sharing our expertise with our clients and future clients. This reflects the first part of our Advise Agree Deliver motto!
Clinical trials from CRA s point of view
 Clinical trials from CRA s point of view Dr Jacek S Gregorczyk MPharmS, PhD Clinical Research Associate (CRA) at MDS Pharma Services UK office - a global CRO REGULATION of CLINICAL TRIALS World Medical
Clinical trials from CRA s point of view Dr Jacek S Gregorczyk MPharmS, PhD Clinical Research Associate (CRA) at MDS Pharma Services UK office - a global CRO REGULATION of CLINICAL TRIALS World Medical
Joint Research Office of Doncaster & Bassetlaw
 Page 1 of 11 Hospitals NHS, Rotherham Doncaster & South Humber NHS and NHS Signatures: Role Name Function Date (DD-MM-YYYY) Signature Author Amy Beckitt Clinical Research Development Manager Reviewer Dr
Page 1 of 11 Hospitals NHS, Rotherham Doncaster & South Humber NHS and NHS Signatures: Role Name Function Date (DD-MM-YYYY) Signature Author Amy Beckitt Clinical Research Development Manager Reviewer Dr
Competent federal higher authority ("Bundesoberbehörde- BOB")
") Nutrition/InterventionaI - GERMANY Competent authority Contact Details Contact Name 1 Competent federal higher authority ("Bundesoberbehörde- BOB") Contact Name 2 Federal Institute for Drugs and Medical
Nutrition/InterventionaI - GERMANY Competent authority Contact Details Contact Name 1 Competent federal higher authority ("Bundesoberbehörde- BOB") Contact Name 2 Federal Institute for Drugs and Medical
PROCEDURE OF FILING A REQUEST TO THE ETHICS COMMITEE OF THE UNIVERSITY CLINICAL CENTRE OF THE REPUBLIC OF SRPSKA
 PROCEDURE OF FILING A REQUEST TO THE ETHICS COMMITEE OF THE UNIVERSITY CLINICAL CENTRE OF THE REPUBLIC OF SRPSKA All requests, reports and letters to the Ethics Committee of the UCC RS (hereinafter referred
PROCEDURE OF FILING A REQUEST TO THE ETHICS COMMITEE OF THE UNIVERSITY CLINICAL CENTRE OF THE REPUBLIC OF SRPSKA All requests, reports and letters to the Ethics Committee of the UCC RS (hereinafter referred
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
Good Clinical Practice
 Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
APPLICATION FORM. Application for a Recognised Research Ethics Committee (REC) Opinion on a Clinical Trial on a Medicinal Product for Human Use.
 Opinion on a Clinical Trial on a Medicinal Product for Human Use.") APPLICATION FORM Application for a Recognised Research Ethics Committee (REC) Opinion on a Clinical Trial on a Medicinal Product for Human Use. Form 1 This application form should be completed and submitted
APPLICATION FORM Application for a Recognised Research Ethics Committee (REC) Opinion on a Clinical Trial on a Medicinal Product for Human Use. Form 1 This application form should be completed and submitted
OFFICE FOR RESEACH PROCEDURE. Sponsor Responsibilities in Investigator Initiated Studies
 OFFICE FOR RESEACH PROCEDURE Sponsor Responsibilities in Investigator Initiated Studies 1. Purpose: To define Sponsor Responsibilities in the conduct of Investigator driven studies. 2. Scope: All phases
OFFICE FOR RESEACH PROCEDURE Sponsor Responsibilities in Investigator Initiated Studies 1. Purpose: To define Sponsor Responsibilities in the conduct of Investigator driven studies. 2. Scope: All phases
How did it evolve? o Public disasters, serious fraud and abuse of human rights. o Trials of War criminals-nuremberg code 1949
 ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
Safety Reporting Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs)
") Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs) Version 1.4 Effective date: 1 December 2011 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision
Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs) Version 1.4 Effective date: 1 December 2011 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
Regulatory and ethics bodies involved in approval process. CA - Submission for authorisation mandatory for
 Medicinal Products for Human Use - CZECH REPUBLIC Competent authority Contact Details Contact Name 1 State Institute for Control of Drugs- SÚKL (Státní ústav pro kontrolu léčiv) Phone +420 272 185 111
