The challenges of software medical device regulation.
|
|
|
- Sylvia Thornton
- 6 years ago
- Views:
Transcription



1 The challenges of software medical device regulation.

2 Introduction A brief history of software device regulation A look at the new device regulations 2
3 Current framework In Vitro Diagnostics Medical Device Directive 98/79/EC Medical Device Directive 93/42/EEC Active Implantable Medical Device Directive 90/385/EEC UK Medical Device Regulations The directives
4 What is a medical device? A thing, for use on humans, that the manufacturer says can be used for: Prevention of disease, Diagnosis, monitoring, treatment or alleviation of disease, an injury or handicap, Compensation for an injury or handicap, Investigation, replacement or modification of the anatomy or of a physiological process, Control of conception And doesn t act principally as a medicine. This intended use is determined by claims on the device, label, instructions or promotional material. 4
5 But I have a disclaimer! It should be noted that general disclaimers (for example this product is not a medical device ) are not acceptable if medical claims are made or implied elsewhere in the product labelling or associated promotional literature. Manufacturer needs to consider use that can be reasonably foreseen prior to placing a product on the market A device does not need to diagnose to be considered to be used for diagnosis. 5
6 Function creep! Adding extra functionality can change qualification as a device. e.g. The addition of QRISK calculator to a basic GP database will make the system a device. Can be managed by CE marking a module. 6

7 Can software be a device? Medical Device Directive - 93/42/EEC Software on its own not specifically included. including the software necessary for its proper application Microsoft releases Windows 3.11, Office 4.0 and MS-DOS 6.0. DOOM released 1994 European guidance - MEDDEV 2.1/1 Distinction of software influencing Software related to the functioning of a medical device may be part of a device or a device in its own right if it is placed on the market separately from the related device. Macintosh System Software - System 1 7
8 Software as a device? 1998 IVD Medical Device Directive 98/79/EC This directive s definition of a medical device does not include software, only including the software necessary for its proper application IVDMD definition does not include mention of software. Win Amending Directive 2007/47/EC (UK March 2010) Specifically adds software into the MDD definition of a medical device. Adds software specific Essential Requirement on validation & verification to the MDD only. iphone (1 st generation) 8
9 General medical devices In Vitro Diagnostics Medical Device Directive 98/79/EC Medical Device Directive 93/42/EEC Active Implantable Medical Device Directive 90/385/EEC UK Medical Device Regulations The directives
10 Recap on the current regulatory system The UK MDR implement the MDD into UK law MDD elements: Articles Essential requirements list Annex I The conformity route annexes II-VII Classification rules IX The rest clinical evaluation & other stuff! 10 The directives
11 Essential requirements Cover safety, design, construction, performance etc. Initially covered risk factors for physical devices. Are not always very specific! Briefly mention software: software must be validated according to the state of the art taking into account the principles of development lifecycle, risk management, validation and verification. Conformance with these can be demonstrated by the use of Harmonised standards. For software : EN 62304: The directives
