Center for Mass Spectrometry and Proteomics Phone (612) (612)
|
|
|
- Joel Harrison
- 6 years ago
- Views:
Transcription
1 Outline Database search types Peptide Mass Fingerprint (PMF) Precursor mass-based Sequence tag Results comparison across programs Manual inspection of results Terminology Mass tolerance MS/MS search FASTA Tandem MS Precursor mass Product ion Theoretical mass Experimental mass Sequence tag de novo sequencing
2 Three Strategies for Protein Inference from Peptide MS or MS/MS Data DATA INPUT: Peptide Mass List (MS1) Peptide MS/MS (MS2) Search TYPE: Peptide Mass Fingerprint (PMF) (historical) Precursor Mass-based 2 Sequencebased (de novo or mass tag) 1 3
3 1 Peptide Mass Fingerprint (PMF) Historical: after 1D or 2D in-gel Digestion of single bands or spots Current applications: limited; decreasing utility Component Input (Data) Target Search Parameters Description Peptide Mass List Mass type (Mr or MH+) Average or Monoisotopic Protein FASTA Sequence Database Proteolytic enzyme # Missed enzyme cleave sites Amino acid modifications Mass tolerance
 Acquire MALDI-TOF Mass Spectrum of peptide mixture A) In gel Trypsin digestion GLSDGEWQQVLNVWGK VEADIAGHGQEVLIR LFTGHPETLEK TEAEMK ASEDLK")
4 Absolute Intensity 1 Peptide Mass Fingerprint: INPUT UNKNOWN PROTEIN (amino acid sequence): GLSDGEWQQVLNVWGKVEADIAGHGQEVLIRLFTG HPETLEKFDKFKHLKTEAEMKASEDLKKHGTVVLT ALGGILKKKGHHEAELKPLAQSHATKHKIPIKYLE FISDAIIHVLHSHPGNFGADAQGAMTKALELFRND IAAKYKELGFQG B) Acquire MALDI-TOF Mass Spectrum of peptide mixture A) In gel Trypsin digestion GLSDGEWQQVLNVWGK VEADIAGHGQEVLIR LFTGHPETLEK TEAEMK ASEDLK HGTVVLTALGGILK GHHEAELKPLAQSHATK YLEFISDAIIHVLHSHPGNFGADAQGAMTK ALELFR NDIAAK ELGFQG m/z C) Experimental Peptide Mass (or m/z) List m/z Peptide NDIAAK ELGFQG ASEDLK TEAEMK ALELFR LFTGHPETLEK HGTVVLTALGGILK VEADIAGHGQEVLIR GLSDGEWQQVLNVWGK GHHEAELKPLAQSHATK YLEFISDAIIHVLHSHPGNFGADAQGAMTK

5 1 PMF TARGET: Protein FASTA Sequence Database >sp P B_HUMAN protein beta/alpha OS=Homo sapiens GN=YWHAB PE=1 SV=3 MTMDKSELVQKAKLAEQAERYDDMAAAMKAVTEQGHELSNEERNLLSVAYKNVVGARRSS WRVISSIEQKTERNEKKQQMGKEYREKIEAELQDICNDVLELLDKYLIPNATQPESKVFY LKMKGDYFRYLSEVASGDNKQTTVSNSQQAYQEAFEISKKEMQPTHPIRLGLALNFSVFY YEILNSPEKACSLAKTAFDEAIAELDTLNEESYKDSTLIMQLLRDNLTLWTSENQGDEGD AGEGEN >sp P B_HUMAN Isoform Short of protein beta/alpha OS=Homo sapiens GN=YWHAB MDKSELVQKAKLAEQAERYDDMAAAMKAVTEQGHELSNEERNLLSVAYKNVVGARRSSWR VISSIEQKTERNEKKQQMGKEYREKIEAELQDICNDVLELLDKYLIPNATQPESKVFYLK MKGDYFRYLSEVASGDNKQTTVSNSQQAYQEAFEISKKEMQPTHPIRLGLALNFSVFYYE ILNSPEKACSLAKTAFDEAIAELDTLNEESYKDSTLIMQLLRDNLTLWTSENQGDEGDAG EGEN >sp P E_HUMAN protein epsilon OS=Homo sapiens GN=YWHAE PE=1 SV=1 MDDREDLVYQAKLAEQAERYDEMVESMKKVAGMDVELTVEERNLLSVAYKNVIGARRASW RIISSIEQKEENKGGEDKLKMIREYRQMVETELKLICCDILDVLDKHLIPAANTGESKVF YYKMKGDYHRYLAEFATGNDRKEAAENSLVAYKAASDIAMTELPPTHPIRLGLALNFSVF YYEILNSPDRACRLAKAAFDDAIAELDTLSEESYKDSTLIMQLLRDNLTLWTSDMQGDGE EQNKEALQDVEDENQ >sp Q F_HUMAN protein eta OS=Homo sapiens GN=YWHAH PE=1 SV=4 MGDREQLLQRARLAEQAERYDDMASAMKAVTELNEPLSNEDRNLLSVAYKNVVGARRSSW RVISSIEQKTMADGNEKKLEKVKAYREKIEKELETVCNDVLSLLDKFLIKNCNDFQYESK VFYLKMKGDYYRYLAEVASGEKKNSVVEASEAAYKEAFEISKEQMQPTHPIRLGLALNFS VFYYEIQNAPEQACLLAKQAFDDAIAELDTLNEDSYKDSTLIMQLLRDNLTLWTSDQQDE EAGEGN >sp P G_HUMAN protein gamma OS=Homo sapiens GN=YWHAG PE=1 SV=2 MVDREQLVQKARLAEQAERYDDMAAAMKNVTELNEPLSNEERNLLSVAYKNVVGARRSSW RVISSIEQKTSADGNEKKIEMVRAYREKIEKELEAVCQDVLSLLDNYLIKNCSETQYESK VFYLKMKGDYYRYLAEVATGEKRATVVESSEKAYSEAHEISKEHMQPTHPIRLGLALNYS VFYYEIQNAPEQACHLAKTAFDDAIAELDTLNEDSYKDSTLIMQLLRDNLTLWTSDQQDD DGGEGNN >sp P S_HUMAN protein sigma OS=Homo sapiens GN=SFN PE=1 SV=1 MERASLIQKAKLAEQAERYEDMAAFMKGAVEKGEELSCEERNLLSVAYKNVVGGQRAAWR VLSSIEQKSNEEGSEEKGPEVREYREKVETELQGVCDTVLGLLDSHLIKEAGDAESRVFY LKMKGDYYRYLAEVATGDDKKRIIDSARSAYQEAMDISKKEMPPTNPIRLGLALNFSVFH YEIANSPEEAISLAKTTFDEAMADLHTLSEDSYKDSTLIMQLLRDNLTLWTADNAGEEGG EAPQEPQS etc UniProt (example) The Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data. UniProt is a collaboration between the European Bioinformatics Institute (EBI), the Swiss Institute of Bioinformatics (SIB) and the Protein Information Resource (PIR). Across the three institutes close to 150 people are involved through different tasks such as database curation, software development and support. Example: 20,292 entries for taxonomy: "Homo sapiens (Human) [9606] AND reviewed:yes
 for a Single Protein: Trypsin = Enzyme Missed Cleave Sites = 0 http://www.expasy.")
6 1 PMF Search Parameters Review: Trypsin Proteolytic Enzyme and 0 Missed Cleave Site (#MC) modifications Example: Theoretical Trypsin Digest Peptide Mass List (truncated) for a Single Protein: Trypsin = Enzyme Missed Cleave Sites = 0
 artif modifications modifications http://www.expasy.org/tools/peptide-mass.")