Medicinal Products for Human Use - CZECH REPUBLIC Competent authority Contact Details Contact Name 1 State Institute for Control of Drugs- SÚKL (Státní ústav pro kontrolu léčiv) Phone +420 272 185 111
The EFGCP Report on The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria
 Bulgaria") The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria Question 1: What laws or regulations apply to an application for conducting a clinical
The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria Question 1: What laws or regulations apply to an application for conducting a clinical
STANDARD OPERATING PROCEDURE FOR RESEARCH. 17. Study Close Down
 Basildon and Thurrock University Hospitals NHS FT Research & Development APPROVED STANDARD OPERATING PROCEDURE FOR RESEARCH 1. BACKGROUND 17. Study Close Down According to ICH Good Clinical Practice (GCP)
Basildon and Thurrock University Hospitals NHS FT Research & Development APPROVED STANDARD OPERATING PROCEDURE FOR RESEARCH 1. BACKGROUND 17. Study Close Down According to ICH Good Clinical Practice (GCP)
RSC/CT Det. no. 1/2013
 RSC/CT Det. no. 1/2013 Protocol 1631-P The English version of this Determination was prepared in order to help comprehension by non-italian mother tongue users, but is NOT an official document. Please
RSC/CT Det. no. 1/2013 Protocol 1631-P The English version of this Determination was prepared in order to help comprehension by non-italian mother tongue users, but is NOT an official document. Please
SAE Reporting Timelines, Causality Assessment And Compensation. Pawandeep Kaur Associate Medical Director CDSA
 SAE Reporting Timelines, Causality Assessment And Compensation Pawandeep Kaur Associate Medical Director CDSA Objective Background Important definitions SAE Reporting Timelines Causality Assessment Compensation
SAE Reporting Timelines, Causality Assessment And Compensation Pawandeep Kaur Associate Medical Director CDSA Objective Background Important definitions SAE Reporting Timelines Causality Assessment Compensation
Marie-Claire Rickard (RG and GCP Manager) Rachel Fay (RG and GCP Manager) Elizabeth Clough (R&D Governance Operations Manager)
 Rachel Fay (RG and GCP Manager) Elizabeth Clough (R&D Governance Operations Manager)") Standard Operating Procedures (SOP) for: For researchers: Pharmacovigilance and Safety Reporting for Sponsored CTIMPs/ATMP SOP Number: 026a Version Number: 12.0 Effective Date: 21/4/16 Review Date: 21/4/18
Standard Operating Procedures (SOP) for: For researchers: Pharmacovigilance and Safety Reporting for Sponsored CTIMPs/ATMP SOP Number: 026a Version Number: 12.0 Effective Date: 21/4/16 Review Date: 21/4/18
NIS Considerations - France An overview of the considerations when conducting Noninterventional
 NIS Considerations - France An overview of the considerations when conducting Noninterventional Studies in France Stuart McCully CHCUK Ltd NIS-C-FR-2014 (Mar 2014) Dr Stuart McCully 2014 NIS-C-FR-2014
NIS Considerations - France An overview of the considerations when conducting Noninterventional Studies in France Stuart McCully CHCUK Ltd NIS-C-FR-2014 (Mar 2014) Dr Stuart McCully 2014 NIS-C-FR-2014
Investigator Site File Index (CTIMP)
") Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol Current version plus all previous versions (or provide file note to detail location of previous
Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol Current version plus all previous versions (or provide file note to detail location of previous
Standard Operating Procedure
 Standard Operating Procedure Title: Clinical Site Monitoring Status: PRIVATE Author Name: Audrey Strader Approver Name: Christine Kubiak Document no.: CSM 02 Effective date: 11/01/2016 Review Date (if
Standard Operating Procedure Title: Clinical Site Monitoring Status: PRIVATE Author Name: Audrey Strader Approver Name: Christine Kubiak Document no.: CSM 02 Effective date: 11/01/2016 Review Date (if
Once notified of the end of trial, a Research Manager, on behalf of the Sponsor, will contact the CI to arrange a close down monitoring visit.