12 Classification rules Risk based classification system: I, IIa, IIb, III Rules based mainly on physical hazards / interactions: E.g. invasive devices, active devices No specific software rules. Software is considered to be an active device Implementing rule for software that drives or influences a device. Most standalone software will be class I unless performing direct diagnosis. 12 The directives



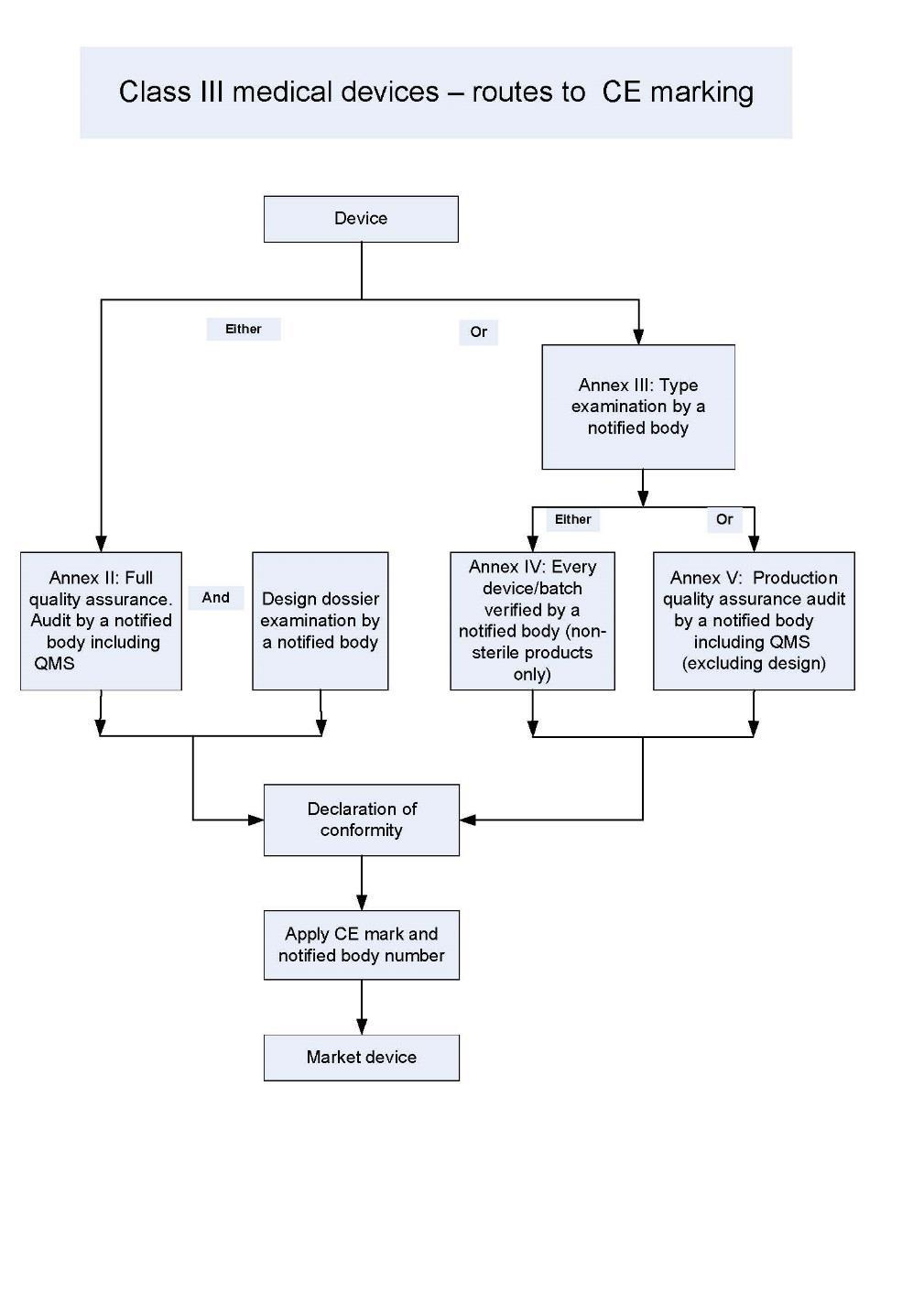
13 Conformity routes Depends on Class. Class I device manufacturers self declare. 13 The directives

14 Guidance: MEDDEVs (MEDical DEVice guidance) Scope, field of application, definition Standalone Software Classification of MD Clinical investigation, clinical evaluation Notified bodies Market surveillance - Vigilance Borderlines 14 The directives
15 The Borderlines & classification manual European committee that looks at borderline and classification issues for devices. Publishes its determinations. Contains a few software device examples: PACS A mobile application for processing ECGs A mobile application for the communication between patient and caregivers while giving birth A mobile medical application for viewing the anatomy of the human body Qualification of software for interpretation of a guideline Qualification and classification of software for delivery and management of cognitive remediation and rehabilitation programs Classification of software for information management and patient monitoring Mobile application for managing pictures of moles Mobile application for the assessment of moles 15 The directives
16 Revision of the directives Starting in 2012: extension of the scope of legislation, better supervision of independent assessment bodies, clear rights for economic operators, and stronger requirements for clinical evidence. Guidance written into the regulations. 16 The regulations
17 MDRs 2017 (Now in force) General and Active Implantable Medical Device Regulations 26 May 2020 (+ 2 if certified UK Law In Vitro Diagnostics Medical Device Regulations 26 May 2022 (+ 4 if certified) 17 The regulations
18 The new regulations Will apply to medical devices placed on the market or put into service from 26 May Will include any updates to existing software devices. (A new device will have been put into service.) Certified devices (by a Notified body) have up to an extra 2 years to be in compliance. 18 The regulations
19 Changes for software? In house developed devices will now be regulated a bit! Essential requirements updated and will now specifically cover security and unauthorised access. There is a specific software rule (Rule 11). Software used to take decisions with a diagnostic or therapeutic purpose will be at least class 2a. If your device is up classified from a class 1 device under the regs, you must remove it from the market until it is certified by a NB. Introduction of UDI labelling. Guidance has been incorporated into the regulations. 19 The regulations
20 In-house conditions Don t need to fully CE mark as long as: not transferred to another legal entity, manufacture and use of the devices occur under appropriate quality management systems, the health institution justifies that the target patient group's specific needs cannot be met, or cannot be met at the appropriate level of performance by an equivalent device available on the market, Appropriate documentation a technical file; the health institution has a publicly available declaration. 20 Article 5(5)
21 Rule 11 - Software Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause: death or an irreversible [serious] deterioration of a person's state of health, in which case it is in class III; or a serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb. Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. All other software is classified as class I. 21 Annex VIII