7 1 PMF Search Parameters: Trypsin Proteolytic Enzyme and 1 Missed Cleave Site MSO = Methionine Oxidation (amino acid modification) artif modifications modifications
8 1 PMF Parameter: Peptide Mass Deviation (+/- m/z) High resolution measurement / Range: Mouse proteins with Trypsin peptide in this range Low resolution measurement 631 +/- 1 Range: Mouse proteins with Trypsin peptide in this range m/z
9 1 Mass Measurement Error Calculation Error Expression Fractional Error Expression Multiply by: ppm / Theoretical value 1,000,000 % / Theoretical value 100 where Δ = Experimental (or observed) Theoretical m/z value Example: Experimental/observed value (i.e., the data acquired by the mass spectrometer) = m/z Theoretical value (calculated from periodic table, after the peak is identified) = m/z Delta (Δ) = Error Expression Error Equation Error ppm (0.079/ )*1,000, ppm % (0.079/ )* % A USEFUL REFERENCE: Brenton AG, Godfrey AR. Accurate mass measurement: terminology and treatment of data. J Am Soc Mass Spectrom Nov;21(11):

10 1 MASCOT Peptide Mass Fingerprint Search

11 1 MASCOT PMF Search Parameter Page
12 1 PMF Search QUERY (Experimental Data) m/z Peptide NDIAAK ELGFQG ASEDLK TEAEMK ALELFR LFTGHPETLEK HGTVVLTALGGILK VEADIAGHGQEVLIR GLSDGEWQQVLNVWGK GHHEAELKPLAQSHATK YLEFISDAIIHVLHSHPGNFGADAQGAMTK Search Score Compare QUERY to THEORETICAL PEPTIDE MASS LIST for each protein in the database Parameters: Enzyme Missed cleave site Amino acid mods Mass tolerance Probability-based MOWSE Score (often, the protein with highest number of peptide matches has the highest score)
13 1
14 1
15 1 PMF Search: Results Interpretation Is the protein ID experimentally rational? Does the MW of protein in search results match MW determined by SDS-PAGE? Does the pi of protein in search results match pi determined by 2D-PAGE? Perform MS/MS if no protein match
16 Three Strategies for Protein Inference from Peptide MS or MS/MS Data DATA INPUT: Peptide Mass List (MS1) Peptide MS/MS (MS2) Search TYPE: Peptide Mass Fingerprint (PMF) (historical) Precursor Mass-based 2 Sequencebased (de novo or mass tag) 1 3
17 2 Tandem MS: Precursor Mass-based Search Component Input (Data) Target Search Parameters Description Precursor Mass & Product Ion Masses Charge state Average or Monoisotopic Protein FASTA Sequence Database Proteolytic enzyme # Missed enzyme cleave sites Amino acid modifications Mass tolerances: Peptide/Precursor Mass Product Ions Masses
18 2 Tandem MS: Precursor Mass-based Search 1. Each MS/MS spectra has 2 data-rich Components 2. Database search for each MS/MS spectra has 2 STEPS
19 2 Tandem MS: Precursor Mass-based Search REVIEW: Each MS/MS Spectra has Two Data-Rich Components In te n s ity, c o u n ts Example: m/z = Precursor m/z +TOF MS: Experiment 1, min from bruce w 2-5 humjak3 tryp.wiff a= e-004, t0= e COMPONENT 1: Intact Peptide Max counts m/z, amu In te n s ity, c o u n ts +TOF Product (612.8): Experiment 2, min from bruce w 2-5 humjak3 tryp.wiff a= e-004, t0= e a COMPONENT 2: Product Ion Mass (m/z) values and Intensities after Peptide Fragmentation a2b2 y b y y a3-nh3(2+) y y4 y7(2+) a4 y8(2+) b4 a5 y y y Max counts m/z, amu y9
20 Tandem MS: Precursor Mass-based Search STEP 1: Find Theoretical Peptides (from in silico Protein Digest) within user specified Precursor Mass Tolerance from MS1 spectrum Mass [M + H] +1 Protein GenBank ID# Peptide gi SRNLGPSTSR gi RASVDIGISR gi MoxRTFMoxISR gi QTYENYTR gi REEMHESR gi IDLVSMoxHSR gi IFLPGHYAR gi FMLPGDTHR gi NMoxRQHDTR gi EVPETKDTR PEPTIDE CANDIDATES 2 evaluate 1 st candidate (next slide) etc etc etc
21 2 Tandem MS: Precursor Mass-based Search STEP 2: Generate Theoretical Product Ion Table for Peptide Candidate SRNLGPSTSR Theoretical Product Ion Table evaluate 1 st candidate Residue a b b-h2o b-nh3 y y-h2o y-nh3 S, Ser R, Arg N, Asn L, Leu G, Gly P, Pro S, Ser T, Thr S, Ser R, Arg
22 2 Tandem MS: Precursor Mass-based Search Compare theoretical product ions to experimental product Ions from MS2 spectrum
23 Intensity 2 REVIEW: b-type fragment ions NH3+ A E P T I R COOH NH2 NH2 A E P T A E P NH2 A E NH2 A m/z
24 Intensity 2 REVIEW: y-type fragment ions NH3+ A E P T I R COOH HOOC HOOC R I T P R I T HOOC R I HOOC R m/z
25 Intensity 2 REVIEW: b- and y-type Fragment ions: PEPTIDE TANDEM MASS SPECTRUM NH3+ A E P T I R COOH b y b y b y b y b y m/z
26 2 Tandem MS: Precursor Mass-based Search SUMMARY- for Each MS/MS Spectrum: Generate theoretical product ion tables for all peptide candidates Compare theoretical product ions to experimental product Ions from MS2 spectrum Score all candidate peptides Rank peptides by Score
27 2 Tandem MS: Precursor Mass-based Search STEP 1 Find Theoretical Peptide Matches from in silico Protein Digest in the range: Experimental Precursor Mass +/- Mass Tolerance STEP 2 For each candidate peptide from Step 1: Compare Theoretical Product Ions (b & y, etc) to Experimental Product ions (data) STEP 3 SCORE: many software programs/ algorithms Peptide rank PROTEIN Report and Grouping: many variations
28 2 Tandem MS: Precursor Massbased search Software at UM Sequest X!Tandem MASCOT
29 Three Strategies for Protein Inference from Peptide MS or MS/MS Data DATA INPUT: Peptide Mass List (MS1) Peptide MS/MS (MS2) Search TYPE: Peptide Mass Fingerprint (PMF) (historical) Precursor Mass-based 2 Sequencebased (de novo or mass tag) 1 3
30 3 Sequence-based Searches: WHEN? Unsequenced genome (protein(s) of interest are not in the database) HOMOLOGY-based search Search for amino acid MUTATIONS Search for LARGE NUMBERS of Post Translational Modifications (PTM s)
31 3 Tandem MS: Sequence-based Search STEP 1: Obtain [full or partial] amino acid sequence string directly from the spectrum by de novo Sequencing de novo (Latin) "from the beginning"
32 3 Tandem MS: Sequence-based Search STEP 1: de novo sequencing: amino acid sequence is determined from delta mass values for a series of successive peptide b- or y-type product ions from a Product Ion (MS/MS) spectrum EXAMPLE de novo sequencing: L 113 V 99 A 71 D 115 L 113 E 129 delta masses between peaks correspond to amino acid residue masses See amino acid residue masses:
 rank candidate amino acid sequence segments. STV is related to the number of taglet hits per region. Slide content: courtesy of Sean L.")