 1. INTRODUCTION This SOP has been produced in accordance with the requirements of The Medicines for Human Use (Clinical Trials) Regulations 2004, Medicines for Human Use (Clinical Trials) Amendment Regulations
1. INTRODUCTION This SOP has been produced in accordance with the requirements of The Medicines for Human Use (Clinical Trials) Regulations 2004, Medicines for Human Use (Clinical Trials) Amendment Regulations
Safety monitoring and reporting in clinical trials involving therapeutic goods
 Safety monitoring and reporting in clinical trials involving therapeutic goods November 2016 Publication Details Publication title: Guidance:. Published: November 2016 Publisher: National Health and Medical
Safety monitoring and reporting in clinical trials involving therapeutic goods November 2016 Publication Details Publication title: Guidance:. Published: November 2016 Publisher: National Health and Medical
ETHICS COMMITTEE. TITLE: Safety Reporting STANDARD OPERATING PROCEDURE SOP006
 1 PURPOSE The purpose of this SOP is to describe the Peter MacCallum Cancer Centre (Peter Mac) Ethics Committee process for submission and handling of safety reports arising out of research reviewed by
1 PURPOSE The purpose of this SOP is to describe the Peter MacCallum Cancer Centre (Peter Mac) Ethics Committee process for submission and handling of safety reports arising out of research reviewed by
Implementation of the EU-law. Tom Van Paepegem Quality co-ordinator D.R.U.G.
 Implementation of the EU-law Tom Van Paepegem Quality co-ordinator D.R.U.G. History (1) World War II: Experiments by Nazi-doctors 1946: Nüremberg process 1947: Nüremberg codeddd History (2) 10 basic ethical
Implementation of the EU-law Tom Van Paepegem Quality co-ordinator D.R.U.G. History (1) World War II: Experiments by Nazi-doctors 1946: Nüremberg process 1947: Nüremberg codeddd History (2) 10 basic ethical
ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL
 Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
STANDARD OPERATING PROCEDURE
 Research and Development STANDARD OPERATING PROCEDURE PHARMACOVIGILANCE S22 Post holder responsible for SOP: Anoushka Tepielow Position: Assistant R&D Manager Author: Anoushka Tepielow Position: Assistant
Research and Development STANDARD OPERATING PROCEDURE PHARMACOVIGILANCE S22 Post holder responsible for SOP: Anoushka Tepielow Position: Assistant R&D Manager Author: Anoushka Tepielow Position: Assistant
Central Committee for Research Involving Human Subjects/ Centrale Commissie Mensgebonden Onderzoek (CCMO) NB! if CCMO acts as CA:
 NB! if CCMO acts as CA:") Medicinal Products for Human Use - NETHERLANDS Competent authority Contact Details Contact Name 1 Central Committee for Research Involving Human Subjects/ Centrale Commissie Mensgebonden Onderzoek (CCMO)
Medicinal Products for Human Use - NETHERLANDS Competent authority Contact Details Contact Name 1 Central Committee for Research Involving Human Subjects/ Centrale Commissie Mensgebonden Onderzoek (CCMO)
Standard Operating Procedure
 Standard Operating Procedure Number: UM/UoM Pharmacovigilance_Reporting Adverse Events/SOP10(i)/6.0 Title: Standard Operating Procedure for Identifying, Recording and Reporting Adverse Events for Clinical
Standard Operating Procedure Number: UM/UoM Pharmacovigilance_Reporting Adverse Events/SOP10(i)/6.0 Title: Standard Operating Procedure for Identifying, Recording and Reporting Adverse Events for Clinical
The compassionate use of medicinal products. An example: the French ATU system. 0ff label use in France
 0 The compassionate use of medicinal products. An example: the French ATU system 0ff label use in France C. Bélorgey Head of Department of evaluation of Clinical Trials and medicinal products of special
0 The compassionate use of medicinal products. An example: the French ATU system 0ff label use in France C. Bélorgey Head of Department of evaluation of Clinical Trials and medicinal products of special
Objectives Discuss the importance of proper data collection. Identify the types of data collected for clinical trials. List potential source documents
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
SWOG
 SWOG http://swog.org Page 1 of 5 pages Original Release Date: July 1985 Departments Affected: All Revision Date: April 2018 Introduction SERIOUS ADVERSE EVENTS The timely reporting of serious adverse events
SWOG http://swog.org Page 1 of 5 pages Original Release Date: July 1985 Departments Affected: All Revision Date: April 2018 Introduction SERIOUS ADVERSE EVENTS The timely reporting of serious adverse events
KLH-21 version 7 With effect from the date of issue (effective date 20th July 2018), this guideline supersedes guideline KLH-21, version 6.