22 Rule 11 IMDRF approach? To treat or to diagnose Treating and diagnosing infers that the information provided by the SaMD will be used to take an immediate or near term action: To drive clinical management Driving clinical management infers that the information provided by the SaMD will be used to aid in treatment, aid in diagnoses, to triage or identify early signs of a disease or condition will be used to guide next diagnostics or next treatment interventions: To Inform clinical management Informing clinical management infers that the information provided by the SaMD will not trigger an immediate or near term action: Critical situation or condition Situations or conditions where accurate and/or timely diagnosis or treatment action is vital to avoid death, longterm disability or other serious deterioration of health of an individual patient or to mitigating impact to public health. IV III II Serious situation or condition Situations or conditions where accurate diagnosis or treatment is of vital importance to avoid unnecessary interventions (e.g., biopsy) or timely interventions are important to mitigate long term irreversible consequences on an individual patient s health condition or public health. III II I Non-Serious situation or condition Situations or conditions where an accurate diagnosis and treatment is important but not critical for interventions to mitigate long term irreversible consequences on an individual patient's health condition or public health. II I I 22
23 Rule 11? Significance of incorrect decision. Related to timescale of implementation of the diagnosis/treatment. High Prompt diagnosis or treatment Medium, Drives clinical management Low, Informs clinical management. (Everything else) Possible outcome of incorrect decision Critical situation or condition III IIb Serious situation or condition IIb IIa Non-serious situation or condition (everything else) 23
 GTIN = manufacturer and device identification (ii) Production identifier (PI) - dynamic data, including: (17) Expiry date (10) Lot number or (21) Serial")
24 Unique Device Identification (UDI) 2D Bar code example Product code (device identification) Lot number Expiry date UDI two parts: (i) Device identifier (DI) - static data (01) GTIN = manufacturer and device identification (ii) Production identifier (PI) - dynamic data, including: (17) Expiry date (10) Lot number or (21) Serial number 24
25 UDI Software requirements UDI Device Identifier (model) & UDI Production Identifier (batch) A new UDI-DI shall be required whenever there is a modification that changes: (a) the original performance; (b) the safety or the intended use of the software; (c) interpretation of data. new or modified algorithms, database structures, operating platform, architecture or new user interfaces or new channels for interoperability Minor software revisions shall require a new UDI-PI and not a new UDI-DI. E.g. bug fixes (enhancements that are not for safety purposes, security patches or operating efficiency) 25 Annex VI 6.5

26 Importers & distributors Supply chain responsibilities: Shall verify that requirements are met: the device has been CE marked and that the EU declaration of conformity of the device has been drawn up; The device is accompanied by the information to be supplied by the manufacturer Manufacturer and Authorised representative identified Check that UDI requirement met Importers to add their details to the packaging/documentation. Contact manufacturer/ca if they believe the device is not in compliance. Record and pass on details of any complaints. 26 Articles 13,14 & 16

27 27 MDR applies from 26/5/2020!