33 3 Tandem MS: Sequence-based Search STEP 2: QUERY THE PROTEINS Example: AB Sciex Paragon Algorithm For each MS/MS spectrum, multiple, short sequences (taglets) are generated, scored, and matched against protein sequences. Sequence Temperature Values (STV) rank candidate amino acid sequence segments. STV is related to the number of taglet hits per region. Slide content: courtesy of Sean L. Seymour, AB Sciex
34 3 Tandem MS: Sequence-based Search STEP 3: Candidate peptide sequences within protein sequences are identified with Precursor mass AND sequence tag information Include: Enzyme specificity Mass tolerance Amino acid modifications Amino Acid mutations STEP 4: Candidates are Ranked and SCORED
35 3 Tandem MS: Sequence-based Search STEP 3b: Error-tolerant Sequence Tag Reconciliation Includes Amino Acid Mutation Search Component Combination: sequence tag & mass match Slide content: adapted from David L. Tabb, Vanderbilt University: Bioinformatics 2009, Baltimore MD
36 3 MS/MS Sequence-based Software at UM ProteinPilot (AB Sciex) PEAKS (BSI) Direct Tag (D Tabb Lab) MASCOT (Matrix Science LTD) Error Tolerant MS/MS Sequence Tag
37 3 MS/MS Sequence-based Software PepNovo (open source de novo) Frank et al, Anal Chem (2005) 77: PEAKS Studio (Commercial de novo) Ma et al, RCMS (2003) 17: 2337:2342 Lutefisk (early de novo tool) Taylor et al, RCMS (1997) 11: Direct Tag (open source tag inference) Tabb et al, J Proteome Res (2008) 7: InsPecT (open source tag identifier) Tanner et al, Anal Chem (2005) 77:

38 Maximize Identifications with Multiple Software Programs Single Dataset analyzed with multiple software programs: Blue = number of peptide matches Red = number of protein matches
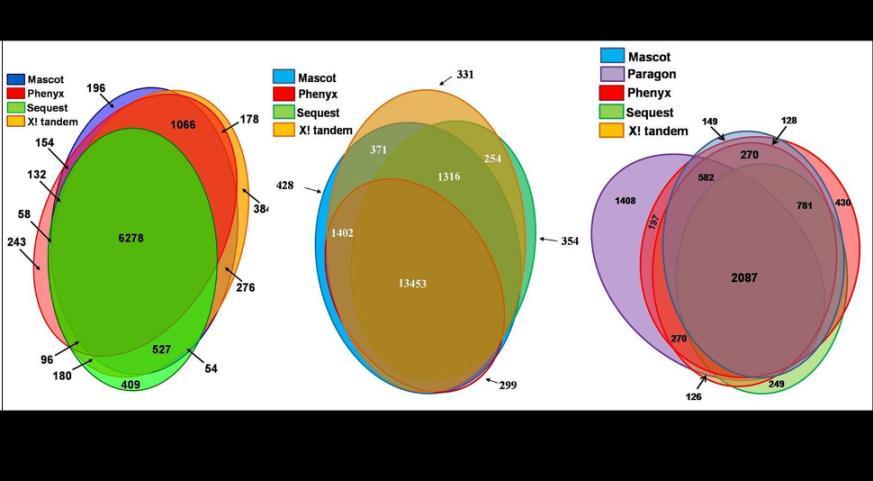
39 Maximize Identifications with Multiple Software Programs
40 Search Algorithm Conclusions 1) Several different search algorithm types exist 2) Each program provides useful results - no unified method exists 3) The same data searched using different algorithms may yield different results (scoring and ranking schemes are distinct)
41 Manual Inspection of Individual MS/MS Spectra Journal requirement (in special cases; see Inspect SINGLE PEPTIDE HITS Site Localization of Post Translational Modifications (PTM s) Detection of unexpected gene products (alternative splice isoforms)
42 STEPS for Manual Inspection of MS/MS Spectra 1) Open RAW data; print spectrum (or, multiple zoomed-in regions of the spectrum) 2) Obtain list of Theoretical Fragment Ions (e.g., open MS Product tool 3) Label printout(s) of spectra with fragment ion types 4) DECISION: a. GOOD MATCH: all/most of the peaks (in the raw data) labeled with fragment ion types b. POOR MATCH: unassigned peaks in the raw data
43 Manual Inspection of MS/MS Spectra Example: RAW data labeled with Fragment Ion Names/Types IT4FYEQFSK IT4 (Fragment Ion Nomenclature Reference: P. Roepstorff & J. Fohlman (2005) Proposal for a Common Nomenclature for Sequence Ions in Mass Spectra of Peptides, Biomedical Spectrometry, 11(11), p 601
PTM Identification and Localization from MS Proteomics Data
 05.10.17 PTM Identification and Localization from MS Proteomics Data Marc Vaudel Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway KG Jebsen Center for Diabetes
05.10.17 PTM Identification and Localization from MS Proteomics Data Marc Vaudel Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway KG Jebsen Center for Diabetes
N- The rank of the specified protein relative to all other proteins in the list of detected proteins.
 PROTEIN SUMMARY file N- The rank of the specified protein relative to all other proteins in the list of detected proteins. Unused (ProtScore) - A measure of the protein confidence for a detected protein,
PROTEIN SUMMARY file N- The rank of the specified protein relative to all other proteins in the list of detected proteins. Unused (ProtScore) - A measure of the protein confidence for a detected protein,
Spectral Counting Approaches and PEAKS
 Spectral Counting Approaches and PEAKS INBRE Proteomics Workshop, April 5, 2017 Boris Zybailov Department of Biochemistry and Molecular Biology University of Arkansas for Medical Sciences 1. Introduction
Spectral Counting Approaches and PEAKS INBRE Proteomics Workshop, April 5, 2017 Boris Zybailov Department of Biochemistry and Molecular Biology University of Arkansas for Medical Sciences 1. Introduction
ProteinPilot Software Overview
 ProteinPilot Software Overview High Quality, In-Depth Protein Identification and Protein Expression Analysis Sean L. Seymour and Christie L. Hunter SCIEX, USA As mass spectrometers for quantitative proteomics
ProteinPilot Software Overview High Quality, In-Depth Protein Identification and Protein Expression Analysis Sean L. Seymour and Christie L. Hunter SCIEX, USA As mass spectrometers for quantitative proteomics
Application Note TOF/MS
 Application Note TOF/MS New Level of Confidence for Protein Identification: Results Dependent Analysis and Peptide Mass Fingerprinting Using the 4700 Proteomics Discovery System Purpose The Applied Biosystems
Application Note TOF/MS New Level of Confidence for Protein Identification: Results Dependent Analysis and Peptide Mass Fingerprinting Using the 4700 Proteomics Discovery System Purpose The Applied Biosystems
Utilizing novel technology for the analysis of therapeutic antibodies and host cell protein contamination
 Utilizing novel technology for the analysis of therapeutic antibodies and host cell protein contamination Kelli Jonakin, Ph.D. Senior Field Application Scientist, Seattle, WA Eric Johansen, Ph.D. Senior
Utilizing novel technology for the analysis of therapeutic antibodies and host cell protein contamination Kelli Jonakin, Ph.D. Senior Field Application Scientist, Seattle, WA Eric Johansen, Ph.D. Senior
Improving Productivity with Applied Biosystems GPS Explorer
 Product Bulletin TOF MS Improving Productivity with Applied Biosystems GPS Explorer Software Purpose GPS Explorer Software is the application layer software for the Applied Biosystems 4700 Proteomics Discovery
Product Bulletin TOF MS Improving Productivity with Applied Biosystems GPS Explorer Software Purpose GPS Explorer Software is the application layer software for the Applied Biosystems 4700 Proteomics Discovery
Proteomics and some of its Mass Spectrometric Applications
 Proteomics and some of its Mass Spectrometric Applications What? Large scale screening of proteins, their expression, modifications and interactions by using high-throughput approaches 2 1 Why? The number
Proteomics and some of its Mass Spectrometric Applications What? Large scale screening of proteins, their expression, modifications and interactions by using high-throughput approaches 2 1 Why? The number
De novo sequencing in the identification of mass data. Wang Quanhui Liu Siqi Beijing Institute of Genomics, CAS
 De novo sequencing in the identification of mass data Wang Quanhui Liu Siqi Beijing Institute of Genomics, CAS The difficulties in mass data analysis Although the techniques of genomic sequencing are being
De novo sequencing in the identification of mass data Wang Quanhui Liu Siqi Beijing Institute of Genomics, CAS The difficulties in mass data analysis Although the techniques of genomic sequencing are being
ProteinPilot Report for ProteinPilot Software
 ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Powerful mass spectrometers like
ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Powerful mass spectrometers like
FACTORS THAT AFFECT PROTEIN IDENTIFICATION BY MASS SPECTROMETRY HAOFEI TIFFANY WANG. (Under the Direction of Ron Orlando) ABSTRACT
 ABSTRACT") FACTORS THAT AFFECT PROTEIN IDENTIFICATION BY MASS SPECTROMETRY by HAOFEI TIFFANY WANG (Under the Direction of Ron Orlando) ABSTRACT Mass spectrometry combined with database search utilities is a valuable
FACTORS THAT AFFECT PROTEIN IDENTIFICATION BY MASS SPECTROMETRY by HAOFEI TIFFANY WANG (Under the Direction of Ron Orlando) ABSTRACT Mass spectrometry combined with database search utilities is a valuable
Peptide and protein identification in mass spectrometry based proteomics. Yafeng Zhu, PhD student Karolinska Institutet, Scilifelab
 Peptide and protein identification in mass spectrometry based proteomics Yafeng Zhu, PhD student Karolinska Institutet, Scilifelab 2017-10-12 Content How is the peptide sequence identified? What is the
Peptide and protein identification in mass spectrometry based proteomics Yafeng Zhu, PhD student Karolinska Institutet, Scilifelab 2017-10-12 Content How is the peptide sequence identified? What is the