, this guideline supersedes guideline KLH-21, version 6.") KLH-21 version 7 Reporting Adverse Reactions to Medicinal Products for Human Use in a Clinical Trial and to Medicinal Products without Marketing Authorisation The guideline is being issued on the basis
KLH-21 version 7 Reporting Adverse Reactions to Medicinal Products for Human Use in a Clinical Trial and to Medicinal Products without Marketing Authorisation The guideline is being issued on the basis
NIS Considerations - Bulgaria
 NIS Considerations - Bulgaria An overview of the considerations when conducting Noninterventional Studies in Bulgaria Stuart McCully CHCUK Ltd NIS-C-BG-2014 1 Table of Contents Disclaimer 5 Copyright 5
NIS Considerations - Bulgaria An overview of the considerations when conducting Noninterventional Studies in Bulgaria Stuart McCully CHCUK Ltd NIS-C-BG-2014 1 Table of Contents Disclaimer 5 Copyright 5
VERSION: 21 st June Date of Publication: 15 th March C/ CAMPEZO, 1 EDIFICIO MADRID Tel.: Fax:
 Memorandum on Collaboration and Exchange of Information between the Spanish Agency of Medicinal Products and Medical Devices and Ethics Committees for investigation with medicinal products VERSION: 21
Memorandum on Collaboration and Exchange of Information between the Spanish Agency of Medicinal Products and Medical Devices and Ethics Committees for investigation with medicinal products VERSION: 21
A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN
 A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN Version 1 - June 2017 A Short Guide For CT Application 1 2 A Short Guide For CT Application DEFINITIONS Clinical
A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN Version 1 - June 2017 A Short Guide For CT Application 1 2 A Short Guide For CT Application DEFINITIONS Clinical
Research Ethics and Good Clinical Practice. Dr. Rosie Mayston, Centre for Global Mental Health, Institute of Psychiatry, King s College London
 Research Ethics and Good Clinical Practice Dr. Rosie Mayston, Centre for Global Mental Health, Institute of Psychiatry, King s College London History of Research Ethics Nuremberg Code End of WWII Nazi
Research Ethics and Good Clinical Practice Dr. Rosie Mayston, Centre for Global Mental Health, Institute of Psychiatry, King s College London History of Research Ethics Nuremberg Code End of WWII Nazi
STANDARD OPERATING PROCEDURE SOP 310
 STANDARD OPERATING PROCEDURE SOP 310 DEVELOPMENT OF PARTICIPANT INFORMATION SHEET AND INFORMED CONSENT FORM Version 1.3 Version date 28.03.2017 Effective date 28.03.2017 Number of pages 8 Review date April
STANDARD OPERATING PROCEDURE SOP 310 DEVELOPMENT OF PARTICIPANT INFORMATION SHEET AND INFORMED CONSENT FORM Version 1.3 Version date 28.03.2017 Effective date 28.03.2017 Number of pages 8 Review date April
Recording, Managing and Reporting Adverse Events in the UK
 This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
MEDICINES CONTROL COUNCIL
 Clinical Trial Application Form MEDICINES CONTROL COUNCIL APPLICATION TO CONDUCT A CLINICAL TRIAL Study title Protocol No. Version No. Study Drug MCC Ref. no. (if applicable) MCC Ref number(s) of comparator
Clinical Trial Application Form MEDICINES CONTROL COUNCIL APPLICATION TO CONDUCT A CLINICAL TRIAL Study title Protocol No. Version No. Study Drug MCC Ref. no. (if applicable) MCC Ref number(s) of comparator
3 RESPONSIBILITIES AND AUTHORITIES
 Amendment submission to Ethics Committee and competent authorities (FAMHP) N : CEHF-SOP-015 / REV008 N ENGLISH VERSION : 061 "Please do take into account that this is a translation of the original French
Amendment submission to Ethics Committee and competent authorities (FAMHP) N : CEHF-SOP-015 / REV008 N ENGLISH VERSION : 061 "Please do take into account that this is a translation of the original French