28 Crown copyright 2017 About copyright All material created by the Medicines and Healthcare Products Regulatory Agency, including materials featured within these Medicines and Healthcare Products Regulatory Agency presentation notes and delegate pack, is subject to Crown copyright protection. We control the copyright to our work (which includes all information, database rights, logos and visual images), under a delegation of authority from the Controller of Her Majesty s Stationery Office (HMSO). The Medicines and Healthcare Products Regulatory Agency authorises you to make one free copy, by downloading to printer or to electronic, magnetic or optical storage media, of these presentations for the purposes of private research, study and reference. Any other copy or use of Crown copyright materials featured on this site, in any form or medium is subject to the prior approval of the Medicines and Healthcare Products Regulatory Agency. Further information, including an application form for requests to reproduce our material can be found at Material from other organisations The permission to reproduce Crown copyright protected material does not extend to any material in this pack which is subject to a separate licence or is the copyright of a third party. Authorisation to reproduce such material must be obtained from the copyright holders concerned
GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES
 MDEG - 2007-12 - II-3.3 MSOGClassIGuidance_Final GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES Foreword These guidance notes do not aim to be a definite interpretation of National Laws and/or
MDEG - 2007-12 - II-3.3 MSOGClassIGuidance_Final GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES Foreword These guidance notes do not aim to be a definite interpretation of National Laws and/or
Due diligence in the European medical devices industry
 Due diligence in the European medical devices industry Alison Dennis, Reed Smith LLP www.practicallaw.com/0-205-5707 As the medical devices industry is highly regulated, determining a target company's
Due diligence in the European medical devices industry Alison Dennis, Reed Smith LLP www.practicallaw.com/0-205-5707 As the medical devices industry is highly regulated, determining a target company's
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS
 Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Update on the IVDR. Sue Spencer
 Update on the IVDR Sue Spencer Caution The new regulations are draft the principles have now been agreed but the Annexes are subject to minor changes Further details will be added later pre and post application
Update on the IVDR Sue Spencer Caution The new regulations are draft the principles have now been agreed but the Annexes are subject to minor changes Further details will be added later pre and post application
Ready or Not: The New Medical Device Regulations Are Here!
 Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Changes to the Medical Devices Directive and affect on Manufacturers
 TÜV Product Service Ltd Webinar 18 th November 2009 Changes to the Medical Devices Directive and affect on Manufacturers Henry Sibun Manager, Medical & Health Services UK CONTENTS / 1. Introduction 1.
TÜV Product Service Ltd Webinar 18 th November 2009 Changes to the Medical Devices Directive and affect on Manufacturers Henry Sibun Manager, Medical & Health Services UK CONTENTS / 1. Introduction 1.