Supplemental Materials
 Supplemental Materials MSGFDB Parameters For all MSGFDB searches, the following parameters were used: 30 ppm precursor mass tolerance, enable target-decoy search, enzyme specific scoring (for Arg-C, Asp-N,
Supplemental Materials MSGFDB Parameters For all MSGFDB searches, the following parameters were used: 30 ppm precursor mass tolerance, enable target-decoy search, enzyme specific scoring (for Arg-C, Asp-N,
Automation of MALDI-TOF Analysis for Proteomics
 Application Note #MT-50 BIFLEX III / REFLEX III Automation of MALDI-TOF Analysis for Proteomics D. Suckau, C. Köster, P.Hufnagel, K.-O. Kräuter and U. Rapp, Bruker Daltonik GmbH, Bremen, Germany Introduction
Application Note #MT-50 BIFLEX III / REFLEX III Automation of MALDI-TOF Analysis for Proteomics D. Suckau, C. Köster, P.Hufnagel, K.-O. Kräuter and U. Rapp, Bruker Daltonik GmbH, Bremen, Germany Introduction
Spectrum Mill MS Proteomics Workbench. Comprehensive tools for MS proteomics
 Spectrum Mill MS Proteomics Workbench Comprehensive tools for MS proteomics Meeting the challenge of proteomics data analysis Mass spectrometry is a core technology for proteomics research, but large-scale
Spectrum Mill MS Proteomics Workbench Comprehensive tools for MS proteomics Meeting the challenge of proteomics data analysis Mass spectrometry is a core technology for proteomics research, but large-scale
About OMICS Group Conferences
 About OMICS Group OMICS Group International is an amalgamation of Open Access publications and worldwide international science conferences and events. Established in the year 2007 with the sole aim of
About OMICS Group OMICS Group International is an amalgamation of Open Access publications and worldwide international science conferences and events. Established in the year 2007 with the sole aim of
Proteomics software at MSI. Pratik Jagtap Minnesota Supercomputing institute
 Proteomics software at MSI. Pratik Jagtap Minnesota Supercomputing institute http://www.mass.msi.umn.edu/ Proteomics software at MSI. proteomics : emerging technology proteomics workflow search algorithms
Proteomics software at MSI. Pratik Jagtap Minnesota Supercomputing institute http://www.mass.msi.umn.edu/ Proteomics software at MSI. proteomics : emerging technology proteomics workflow search algorithms
Protein Valida-on (Sta-s-cal Inference) and Protein Quan-fica-on. Center for Mass Spectrometry and Proteomics Phone (612) (612)
 and Protein Quan-fica-on. Center for Mass Spectrometry and Proteomics Phone (612) (612)") Protein Valida-on (Sta-s-cal Inference) and Protein Quan-fica-on Terminology Pep-de Spectrum Match Target / Decoy False discovery rate Shared pep-de Parsimony One hit wonders SPECTRUM Rela-ve Abundance
Protein Valida-on (Sta-s-cal Inference) and Protein Quan-fica-on Terminology Pep-de Spectrum Match Target / Decoy False discovery rate Shared pep-de Parsimony One hit wonders SPECTRUM Rela-ve Abundance
ProteinPilot Software for Protein Identification and Expression Analysis
 ProteinPilot Software for Protein Identification and Expression Analysis Providing expert results for non-experts and experts alike ProteinPilot Software Overview New ProteinPilot Software transforms protein
ProteinPilot Software for Protein Identification and Expression Analysis Providing expert results for non-experts and experts alike ProteinPilot Software Overview New ProteinPilot Software transforms protein
1638 Molecular & Cellular Proteomics 6.9
 Supplemental Material can be found at: http://www.mcponline.org/cgi/content/full/t600050-mcp200/ DC1 Technology The Paragon Algorithm, a Next Generation Search Engine That Uses Sequence Temperature Values
Supplemental Material can be found at: http://www.mcponline.org/cgi/content/full/t600050-mcp200/ DC1 Technology The Paragon Algorithm, a Next Generation Search Engine That Uses Sequence Temperature Values
Highly Confident Peptide Mapping of Protein Digests Using Agilent LC/Q TOFs
 Technical Overview Highly Confident Peptide Mapping of Protein Digests Using Agilent LC/Q TOFs Authors Stephen Madden, Crystal Cody, and Jungkap Park Agilent Technologies, Inc. Santa Clara, California,
Technical Overview Highly Confident Peptide Mapping of Protein Digests Using Agilent LC/Q TOFs Authors Stephen Madden, Crystal Cody, and Jungkap Park Agilent Technologies, Inc. Santa Clara, California,
Practical Tips. : Practical Tips Matrix Science
 Practical Tips : Practical Tips 2006 Matrix Science 1 Peak detection Especially critical for Peptide Mass Fingerprints A tryptic digest of an average protein (30 kda) should produce of the order of 50
Practical Tips : Practical Tips 2006 Matrix Science 1 Peak detection Especially critical for Peptide Mass Fingerprints A tryptic digest of an average protein (30 kda) should produce of the order of 50
Bioinformatics for proteomics
 Purdue-UAB Botanicals Center for Age-Related Disease Bioinformatics for proteomics Stephen Barnes, PhD Purdue-UAB Botanical Center Workshop 2002 Mass Spectrometry Methods in Botanicals Research Objectives
Purdue-UAB Botanicals Center for Age-Related Disease Bioinformatics for proteomics Stephen Barnes, PhD Purdue-UAB Botanical Center Workshop 2002 Mass Spectrometry Methods in Botanicals Research Objectives
Advances in analytical biochemistry and systems biology: Proteomics
 Advances in analytical biochemistry and systems biology: Proteomics Brett Boghigian Department of Chemical & Biological Engineering Tufts University July 29, 2005 Proteomics The basics History Current
Advances in analytical biochemistry and systems biology: Proteomics Brett Boghigian Department of Chemical & Biological Engineering Tufts University July 29, 2005 Proteomics The basics History Current
基于质谱的蛋白质药物定性定量分析技术及应用
 基于质谱的蛋白质药物定性定量分析技术及应用 蛋白质组学质谱数据分析 单抗全蛋白测序及鉴定 Bioinformatics Solutions Inc. Waterloo, ON, Canada 上海 中国 2017 1 3 PEAKS Studio GUI Introduction Overview of PEAKS Studio GUI Project View Tasks, running info,
基于质谱的蛋白质药物定性定量分析技术及应用 蛋白质组学质谱数据分析 单抗全蛋白测序及鉴定 Bioinformatics Solutions Inc. Waterloo, ON, Canada 上海 中国 2017 1 3 PEAKS Studio GUI Introduction Overview of PEAKS Studio GUI Project View Tasks, running info,
Algorithm for Matching Additional Spectra
 Improved Methods for Comprehensive Sample Analysis Using Protein Prospector Peter R. Baker 1, Katalin F. Medzihradszky 1 and Alma L. Burlingame 1 1 Mass Spectrometry Facility, Dept. of Pharmaceutical Chemistry,
Improved Methods for Comprehensive Sample Analysis Using Protein Prospector Peter R. Baker 1, Katalin F. Medzihradszky 1 and Alma L. Burlingame 1 1 Mass Spectrometry Facility, Dept. of Pharmaceutical Chemistry,
MIAPE: Mass Spectrometry Informatics
 MIAPE: Mass Spectrometry Informatics Pierre-Alain Binz[1,2]*, Robert Barkovich[3], Ronald C. Beavis[4], David Creasy[5], David M. Horn[6], Randall K. Julian Jr.[7], Sean L. Seymour[8], Chris F. Taylor[9],
MIAPE: Mass Spectrometry Informatics Pierre-Alain Binz[1,2]*, Robert Barkovich[3], Ronald C. Beavis[4], David Creasy[5], David M. Horn[6], Randall K. Julian Jr.[7], Sean L. Seymour[8], Chris F. Taylor[9],
Quantitative mass spec based proteomics
 Quantitative mass spec based proteomics Tuula Nyman Institute of Biotechnology tuula.nyman@helsinki.fi THE PROTEOME The complete protein complement expressed by a genome or by a cell or a tissue type (M.
Quantitative mass spec based proteomics Tuula Nyman Institute of Biotechnology tuula.nyman@helsinki.fi THE PROTEOME The complete protein complement expressed by a genome or by a cell or a tissue type (M.
New Approaches to Quantitative Proteomics Analysis
 New Approaches to Quantitative Proteomics Analysis Chris Hodgkins, Market Development Manager, SCIEX ANZ 2 nd November, 2017 Who is SCIEX? Founded by Dr. Barry French & others: University of Toronto Introduced
New Approaches to Quantitative Proteomics Analysis Chris Hodgkins, Market Development Manager, SCIEX ANZ 2 nd November, 2017 Who is SCIEX? Founded by Dr. Barry French & others: University of Toronto Introduced
A New Strategy for Quantitative Proteomics Using Isotope-Coded Protein Labels
 ICPL TM -Isotope Coded Label A New Strategy for Quantitative Proteomics Using Isotope-Coded Labels Alexander Schmidt Department for Max-Planck-Institute of Biochemistry Spotfire User Conference 2004, Cologne
ICPL TM -Isotope Coded Label A New Strategy for Quantitative Proteomics Using Isotope-Coded Labels Alexander Schmidt Department for Max-Planck-Institute of Biochemistry Spotfire User Conference 2004, Cologne
Nature Biotechnology: doi: /nbt Supplementary Figure 1. The workflow of Open-pFind.