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES
 IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
GUIDELINES ON MEDICAL DEVICES
 EUROPEAN COMMISSION DG Internal Market, Industry, Entrepreneurship and SMEs Consumer, Environmental and Health Technologies Health technology and Cosmetics MEDDEV 2.7/3 revision 3 May 2015 GUIDELINES ON
EUROPEAN COMMISSION DG Internal Market, Industry, Entrepreneurship and SMEs Consumer, Environmental and Health Technologies Health technology and Cosmetics MEDDEV 2.7/3 revision 3 May 2015 GUIDELINES ON
RESEARCH ETHICS BOARD GUIDELINES SUBMISSION OF RESEARCH PROJECTS
 1. Role of the Research Ethics Board The mandate of the Research Ethics Board is to safeguard the rights, safety and well being of all research participants. The REB reviews and approves research projects
1. Role of the Research Ethics Board The mandate of the Research Ethics Board is to safeguard the rights, safety and well being of all research participants. The REB reviews and approves research projects
Meeting the Clinical Evaluation Requirements for CE Marking: Challenges for Innovative Start-ups Sarah Sorrel, MedPass International SAS
 Meeting the Clinical Evaluation Requirements for CE Marking: Challenges for Innovative Start-ups Sarah Sorrel, MedPass International SAS TÜV SÜD America Clinical Workshop Carlsbad, CA - January 27, 2015
Meeting the Clinical Evaluation Requirements for CE Marking: Challenges for Innovative Start-ups Sarah Sorrel, MedPass International SAS TÜV SÜD America Clinical Workshop Carlsbad, CA - January 27, 2015
GUIDELINES ON MEDICAL DEVICES
 Ref. Ares(2016)1982736-26/04/2016 EUROPEAN COMMISSION DG Internal Market, Industry, Entrepreneurship and SMEs Consumer, Environmental and Health Technologies Health technology and Cosmetics MEDDEV 2.7/3
Ref. Ares(2016)1982736-26/04/2016 EUROPEAN COMMISSION DG Internal Market, Industry, Entrepreneurship and SMEs Consumer, Environmental and Health Technologies Health technology and Cosmetics MEDDEV 2.7/3
PROPOSED DOCUMENT. Global Harmonization Task Force. Title: Reportable Events During Pre-Market Clinical Investigations
 SG2&5(PD1)N5R10 PROPOSED DOCUMENT Global Harmonization Task Force Title: Reportable Events During Pre-Market Clinical Investigations Authoring Group: Study Groups 2 and 5 Date: 7 June 2011 CONTENTS Reportable
SG2&5(PD1)N5R10 PROPOSED DOCUMENT Global Harmonization Task Force Title: Reportable Events During Pre-Market Clinical Investigations Authoring Group: Study Groups 2 and 5 Date: 7 June 2011 CONTENTS Reportable
1.4 Applicable Regulatory Requirement(s) Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.
 Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.") 1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
STUDY DOCUMENTS. DOCUMENT NO.: CR007 v4.0. Marise Buçukoğlu ISSUE DATE: 07 MAR 2017 EFFECTIVE DATE: 21 MAR INTRODUCTION
 STUDY DOCUMENTS DOCUMENT NO.: CR007 v4.0 AUTHOR: Marise Buçukoğlu ISSUE DATE: 07 MAR 2017 1 INTRODUCTION 1.1 The Academic & Clinical Central Office for Research & Development (ACCORD) is a joint office
STUDY DOCUMENTS DOCUMENT NO.: CR007 v4.0 AUTHOR: Marise Buçukoğlu ISSUE DATE: 07 MAR 2017 1 INTRODUCTION 1.1 The Academic & Clinical Central Office for Research & Development (ACCORD) is a joint office
Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers
 ZP-21 Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers This guideline supersedes guideline SÚKL PZT-16 as of November 1, 2004. The purpose of
ZP-21 Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers This guideline supersedes guideline SÚKL PZT-16 as of November 1, 2004. The purpose of
Impact of the transposition of the European Clinical Trials Directive. CEMO, Paris 17 November 2004