Implications of the new MDR from a Product Testing and Certification Perspective
 Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
PSMF in Practice Your Questions Answered! GPvP Symposium, 14 March 2014 Jonathan Rowell, Senior GPvP Inspector
 PSMF in Practice Your Questions Answered! GPvP Symposium, 14 March 2014 Jonathan Rowell, Senior GPvP Inspector Content Answer industry questions related to the PSMF MHRA inspector s preparation: How we
PSMF in Practice Your Questions Answered! GPvP Symposium, 14 March 2014 Jonathan Rowell, Senior GPvP Inspector Content Answer industry questions related to the PSMF MHRA inspector s preparation: How we
GENERAL AND ORGANISATIONAL REQUIREMENTS
 NBOG working document applicable for MDR and IVDR WD 2017-1 Draft list of documents to be submitted in the application for designation as a notified body under Regulation (EU) 2017/745 and Regulation (EU)
NBOG working document applicable for MDR and IVDR WD 2017-1 Draft list of documents to be submitted in the application for designation as a notified body under Regulation (EU) 2017/745 and Regulation (EU)
IVD Regulation 2017/746
 IVD Regulation 2017/746 Dr. Anne Van Nerom Famhp 2017-06-13 Recast-symposium Auditorium Storck (Eurostation II) Rue Juliette Wytsmanstraat 14 1050 Brussels Belgium T +32 2 642 51 11 F +32 2 642 50 01 email:
IVD Regulation 2017/746 Dr. Anne Van Nerom Famhp 2017-06-13 Recast-symposium Auditorium Storck (Eurostation II) Rue Juliette Wytsmanstraat 14 1050 Brussels Belgium T +32 2 642 51 11 F +32 2 642 50 01 email:
Guide for Ethics Committees on Clinical Investigation of Medical Devices
 Guide for Ethics Committees on Clinical Investigation of Medical Devices AUT-G0044-2 04 JUNE 2010 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes
Guide for Ethics Committees on Clinical Investigation of Medical Devices AUT-G0044-2 04 JUNE 2010 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes
Technical Documentation
 Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994
 Regulations, 1994") Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994 SUR-G0006-2 27 AUGUST 2010 This guide does not purport to be an interpretation of law and/or
Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994 SUR-G0006-2 27 AUGUST 2010 This guide does not purport to be an interpretation of law and/or
MHRA GDP Symposium. Novotel London West, London 8 & 10 December #GMDPevents
 MHRA GDP Symposium Novotel London West, London 8 & 10 December 2015 Complex Business Models - Outsourced Activities Presented by: Jacqueline Masayi, GDP Inspector Background Increased complexities of the
MHRA GDP Symposium Novotel London West, London 8 & 10 December 2015 Complex Business Models - Outsourced Activities Presented by: Jacqueline Masayi, GDP Inspector Background Increased complexities of the
CE Marking for Medical devices
 CE Marking of Medical Devices Bart Mersseman Head of Notified Body SGS Belgium Thursday Octobere 12 th 2017 Vlaamse dag van de CE-markering CE Marking for Medical devices Prologue: Legislation in Europe
CE Marking of Medical Devices Bart Mersseman Head of Notified Body SGS Belgium Thursday Octobere 12 th 2017 Vlaamse dag van de CE-markering CE Marking for Medical devices Prologue: Legislation in Europe
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior
 Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE
 VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE Post-Market Surveillance dal punto di vista dell O.N in vista dei nuovi Regolamenti (MDR/IVDR) Francesco
VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE Post-Market Surveillance dal punto di vista dell O.N in vista dei nuovi Regolamenti (MDR/IVDR) Francesco
Post Market Surveillance (including PMCF): common non compliances
: common non compliances") Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
MHRA GDP Symposium. Novotel London West, London 8 & 10 December #GMDPevents
 MHRA GDP Symposium Novotel London West, London 8 & 10 December 2015 Deficiencies in GDP Phil Neale, GDP Operations Manager Tony Orme, GDP Senior Inspector. Finding deficiencies in GDP is not only for those
MHRA GDP Symposium Novotel London West, London 8 & 10 December 2015 Deficiencies in GDP Phil Neale, GDP Operations Manager Tony Orme, GDP Senior Inspector. Finding deficiencies in GDP is not only for those