 Supplementary Figure 1 The workflow of Open-pFind. The MS data are first preprocessed by pparse, and then the MS/MS data are searched by the open search module. Next, the MS/MS data are re-searched by
Supplementary Figure 1 The workflow of Open-pFind. The MS data are first preprocessed by pparse, and then the MS/MS data are searched by the open search module. Next, the MS/MS data are re-searched by
Proteomics: A Challenge for Technology and Information Science. What is proteomics?
 Proteomics: A Challenge for Technology and Information Science CBCB Seminar, November 21, 2005 Tim Griffin Dept. Biochemistry, Molecular Biology and Biophysics tgriffin@umn.edu What is proteomics? Proteomics
Proteomics: A Challenge for Technology and Information Science CBCB Seminar, November 21, 2005 Tim Griffin Dept. Biochemistry, Molecular Biology and Biophysics tgriffin@umn.edu What is proteomics? Proteomics
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture X roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture X roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
RockerBox. Filtering massive Mascot search results at the.dat level
 RockerBox Filtering massive Mascot search results at the.dat level Challenges Big experiments High amount of data Large raw and.dat files (> 2GB) How to handle our results?? The 2.2 peptide summary could
RockerBox Filtering massive Mascot search results at the.dat level Challenges Big experiments High amount of data Large raw and.dat files (> 2GB) How to handle our results?? The 2.2 peptide summary could
Proteomics And Cancer Biomarker Discovery. Dr. Zahid Khan Institute of chemical Sciences (ICS) University of Peshawar. Overview. Cancer.
 University of Peshawar. Overview. Cancer.") Proteomics And Cancer Biomarker Discovery Dr. Zahid Khan Institute of chemical Sciences (ICS) University of Peshawar Overview Proteomics Cancer Aims Tools Data Base search Challenges Summary 1 Overview
Proteomics And Cancer Biomarker Discovery Dr. Zahid Khan Institute of chemical Sciences (ICS) University of Peshawar Overview Proteomics Cancer Aims Tools Data Base search Challenges Summary 1 Overview
Využití cílené proteomiky pro kontrolu falšování potravin: identifikace peptidových markerů v mase pomocí LC- Q Exactive MS/MS
 Využití cílené proteomiky pro kontrolu falšování potravin: identifikace peptidových markerů v mase pomocí LC- Q Exactive MS/MS Michal Godula Ph.D. Thermo Fisher Scientific The world leader in serving science
Využití cílené proteomiky pro kontrolu falšování potravin: identifikace peptidových markerů v mase pomocí LC- Q Exactive MS/MS Michal Godula Ph.D. Thermo Fisher Scientific The world leader in serving science
Proteomics Informatics (BMSC-GA 4437)
") Proteomics Informatics (BMSC-GA 4437) Instructor David Fenyö Contact information David@FenyoLab.org htt://fenyolab.org/resentations/proteomics_informatics_2013/ Learning Objectives Be able analyze a roteomics
Proteomics Informatics (BMSC-GA 4437) Instructor David Fenyö Contact information David@FenyoLab.org htt://fenyolab.org/resentations/proteomics_informatics_2013/ Learning Objectives Be able analyze a roteomics
How to view Results with Scaffold. Proteomics Shared Resource
 How to view Results with Scaffold Proteomics Shared Resource Starting out Download Scaffold from http://www.proteomes oftware.com/proteom e_software_prod_sca ffold_download.html Follow installation instructions
How to view Results with Scaffold Proteomics Shared Resource Starting out Download Scaffold from http://www.proteomes oftware.com/proteom e_software_prod_sca ffold_download.html Follow installation instructions
Protein Grouping, FDR Analysis and Databases.
 Protein Grouping, FDR Analysis and Databases. March 15th 2012 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Protein Grouping, FDR Analysis and Databases Overview. Protein Grouping : Concept
Protein Grouping, FDR Analysis and Databases. March 15th 2012 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Protein Grouping, FDR Analysis and Databases Overview. Protein Grouping : Concept
Quantification of Isotope Encoded Proteins in 2D Gels
 Quantification of Isotope Encoded Proteins in 2D Gels Using Surface Enhanced Resonance Raman Giselle M. Knudsen 1, Brandon M. Davis 2, Shirshendu K. Deb 1, Yvette Loethen 2, Ravindra Gudihal 1, Pradeep
Quantification of Isotope Encoded Proteins in 2D Gels Using Surface Enhanced Resonance Raman Giselle M. Knudsen 1, Brandon M. Davis 2, Shirshendu K. Deb 1, Yvette Loethen 2, Ravindra Gudihal 1, Pradeep
Mass Spectrometry Based Proteomics Data Analysis Using GalaxyP
 Mass Spectrometry Based Proteomics Data Analysis Using GalaxyP GCC 2015 GalaxyP Workshop July 6th, 2015 Norwich, UK Presenters: Tim Griffin, Pratik Jagtap and James Johnson Documentation: Kevin Murray,
Mass Spectrometry Based Proteomics Data Analysis Using GalaxyP GCC 2015 GalaxyP Workshop July 6th, 2015 Norwich, UK Presenters: Tim Griffin, Pratik Jagtap and James Johnson Documentation: Kevin Murray,
LTQ Orbitrap XL Hybrid FT Mass Spectrometer Unrivaled Performance and Flexibility
 m a s s s p e c t r o m e t r y LTQ Orbitrap XL Hybrid FT Mass Spectrometer Unrivaled Performance and Flexibility Part of Thermo Fisher Scientific LTQ Orbitrap XL OFFERING OUTSTANDING MASS ACCURACY, RESOLVING
m a s s s p e c t r o m e t r y LTQ Orbitrap XL Hybrid FT Mass Spectrometer Unrivaled Performance and Flexibility Part of Thermo Fisher Scientific LTQ Orbitrap XL OFFERING OUTSTANDING MASS ACCURACY, RESOLVING
Innovations for Protein Research. Protein Research. Powerful workflows built on solid science
 Innovations for Protein Research Protein Research Powerful workflows built on solid science Your partner in discovering innovative solutions for quantitative protein and biomarker research Applied Biosystems/MDS
Innovations for Protein Research Protein Research Powerful workflows built on solid science Your partner in discovering innovative solutions for quantitative protein and biomarker research Applied Biosystems/MDS
Ariadne tutorial 1: RNA identification
 Ariadne tutorial 1: RNA identification 0. Introduction In this tutorial, we are going to introduce how to characterize RNA MS/MS data by using the Ariadne server via the Internet. How do you start searching
Ariadne tutorial 1: RNA identification 0. Introduction In this tutorial, we are going to introduce how to characterize RNA MS/MS data by using the Ariadne server via the Internet. How do you start searching
ENHANCED PROTEIN AND PEPTIDE CHARACTERIZATION
 Agilent MassHunter BioConfirm Software ENHANCED PROTEIN AND PEPTIDE CHARACTERIZATION Spectrum Algorithm Peak Modeling Deconvolution Mass 16951.778 Sequence Name Myoglobin (horse) Target mass 16951.673
Agilent MassHunter BioConfirm Software ENHANCED PROTEIN AND PEPTIDE CHARACTERIZATION Spectrum Algorithm Peak Modeling Deconvolution Mass 16951.778 Sequence Name Myoglobin (horse) Target mass 16951.673
d Yield of peptide (µm)
") a Pro template DN native trn mix pre-charged Pro-tRN Pro T7 RN polymerase [ 4 ]sp Met Lys ly sprs MetRS LysRS lyrs E. coli ribosome Tyr TyrRS 5 amino acids and 5 aminoacyl-trn synthetases other protein
a Pro template DN native trn mix pre-charged Pro-tRN Pro T7 RN polymerase [ 4 ]sp Met Lys ly sprs MetRS LysRS lyrs E. coli ribosome Tyr TyrRS 5 amino acids and 5 aminoacyl-trn synthetases other protein
Ensure your Success with Agilent s Biopharma Workflows
 Ensure your Success with Agilent s Biopharma Workflows Steve Madden Software Product Manager Agilent Technologies, Inc. June 5, 2018 Agenda Agilent BioPharma Workflow Platform for LC/MS Intact Proteins
Ensure your Success with Agilent s Biopharma Workflows Steve Madden Software Product Manager Agilent Technologies, Inc. June 5, 2018 Agenda Agilent BioPharma Workflow Platform for LC/MS Intact Proteins
An Improved Approach for Glycan Structure Identification from HCD MS/MS Spectra
 Introduction Method Experiments An Improved Approach for Glycan Structure Identification from HCD MS/MS Spectra Weiping Sun, Yi Liu, Gilles Lajoie, Bin Ma and Kaizhong Zhang Department of Computer Science,
Introduction Method Experiments An Improved Approach for Glycan Structure Identification from HCD MS/MS Spectra Weiping Sun, Yi Liu, Gilles Lajoie, Bin Ma and Kaizhong Zhang Department of Computer Science,
Proteomics. Proteomics is the study of all proteins within organism. Challenges
 Proteomics Proteomics is the study of all proteins within organism. Challenges 1. The proteome is larger than the genome due to alternative splicing and protein modification. As we have said before we
Proteomics Proteomics is the study of all proteins within organism. Challenges 1. The proteome is larger than the genome due to alternative splicing and protein modification. As we have said before we
Agilent Software Tools for Mass Spectrometry Based Multi-omics Studies
 Agilent Software Tools for Mass Spectrometry Based Multi-omics Studies Technical Overview Introduction The central dogma for biological information flow is expressed as a series of chemical conversions
Agilent Software Tools for Mass Spectrometry Based Multi-omics Studies Technical Overview Introduction The central dogma for biological information flow is expressed as a series of chemical conversions
Supporting Information for
 Supporting Information for CharmeRT: Boosting peptide identifications by chimeric spectra identification and retention time prediction Viktoria Dorfer*,,a, Sergey Maltsev,b, Stephan Winkler a, Karl Mechtler*,b,c.