 Impact of the transposition of the European Clinical Trials Directive CEMO, Paris 17 November 2004 Dr Martine Dehlinger-Kremer VP Regulatory Affairs International Agenda Overview of key areas of Directive
Impact of the transposition of the European Clinical Trials Directive CEMO, Paris 17 November 2004 Dr Martine Dehlinger-Kremer VP Regulatory Affairs International Agenda Overview of key areas of Directive
The EFGCP Report on The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Ireland
 Ireland") The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Ireland Question 1: What laws or regulations apply to an application for conducting a clinical
The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Ireland Question 1: What laws or regulations apply to an application for conducting a clinical
Guidance for applicants for ethics committee opinion on the conduct of a clinical trial of pharmaceuticals
 KLH-EC-01 APPLICATION FOR ETHICS COMMITTEE OPINION ON THE CONDUCT OF A CLINICAL TRIAL IN THE CZECH REPUBLIC requirements governing the documentation to be submitted This guideline is being published in
KLH-EC-01 APPLICATION FOR ETHICS COMMITTEE OPINION ON THE CONDUCT OF A CLINICAL TRIAL IN THE CZECH REPUBLIC requirements governing the documentation to be submitted This guideline is being published in
Clinical Trials application process, legislation & guidelines
 Clinical Trials application process, legislation & guidelines IMB Clinical Trials Seminar 19 th June 2012 Elaine Breslin MB BCh (NUI), PhD, FRCPI Clinical Assessment Manager 19/06/2012 Slide 1 IMB Mission
Clinical Trials application process, legislation & guidelines IMB Clinical Trials Seminar 19 th June 2012 Elaine Breslin MB BCh (NUI), PhD, FRCPI Clinical Assessment Manager 19/06/2012 Slide 1 IMB Mission
Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland
 Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland AUT-G0095-1 15 AUGUST 2014 This guide does not purport to be an interpretation of law and/or regulations and is for
Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland AUT-G0095-1 15 AUGUST 2014 This guide does not purport to be an interpretation of law and/or regulations and is for
INSPECTION OF INDEPENDENT ETHICS COMMITTEES (IEC) The Italian Experience
 The Italian Experience") INSPECTION OF INDEPENDENT ETHICS COMMITTEES (IEC) The Italian Experience Umberto Filibeck Former Head of AIFA GCP Inspectorate and GCP Promotion Unit UNICRI Consultant for Projects on GCP of CTs in developing
INSPECTION OF INDEPENDENT ETHICS COMMITTEES (IEC) The Italian Experience Umberto Filibeck Former Head of AIFA GCP Inspectorate and GCP Promotion Unit UNICRI Consultant for Projects on GCP of CTs in developing
N : AAHRPP-SOP-057 / REV 003 N ENGLISH VERSION :188
 SOP FOR THE SUBMISSION OF THE EXPERIMENT ANNUAL REPORT TO THE ETHICS COMMITTEE N : AAHRPP-SOP-057 / REV 003 N ENGLISH VERSION :188 "Please do take into account that this is a translation of the original
SOP FOR THE SUBMISSION OF THE EXPERIMENT ANNUAL REPORT TO THE ETHICS COMMITTEE N : AAHRPP-SOP-057 / REV 003 N ENGLISH VERSION :188 "Please do take into account that this is a translation of the original
GUIDELINES FOR CONDUCTING CLINICAL TRIALS.
 GUIDELINES FOR CONDUCTING CLINICAL TRIALS. FOREWORD The purpose of Drug Registration is to ensure that a pharmaceutical product has been adequately tested and evaluated for safety, efficacy and quality,
GUIDELINES FOR CONDUCTING CLINICAL TRIALS. FOREWORD The purpose of Drug Registration is to ensure that a pharmaceutical product has been adequately tested and evaluated for safety, efficacy and quality,
Life cycle of a clinical trial. IMB Clinical Trial Seminar June 19 th Dr. Agnieszka Przybyszewska, MD, MSc, PhD, PGDipPharmMed.