The Top 5 Mistakes in Making a Declaration of Conformity for CE Marking
 Intertek Cleeve Road, Leatherhead, Surrey KT22 7SB UK info.uk@intertek.com 01372 370900 www.intertek.com Contents Introduction... 2 The top 5 mistakes in making a Declaration of Conformity... 3 1. Not
Intertek Cleeve Road, Leatherhead, Surrey KT22 7SB UK info.uk@intertek.com 01372 370900 www.intertek.com Contents Introduction... 2 The top 5 mistakes in making a Declaration of Conformity... 3 1. Not
HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737)
") MDA/GL No 1: July 2013 Guidelines for implementation of medical device regulatory system HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737) [Appendix 4 Schedule 3 Medical
MDA/GL No 1: July 2013 Guidelines for implementation of medical device regulatory system HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737) [Appendix 4 Schedule 3 Medical
MEDICAL DEVICE GUIDANCE
 Effective from 1 January 2013 MEDICAL DEVICE GUIDANCE GN-15: Guidance on Medical Device Product Registration Revision 5 CONTENTS 1. INTRODUCTION... 4 1.1. Scope... 5 1.2. Definitions... 5 2. RISK CLASSIFICATION
Effective from 1 January 2013 MEDICAL DEVICE GUIDANCE GN-15: Guidance on Medical Device Product Registration Revision 5 CONTENTS 1. INTRODUCTION... 4 1.1. Scope... 5 1.2. Definitions... 5 2. RISK CLASSIFICATION
MEDICAL DEVICES : Guidance document
 EUROPEAN COMMISSION DG ENTERPRISE Directorate G Unit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES : Guidance document MEDDEV 2. 1/4 March 1994 GUIDELINES RELATING TO THE APPLICATION
EUROPEAN COMMISSION DG ENTERPRISE Directorate G Unit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES : Guidance document MEDDEV 2. 1/4 March 1994 GUIDELINES RELATING TO THE APPLICATION
European Medical Device Regulations (MDR): What To Expect
: What To Expect") European Medical Device Regulations (MDR): What To Expect MDQC March 2016 Paul Brooks Senior Vice President Healthcare Solutions Copyright 2015 BSI. All rights reserved. Sources for MDR Update Commission
European Medical Device Regulations (MDR): What To Expect MDQC March 2016 Paul Brooks Senior Vice President Healthcare Solutions Copyright 2015 BSI. All rights reserved. Sources for MDR Update Commission
The New EU IVDR. Overview of the Main Changes & Clinical Data Requirements. Advance Regulatory Consulting Ltd.
 Overview of the Main Changes & Clinical Data Requirements Advance Regulatory Consulting Ltd. : Timeline: Entry in to force Q2 (Apr) 2017 Adoption: +6m NB s apply for designation IVDs classified as Class
Overview of the Main Changes & Clinical Data Requirements Advance Regulatory Consulting Ltd. : Timeline: Entry in to force Q2 (Apr) 2017 Adoption: +6m NB s apply for designation IVDs classified as Class
GE/GN8640. Risk Evaluation and Assessment. Guidance on Planning an Application of the Common Safety Method on. Rail Industry Guidance Note
 GN Published by: Block 2 Angel Square 1 Torrens Street London EC1V 1NY Copyright 2014 Rail Safety and Standards Board Limited GE/GN8640 Method on Risk Evaluation and Assessment Issue One; June 2014 Rail
GN Published by: Block 2 Angel Square 1 Torrens Street London EC1V 1NY Copyright 2014 Rail Safety and Standards Board Limited GE/GN8640 Method on Risk Evaluation and Assessment Issue One; June 2014 Rail
MDR. Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation
 MDR Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation Suzanne Halliday, D.Phil. Jaishankar Kutty, Ph.D. Ronald Rakos, Ph.D BSI Roadshow, October 2017
MDR Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation Suzanne Halliday, D.Phil. Jaishankar Kutty, Ph.D. Ronald Rakos, Ph.D BSI Roadshow, October 2017
Regulations governing the application of medical accelerators
 Regulations governing the application of medical accelerators in 50 minutes. marko.mehle@cosylab.com 2 1.The wonderland of STANDARDS AND REGULATIONS 3 Laws and standards Medical devices (and systems) are
Regulations governing the application of medical accelerators in 50 minutes. marko.mehle@cosylab.com 2 1.The wonderland of STANDARDS AND REGULATIONS 3 Laws and standards Medical devices (and systems) are
MedDev and NB-MED Recommendations
 Titel of 2.1 Scope, field of application, explanation of terms Guidelines relating to the application of AIMD and MDD: Definition of medical devices, accessory and manufacturer Guidelines relating to the