Supporting Information for CharmeRT: Boosting peptide identifications by chimeric spectra identification and retention time prediction Viktoria Dorfer*,,a, Sergey Maltsev,b, Stephan Winkler a, Karl Mechtler*,b,c.
Put the PRO in Protein Characterization
 Put the PRO in Protein Characterization Aaron Boice LC/Q-TOF Product Manager 5-June-2017 Agenda The Right Tools for the Job Tackling Intact Proteins Break it Down Peptides and PTMs Sweet Success Released
Put the PRO in Protein Characterization Aaron Boice LC/Q-TOF Product Manager 5-June-2017 Agenda The Right Tools for the Job Tackling Intact Proteins Break it Down Peptides and PTMs Sweet Success Released
BIOINFORMATICS ORIGINAL PAPER
 BIOINFORMATICS ORIGINAL PAPER Vol 25 no 22 29, pages 2969 2974 doi:93/bioinformatics/btp5 Data and text mining Improving peptide identification with single-stage mass spectrum peaks Zengyou He and Weichuan
BIOINFORMATICS ORIGINAL PAPER Vol 25 no 22 29, pages 2969 2974 doi:93/bioinformatics/btp5 Data and text mining Improving peptide identification with single-stage mass spectrum peaks Zengyou He and Weichuan
Protein Reports CPTAC Common Data Analysis Pipeline (CDAP)
") Protein Reports CPTAC Common Data Analysis Pipeline (CDAP) v. 4/13/2015 Summary The purpose of this document is to describe the protein reports generated as part of the CPTAC Common Data Analysis Pipeline
Protein Reports CPTAC Common Data Analysis Pipeline (CDAP) v. 4/13/2015 Summary The purpose of this document is to describe the protein reports generated as part of the CPTAC Common Data Analysis Pipeline
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/1144622/dc1 Supporting Online Material for Genome Transplantation in Bacteria: Changing One Species to Another Carole Lartigue, John I. Glass, * Nina Alperovich, Rembert
www.sciencemag.org/cgi/content/full/1144622/dc1 Supporting Online Material for Genome Transplantation in Bacteria: Changing One Species to Another Carole Lartigue, John I. Glass, * Nina Alperovich, Rembert
ProMass HR Applications!
 ProMass HR Applications! ProMass HR Features Ø ProMass HR includes features for high resolution data processing. Ø ProMass HR includes the standard ProMass deconvolution algorithm as well as the full Positive
ProMass HR Applications! ProMass HR Features Ø ProMass HR includes features for high resolution data processing. Ø ProMass HR includes the standard ProMass deconvolution algorithm as well as the full Positive
Bioinformatic Tools. So you acquired data.. But you wanted knowledge. So Now What?
 Bioinformatic Tools So you acquired data.. But you wanted knowledge So Now What? We have a series of questions What the Heck is That Ion? How come my MW does not match? How do I make a DB to search against?
Bioinformatic Tools So you acquired data.. But you wanted knowledge So Now What? We have a series of questions What the Heck is That Ion? How come my MW does not match? How do I make a DB to search against?
Informatics on MS-Based Proteomics
 Informatics on MS-Based Proteomics Yet-Ran Chen 陳逸然 Associate Research Fellow / Associate Professor Agricultural Biotech. Research Center / Institute of Biotechnology & Genomics and Systems Biology Academia
Informatics on MS-Based Proteomics Yet-Ran Chen 陳逸然 Associate Research Fellow / Associate Professor Agricultural Biotech. Research Center / Institute of Biotechnology & Genomics and Systems Biology Academia
How to view Results with. Proteomics Shared Resource
 How to view Results with Scaffold 3.0 Proteomics Shared Resource An overview This document is intended to walk you through Scaffold version 3.0. This is an introductory guide that goes over the basics
How to view Results with Scaffold 3.0 Proteomics Shared Resource An overview This document is intended to walk you through Scaffold version 3.0. This is an introductory guide that goes over the basics
Really high sensitivity mass spectrometry and Discovery and analysis of protein complexes
 Really high sensitivity mass spectrometry and Discovery and analysis of protein complexes The PRIME lab and AMS Importance of protein complexes in biology Methods for isolation of protein complexes In
Really high sensitivity mass spectrometry and Discovery and analysis of protein complexes The PRIME lab and AMS Importance of protein complexes in biology Methods for isolation of protein complexes In
High-throughput Proteomic Data Analysis. Suh-Yuen Liang ( 梁素雲 ) NRPGM Core Facilities for Proteomics and Glycomics Academia Sinica Dec.
 NRPGM Core Facilities for Proteomics and Glycomics Academia Sinica Dec.") High-throughput Proteomic Data Analysis Suh-Yuen Liang ( 梁素雲 ) NRPGM Core Facilities for Proteomics and Glycomics Academia Sinica Dec. 9, 2009 High-throughput Proteomic Data Are Information Rich and Dependent
High-throughput Proteomic Data Analysis Suh-Yuen Liang ( 梁素雲 ) NRPGM Core Facilities for Proteomics and Glycomics Academia Sinica Dec. 9, 2009 High-throughput Proteomic Data Are Information Rich and Dependent
Faster, More Sensitive Peptide ID by Sequence DB Compression. Nathan Edwards Center for Bioinformatics and Computational Biology
 Faster, More Sensitive Peptide ID by Sequence DB Compression Nathan Edwards Center for Bioinformatics and Computational Biology MS/MS Search Engines Fail when peptides are missing from sequence database
Faster, More Sensitive Peptide ID by Sequence DB Compression Nathan Edwards Center for Bioinformatics and Computational Biology MS/MS Search Engines Fail when peptides are missing from sequence database
Web based Bioinformatics Applications in Proteomics. Genbank
 Web based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu February 9, 2010 Genbank Primary nucleic acid sequence database Maintained by NCBI National Center for Biotechnology
Web based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu February 9, 2010 Genbank Primary nucleic acid sequence database Maintained by NCBI National Center for Biotechnology
Supplementary Tables. Note: Open-pFind is embedded as the default open search workflow of the pfind tool. Nature Biotechnology: doi: /nbt.