 Life cycle of a clinical trial IMB Clinical Trial Seminar June 19 th Dr. Agnieszka Przybyszewska, MD, MSc, PhD, PGDipPharmMed. Life cycle of a clinical trial In each MS concerned EUDRA-CT S P O N S O R
Life cycle of a clinical trial IMB Clinical Trial Seminar June 19 th Dr. Agnieszka Przybyszewska, MD, MSc, PhD, PGDipPharmMed. Life cycle of a clinical trial In each MS concerned EUDRA-CT S P O N S O R
SERIOUS ADVERSE EVENTS
 EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
Good Clinical Practice, GCP. Clinical Trials 101 GCIG CCRN QA
 Good Clinical Practice, GCP Clinical Trials 101 GCIG CCRN QA Monica Bacon/Adriana Chavez-Blanco GCIG-CCRN Background: 1949 The Nuremburg Code 1964 Declaration of Helsinki (World Medical Association) 1990
Good Clinical Practice, GCP Clinical Trials 101 GCIG CCRN QA Monica Bacon/Adriana Chavez-Blanco GCIG-CCRN Background: 1949 The Nuremburg Code 1964 Declaration of Helsinki (World Medical Association) 1990
The Victorian Streamlined Review Process and the National Mutual Acceptance (NMA) Streamlined Review Process for Clinical Trials
 Streamlined Review Process for Clinical Trials") The Victorian Streamlined Review Process and the National Mutual Acceptance (NMA) Streamlined Review Process for Clinical Trials Angela Henjak, Alfred Health Streamlined Processes: SERP Victorian streamlined
The Victorian Streamlined Review Process and the National Mutual Acceptance (NMA) Streamlined Review Process for Clinical Trials Angela Henjak, Alfred Health Streamlined Processes: SERP Victorian streamlined
STANDARD OPERATING PROCEDURE. STH Researcher. Investigator Site File
 Research Department STANDARD OPERATING PROCEDURE STH Researcher SOP History CSUH 00/016 SOP Number A116 Created STH Research Department (TL) Reviewed by STH Research Department (AL) 06 August 2009 Superseded
Research Department STANDARD OPERATING PROCEDURE STH Researcher SOP History CSUH 00/016 SOP Number A116 Created STH Research Department (TL) Reviewed by STH Research Department (AL) 06 August 2009 Superseded
Highlights of the proposed Clinical Trials Regulation in Europe
 Highlights of the proposed Clinical Trials Regulation in Europe Dr Daryl Rees 22 January 2013 Proposed Clinical Trials Regulation On 17July 2012, the Commission adopted the proposal for a "Clinical Trials
Highlights of the proposed Clinical Trials Regulation in Europe Dr Daryl Rees 22 January 2013 Proposed Clinical Trials Regulation On 17July 2012, the Commission adopted the proposal for a "Clinical Trials
Developing a European First-in-Human Study: Three Key Decisions
 Developing a European First-in-Human Study: Three Key Decisions By Nicole Feist, BA Clinical A key step in the translational medicine benchtop to bedside process model is the move from research and preclinical
Developing a European First-in-Human Study: Three Key Decisions By Nicole Feist, BA Clinical A key step in the translational medicine benchtop to bedside process model is the move from research and preclinical
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd.
 Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
The installation and control in France of clinical trials relating to medical devices and in vitro diagnostic medical devices
 Notice for Sponsors The installation and control in France of clinical trials relating to medical devices and in vitro diagnostic medical devices This document is intended to help the participants in biomedical
Notice for Sponsors The installation and control in France of clinical trials relating to medical devices and in vitro diagnostic medical devices This document is intended to help the participants in biomedical
Trial Master File. SOP No. SOP 7
 Trial Master File SOP Title Trial Master File SOP No. SOP 7 Author Consultation Departments Date approved Julia Farmery Lincolnshire Clinical Research Facility, Research and Development, Trust consultants
Trial Master File SOP Title Trial Master File SOP No. SOP 7 Author Consultation Departments Date approved Julia Farmery Lincolnshire Clinical Research Facility, Research and Development, Trust consultants
The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective
 The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective Almath Spooner, Irish Medicines Board Pharmaceutical Society of Ireland National Pharmacy Summit, November 2008. Presentation Topics
The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective Almath Spooner, Irish Medicines Board Pharmaceutical Society of Ireland National Pharmacy Summit, November 2008. Presentation Topics
N : CEHF-SOP-019 / REV003 N ENGLISH VERSION : 131