 Supplementary Tables Supplementary Table 1. Detailed information for the six datasets used in this study Dataset Mass spectrometer # Raw files # MS2 scans Reference Dong-Ecoli-QE Q Exactive 5 202,452 /
Supplementary Tables Supplementary Table 1. Detailed information for the six datasets used in this study Dataset Mass spectrometer # Raw files # MS2 scans Reference Dong-Ecoli-QE Q Exactive 5 202,452 /
Basic protein and peptide science for proteomics. Henrik Johansson
 Basic protein and peptide science for proteomics Henrik Johansson Proteins are the main actors in the cell Membranes Transport and storage Chemical factories DNA Building proteins Structure Proteins mediate
Basic protein and peptide science for proteomics Henrik Johansson Proteins are the main actors in the cell Membranes Transport and storage Chemical factories DNA Building proteins Structure Proteins mediate
Workflows and Pipelines for NGS analysis: Lessons from proteomics
 Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
Workflows and Pipelines for NGS analysis: Lessons from proteomics Conference on Applying NGS in Basic research Health care and Agriculture 11 th Sep 2014 Debasis Dash Where are the protein coding genes
Liver Mitochondria Proteomics Employing High-Resolution MS Technology
 Liver Mitochondria Proteomics Employing High-Resolution MS Technology Jenny Ho, 1 Loïc Dayon, 2 John Corthésy, 2 Umberto De Marchi, 2 Antonio Núñez, 2 Andreas Wiederkehr, 2 Rosa Viner, 3 Michael Blank,
Liver Mitochondria Proteomics Employing High-Resolution MS Technology Jenny Ho, 1 Loïc Dayon, 2 John Corthésy, 2 Umberto De Marchi, 2 Antonio Núñez, 2 Andreas Wiederkehr, 2 Rosa Viner, 3 Michael Blank,
Proteomics and Cancer
 Proteomics and Cancer Japan Society for the Promotion of Science (JSPS) Science Dialogue Program at Niitsu Senior High School Niitsu, Niigata September 4th 2006 Vladimir Valera, M.D, PhD JSPS Postdoctoral
Proteomics and Cancer Japan Society for the Promotion of Science (JSPS) Science Dialogue Program at Niitsu Senior High School Niitsu, Niigata September 4th 2006 Vladimir Valera, M.D, PhD JSPS Postdoctoral
Database Searching for Protein Identification and Characterization
 Database Searching for Protein Identification and Characterization John Cottrell Matrix Science http://tinyurl.com/e4brg jcottrell@matrixscience.com 1 Topics Methods of database searching Practical tips
Database Searching for Protein Identification and Characterization John Cottrell Matrix Science http://tinyurl.com/e4brg jcottrell@matrixscience.com 1 Topics Methods of database searching Practical tips
Application Note # ET-20 BioPharma Compass: A fully Automated Solution for Characterization and QC of Intact and Digested Proteins
 Application Note # ET-20 BioPharma Compass: A fully Automated Solution for Characterization and QC of Intact and Digested Proteins BioPharma Compass TM is a fully automated solution for the rapid characterization
Application Note # ET-20 BioPharma Compass: A fully Automated Solution for Characterization and QC of Intact and Digested Proteins BioPharma Compass TM is a fully automated solution for the rapid characterization
Isotopic Resolution of Chromatographically Separated IdeS Subunits Using the X500B QTOF System
 Isotopic Resolution of Chromatographically Separated IdeS Subunits Using the X500B QTOF System Fan Zhang 2, Sean McCarthy 1 1 SCIEX, MA, USA, 2 SCIEX, CA USA Analysis of protein subunits using high resolution
Isotopic Resolution of Chromatographically Separated IdeS Subunits Using the X500B QTOF System Fan Zhang 2, Sean McCarthy 1 1 SCIEX, MA, USA, 2 SCIEX, CA USA Analysis of protein subunits using high resolution
Strategies for Quantitative Proteomics. Atelier "Protéomique Quantitative" La Grande Motte, France - June 26, 2007
 Strategies for Quantitative Proteomics Atelier "Protéomique Quantitative", France - June 26, 2007 Bruno Domon, Ph.D. Institut of Molecular Systems Biology ETH Zurich Zürich, Switzerland OUTLINE Introduction
Strategies for Quantitative Proteomics Atelier "Protéomique Quantitative", France - June 26, 2007 Bruno Domon, Ph.D. Institut of Molecular Systems Biology ETH Zurich Zürich, Switzerland OUTLINE Introduction
Mass Spectrometry and Proteomics - Lecture 6 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 6 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Quantitation techniques Label-free TMT SILAC Peptide identification and FDR 194 Lecture
Mass Spectrometry and Proteomics - Lecture 6 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Quantitation techniques Label-free TMT SILAC Peptide identification and FDR 194 Lecture
Representing Errors and Uncertainty in Plasma Proteomics
 Representing Errors and Uncertainty in Plasma Proteomics David J. States, M.D., Ph.D. University of Michigan Bioinformatics Program Proteomics Alliance for Cancer Genomics vs. Proteomics Genome sequence
Representing Errors and Uncertainty in Plasma Proteomics David J. States, M.D., Ph.D. University of Michigan Bioinformatics Program Proteomics Alliance for Cancer Genomics vs. Proteomics Genome sequence
Confident Protein ID using Spectrum Mill Software
 Welcome to our E-Seminar: Confident Protein ID using Spectrum Mill Software Slide 1 Spectrum Mill Informatics Software Start with batches of raw MS data! Sp ec t ru m Mi ll Biologist-friendly answers!
Welcome to our E-Seminar: Confident Protein ID using Spectrum Mill Software Slide 1 Spectrum Mill Informatics Software Start with batches of raw MS data! Sp ec t ru m Mi ll Biologist-friendly answers!
Shotgun Proteomics: How Confident are you in that Identification? or Statistical Evaluation of Shotgun Proteomic Data
 Shotgun Proteomics: How Confident are you in that Identification? or Statistical Evaluation of Shotgun Proteomic Data Ron Orlando Complex Carbohydrate Research Center University of Georgia Athens, GA 30602
Shotgun Proteomics: How Confident are you in that Identification? or Statistical Evaluation of Shotgun Proteomic Data Ron Orlando Complex Carbohydrate Research Center University of Georgia Athens, GA 30602
Sequence Databases and database scanning
 Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
ichemistry SOLUTIONS Integrated chemistries to boost mass spectrometry workflows
 ichemistry SOLUTIONS Integrated chemistries to boost mass spectrometry workflows ichemistry SOLUTIONS ichemistry solutions ichemistry solutions are the world s only reagents and consumables that are customdesigned
ichemistry SOLUTIONS Integrated chemistries to boost mass spectrometry workflows ichemistry SOLUTIONS ichemistry solutions ichemistry solutions are the world s only reagents and consumables that are customdesigned
Optimization-Based Peptide Mass Fingerprinting for Protein Mixture Identification
 Optimization-Based Peptide Mass Fingerprinting for Protein Mixture Identification Weichuan Yu, Ph.D. Department of Electronic and Computer Engineering The Hong Kong University of Science and Technology
Optimization-Based Peptide Mass Fingerprinting for Protein Mixture Identification Weichuan Yu, Ph.D. Department of Electronic and Computer Engineering The Hong Kong University of Science and Technology
MBios 478: Mass Spectrometry Applications [Dr. Wyrick] Slide #1. Lecture 25: Mass Spectrometry Applications
![MBios 478: Mass Spectrometry Applications [Dr. Wyrick] Slide #1. Lecture 25: Mass Spectrometry Applications](/thumbs/74/71368870.jpg "MBios 478: Mass Spectrometry Applications [Dr. Wyrick] Slide #1. Lecture 25: Mass Spectrometry Applications") MBios 478: Mass Spectrometry Applications [Dr. Wyrick] Slide #1 Lecture 25: Mass Spectrometry Applications Measuring Protein Abundance o ICAT o DIGE Identifying Post-Translational Modifications Protein-protein
MBios 478: Mass Spectrometry Applications [Dr. Wyrick] Slide #1 Lecture 25: Mass Spectrometry Applications Measuring Protein Abundance o ICAT o DIGE Identifying Post-Translational Modifications Protein-protein
Comparability Analysis of Protein Therapeutics by Bottom-Up LC-MS with Stable Isotope-Tagged Reference Standards
 Comparability Analysis of Protein Therapeutics by Bottom-Up LC-MS with Stable Isotope-Tagged Reference Standards 16 September 2011 Abbott Bioresearch Center, Worcester, MA USA Manuilov, A. V., C. H. Radziejewski
Comparability Analysis of Protein Therapeutics by Bottom-Up LC-MS with Stable Isotope-Tagged Reference Standards 16 September 2011 Abbott Bioresearch Center, Worcester, MA USA Manuilov, A. V., C. H. Radziejewski
Cell Signaling Technology
 Cell Signaling Technology PTMScan Direct: Multipathway v2.0 Proteomics Service Group January 14, 2013 PhosphoScan Deliverables Project Overview Methods PTMScan Direct: Multipathway V2.0 (Tables 1,2) Qualitative
Cell Signaling Technology PTMScan Direct: Multipathway v2.0 Proteomics Service Group January 14, 2013 PhosphoScan Deliverables Project Overview Methods PTMScan Direct: Multipathway V2.0 (Tables 1,2) Qualitative
Appendix. Table of contents
 Appendix Table of contents 1. Appendix figures 2. Legends of Appendix figures 3. References Appendix Figure S1 -Tub STIL (41) DVFFYQADDEHYIPR (55) (64) AVLLDLEPR (72) 1 451aa (1071) YLNENQLSQLSVTR (1084)
Appendix Table of contents 1. Appendix figures 2. Legends of Appendix figures 3. References Appendix Figure S1 -Tub STIL (41) DVFFYQADDEHYIPR (55) (64) AVLLDLEPR (72) 1 451aa (1071) YLNENQLSQLSVTR (1084)
11/22/13. Proteomics, functional genomics, and systems biology. Biosciences 741: Genomics Fall, 2013 Week 11
 Proteomics, functional genomics, and systems biology Biosciences 741: Genomics Fall, 2013 Week 11 1 Figure 6.1 The future of genomics Functional Genomics The field of functional genomics represents the
Proteomics, functional genomics, and systems biology Biosciences 741: Genomics Fall, 2013 Week 11 1 Figure 6.1 The future of genomics Functional Genomics The field of functional genomics represents the
Exam MOL3007 Functional Genomics
 Faculty of Medicine Department of Cancer Research and Molecular Medicine Exam MOL3007 Functional Genomics Thursday December 20 th 9.00-13.00 ECTS credits: 7.5 Number of pages (included front-page): 5 Supporting
Faculty of Medicine Department of Cancer Research and Molecular Medicine Exam MOL3007 Functional Genomics Thursday December 20 th 9.00-13.00 ECTS credits: 7.5 Number of pages (included front-page): 5 Supporting
Combination of Isobaric Tagging Reagents and Cysteinyl Peptide Enrichment for In-Depth Quantification
 Combination of Isobaric Tagging Reagents and Cysteinyl Peptide Enrichment for In-Depth Quantification Protein Expression Analysis using the TripleTOF 5600 System and itraq Reagents Vojtech Tambor 1, Christie
Combination of Isobaric Tagging Reagents and Cysteinyl Peptide Enrichment for In-Depth Quantification Protein Expression Analysis using the TripleTOF 5600 System and itraq Reagents Vojtech Tambor 1, Christie
Mass Spectrometry in Proteomics
 Mass Spectrometry in Proteomics Bin Ma Assistant Professor Canada Research Chair in Bioinformatics Department of Computer Science University of Western Ontario bma@csd.uwo.ca From the Genome to the Proteome
Mass Spectrometry in Proteomics Bin Ma Assistant Professor Canada Research Chair in Bioinformatics Department of Computer Science University of Western Ontario bma@csd.uwo.ca From the Genome to the Proteome
Filter-based Protein Digestion (FPD): A Detergent-free and Scaffold-based Strategy for TMT workflows
: A Detergent-free and Scaffold-based Strategy for TMT workflows") Supporting Information Filter-based Protein Digestion (FPD): A Detergent-free and Scaffold-based Strategy for TMT workflows Ekaterina Stepanova 1, Steven P. Gygi 1, *, Joao A. Paulo 1, * 1 Department of
Supporting Information Filter-based Protein Digestion (FPD): A Detergent-free and Scaffold-based Strategy for TMT workflows Ekaterina Stepanova 1, Steven P. Gygi 1, *, Joao A. Paulo 1, * 1 Department of
with Database Search of Tandem Mass Spectra
 MCP Papers in Press. Published on August 14, 2008 as Manuscript M800103-MCP200 Spectral Dictionaries: Integrating De Novo Peptide Sequencing with Database Search of Tandem Mass Spectra Sangtae Kim 1, Nitin
MCP Papers in Press. Published on August 14, 2008 as Manuscript M800103-MCP200 Spectral Dictionaries: Integrating De Novo Peptide Sequencing with Database Search of Tandem Mass Spectra Sangtae Kim 1, Nitin
Biotherapeutic Non-Reduced Peptide Mapping
 Biotherapeutic Non-Reduced Peptide Mapping Routine non-reduced peptide mapping of biotherapeutics on the X500B QTOF System Method details for the routine non-reduced peptide mapping of a biotherapeutic
Biotherapeutic Non-Reduced Peptide Mapping Routine non-reduced peptide mapping of biotherapeutics on the X500B QTOF System Method details for the routine non-reduced peptide mapping of a biotherapeutic
Supplementary information, Figure S1A ShHTL7 interacted with MAX2 but not another F-box protein COI1.
 GR24 (μm) 0 20 0 20 GST-ShHTL7 anti-gst His-MAX2 His-COI1 PVDF staining Supplementary information, Figure S1A ShHTL7 interacted with MAX2 but not another F-box protein COI1. Pull-down assays using GST-ShHTL7
GR24 (μm) 0 20 0 20 GST-ShHTL7 anti-gst His-MAX2 His-COI1 PVDF staining Supplementary information, Figure S1A ShHTL7 interacted with MAX2 but not another F-box protein COI1. Pull-down assays using GST-ShHTL7
with Database Search of Tandem Mass Spectra
 MCP Papers in Press. Published on August 14, 2008 as Manuscript M800103-MCP200 Spectral Dictionaries: Integrating De Novo Peptide Sequencing with Database Search of Tandem Mass Spectra Sangtae Kim 1, Nitin
MCP Papers in Press. Published on August 14, 2008 as Manuscript M800103-MCP200 Spectral Dictionaries: Integrating De Novo Peptide Sequencing with Database Search of Tandem Mass Spectra Sangtae Kim 1, Nitin
Hongwei Xie, Martin Gilar, and John C. Gebler Waters Corporation, Milford, MA, U.S.A. INTRODUCTION EXPERIMENTAL
 Analysis of Deamidation and Oxidation in MONOCLONAL Antibody Using Peptide Mapping with UPLC/MS E Hongwei Xie, Martin Gilar, and John C. Gebler Waters Corporation, Milford, MA, U.S.A. INTRODUCTION Monoclonal
Analysis of Deamidation and Oxidation in MONOCLONAL Antibody Using Peptide Mapping with UPLC/MS E Hongwei Xie, Martin Gilar, and John C. Gebler Waters Corporation, Milford, MA, U.S.A. INTRODUCTION Monoclonal
Instrumental Solutions for Metabolomics
 Instrumental Solutions for Metabolomics Dr. Desislav Donchev US14702384-16012196010132-S01-A01.tif 300 400 500 600 700 800 3100 3200 3300 3400 3500 3600 3700 3800 Color Scale R = [52 1484] G = [26 514]
Instrumental Solutions for Metabolomics Dr. Desislav Donchev US14702384-16012196010132-S01-A01.tif 300 400 500 600 700 800 3100 3200 3300 3400 3500 3600 3700 3800 Color Scale R = [52 1484] G = [26 514]
Nature Biotechnology: doi: /nbt Supplementary Figure 1
 Supplementary Figure 1 The mass accuracy of fragment ions is important for peptide recovery in wide-tolerance searches. The same data as in Figure 1B was searched with varying fragment ion tolerances (FIT).
Supplementary Figure 1 The mass accuracy of fragment ions is important for peptide recovery in wide-tolerance searches. The same data as in Figure 1B was searched with varying fragment ion tolerances (FIT).
Protein De novo Sequencing
 Protein De novo Sequencing by Rong Wang A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics in Computer Science Waterloo, Ontario,
Protein De novo Sequencing by Rong Wang A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics in Computer Science Waterloo, Ontario,
LC/MS/MS Solutions for Biomarker Discovery QSTAR. Elite Hybrid LC/MS/MS System. More performance, more reliability, more answers
 LC/MS/MS Solutions for Biomarker Discovery QSTAR Elite Hybrid LC/MS/MS System More performance, more reliability, more answers More is better and the QSTAR Elite LC/MS/MS system has more to offer. More
LC/MS/MS Solutions for Biomarker Discovery QSTAR Elite Hybrid LC/MS/MS System More performance, more reliability, more answers More is better and the QSTAR Elite LC/MS/MS system has more to offer. More
The best proteomics: combination of upfront reduction of sample complexity, reproducible resolution, and effective follow up
 The best proteomics: combination of upfront reduction of sample complexity, reproducible resolution, and effective follow up Helen Kim, Ph.D. Dept of Pharmacology & Toxicology University of Alabama at
The best proteomics: combination of upfront reduction of sample complexity, reproducible resolution, and effective follow up Helen Kim, Ph.D. Dept of Pharmacology & Toxicology University of Alabama at
Protein Bioinformatics Part I: Access to information
 Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Supplementary Information
 Identifying sources of tick blood meals using unidentified tandem mass spectral libraries Özlem Önder 1, Wenguang Shao 2, Brian Kemps 1, Henry Lam 2,3,*, Dustin Brisson 1,* 1 Department of Biology, University
Identifying sources of tick blood meals using unidentified tandem mass spectral libraries Özlem Önder 1, Wenguang Shao 2, Brian Kemps 1, Henry Lam 2,3,*, Dustin Brisson 1,* 1 Department of Biology, University