 SOP for the end of trial notification to investigator, institution, CEHF and to the competent authorities N : CEHF-SOP-019 / REV003 N ENGLISH VERSION : 131 "Please do take into account that this is a translation
SOP for the end of trial notification to investigator, institution, CEHF and to the competent authorities N : CEHF-SOP-019 / REV003 N ENGLISH VERSION : 131 "Please do take into account that this is a translation
Course Title ID Duration Basic Premium. Biological Evaluation of Medical Devices: A Risk-Based Approach N mins
 Pre-Clinical Basic Level : CMDA Certified Medical Device Associate Biological Evaluation of Medical Devices: A Risk-Based Approach N134 63 mins 186.00 144.00 Introduction to Process Validation N135 75
Pre-Clinical Basic Level : CMDA Certified Medical Device Associate Biological Evaluation of Medical Devices: A Risk-Based Approach N134 63 mins 186.00 144.00 Introduction to Process Validation N135 75
ICH GCP Revision and EU Clinical Trial Regulation
 ICH GCP Revision and EU Clinical Trial Regulation Sinead Curran, GCP/PhV Inspection Manager Irish Research Nurses Network Annual National Conference Friday, 17 November 2017 Overview Guideline for Good
ICH GCP Revision and EU Clinical Trial Regulation Sinead Curran, GCP/PhV Inspection Manager Irish Research Nurses Network Annual National Conference Friday, 17 November 2017 Overview Guideline for Good
Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products
 1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
DISTRIBUTION AND SUPPLY OF PLASMA DERIVED FRACTIONATED BLOOD PRODUCTS AND RECOMBINANT PRODUCTS IN NEW ZEALAND
 REASON FOR CHANGE: Wholesaler s licences are now issued by the Ministry of Health rather than Medsafe; clarify the approval process for the supply of products only to authorised facilities and healthcare
REASON FOR CHANGE: Wholesaler s licences are now issued by the Ministry of Health rather than Medsafe; clarify the approval process for the supply of products only to authorised facilities and healthcare
GUIDELINE REGARDING COLLECTION, VERIFICATION, AND SUBMISSION OF THE REPORTS OF ADVERSE EVENTS / REACTIONS OCCURRING IN CLINICAL DRUG TRIALS
 1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
Standard Operating Procedure
 Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Writing an Assessment Report
 Safeguarding public health Writing an Assessment Report Name: Malcolm Dash Date: Programme Why we need Assessment Reports Writing style Deficiency points Potential Serious Risk to Public Health Targeted
Safeguarding public health Writing an Assessment Report Name: Malcolm Dash Date: Programme Why we need Assessment Reports Writing style Deficiency points Potential Serious Risk to Public Health Targeted
ORPHAN DESIGNATION BY THE EMA. Paillard Juliette M2 AREIPS 15/11/2016
 ORPHAN DESIGNATION BY THE EMA Paillard Juliette M2 AREIPS 15/11/2016 Legal basis 1. Legal background 2. Criteria SUMMARY Procedure 1. Prior to the submission 2. Submission and validation 3. Evaluation
ORPHAN DESIGNATION BY THE EMA Paillard Juliette M2 AREIPS 15/11/2016 Legal basis 1. Legal background 2. Criteria SUMMARY Procedure 1. Prior to the submission 2. Submission and validation 3. Evaluation
Standard Operating Procedure Research Governance
 Research and Enterprise Standard Operating Procedure Research Governance Title: Development, Review and Amendment: Study Protocol SOP Reference Number: QUB-ADRE-002 Date prepared 28 May 2008 Version Number:
Research and Enterprise Standard Operating Procedure Research Governance Title: Development, Review and Amendment: Study Protocol SOP Reference Number: QUB-ADRE-002 Date prepared 28 May 2008 Version Number:
Responsibilities of Investigators for Clinical Trials from Consent to Safety Reporting
 Responsibilities of Investigators for Clinical Trials from Consent to Safety Reporting Young-Hak Kim, MD, PhD Department of Medicine,, University of Ulsan College of Medicine, Seoul, Korea Responsibilities
Responsibilities of Investigators for Clinical Trials from Consent to Safety Reporting Young-Hak Kim, MD, PhD Department of Medicine,, University of Ulsan College of Medicine, Seoul, Korea Responsibilities
Standard Operating Procedure. Safety Reporting for NuTH Sponsored, Medical Device Trials. SOP effective: 03 January 2018 Review date: 03 January 2020
 Standard Operating Procedure SOP number: SOP full title: SOP-JRO-40-001 Safety Reporting for NuTH Sponsored, Medical Device Trials SOP effective: Review date: 03 January 2020 SOP author signature: SIGNED
Standard Operating Procedure SOP number: SOP full title: SOP-JRO-40-001 Safety Reporting for NuTH Sponsored, Medical Device Trials SOP effective: Review date: 03 January 2020 SOP author signature: SIGNED