Titel of 2.1 Scope, field of application, explanation of terms Guidelines relating to the application of AIMD and MDD: Definition of medical devices, accessory and manufacturer Guidelines relating to the
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155
 MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MDR ID: Definition: Applicable:
 MDR Classification: (Reference Medical Device Regulation EU 2017/745, Annex VIII) Product: Product Name 1. DURATION OF USE MDR ID: Definition: Applicable: - Invasive Device: Continue Go to 2. Invasive
MDR Classification: (Reference Medical Device Regulation EU 2017/745, Annex VIII) Product: Product Name 1. DURATION OF USE MDR ID: Definition: Applicable: - Invasive Device: Continue Go to 2. Invasive
Conformity Assessment of Own Brand Labelling
 NBRG/306/06 Co-ordination of Title: Chapter: Conformity Assessment of Own Brand Labelling 2.5.5 Conformity assessment for particular product groups Text: Key words: manufacturer means the natural or legal
NBRG/306/06 Co-ordination of Title: Chapter: Conformity Assessment of Own Brand Labelling 2.5.5 Conformity assessment for particular product groups Text: Key words: manufacturer means the natural or legal
GS1 Guide on Unique Device Identification (UDI) implementation
 implementation") GS1 Guide on Unique Device Identification (UDI) implementation This document aims at providing clarification to questions raised by the industry as well as implementation guidance on the use of GS1 standards.
GS1 Guide on Unique Device Identification (UDI) implementation This document aims at providing clarification to questions raised by the industry as well as implementation guidance on the use of GS1 standards.
AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5. The Declaration of Conformity
 AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5 The Declaration of Conformity 30 October 2003 DISCLAIMER This document is provided for guidance only. It should not be relied upon to address every
AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5 The Declaration of Conformity 30 October 2003 DISCLAIMER This document is provided for guidance only. It should not be relied upon to address every
GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN MAKING AN ASSESSMENT OF CLINICAL INVESTIGATION NOTIFICATION
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
Medical Device Directive
 Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
TERMS AND DEFINITIONS
 TERMS AND DEFINITIONS Advisory notice A notice issued by the organization, subsequent to delivery of the medical device, to provide supplementary information and/or to advise what action should be taken
TERMS AND DEFINITIONS Advisory notice A notice issued by the organization, subsequent to delivery of the medical device, to provide supplementary information and/or to advise what action should be taken
L 96/26 EN Official Journal of the European Union. REGULATION (EC) No 552/2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL.
 No 552/2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL.") L 96/26 EN Official Journal of the European Union REGULATION (EC) No 552/2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 10 March 2004 on the interoperability of the European Air Traffic Management
L 96/26 EN Official Journal of the European Union REGULATION (EC) No 552/2004 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 10 March 2004 on the interoperability of the European Air Traffic Management
Medical Device Epidemiology Introduction
 Medical Device Epidemiology Introduction October 24, 2014 Mary E Ritchey, PhD Pre-Conference Educational Session International Conference on Pharmacoepidemiology Course Overview Topic Time Welcome and
Medical Device Epidemiology Introduction October 24, 2014 Mary E Ritchey, PhD Pre-Conference Educational Session International Conference on Pharmacoepidemiology Course Overview Topic Time Welcome and
OPAC EN Operating Procedure for the Attestation of Conformity of Structural Bearings in compliance with Annex ZA of EN 1337/3/4/5/6/7
 OPAC EN1337-00 Operating Procedure for the Attestation of Conformity to EN1337 Revision 00 Approved by BT3 on 20050525, by Board of directors on 20050610 Operating Procedure for the Attestation of Conformity
OPAC EN1337-00 Operating Procedure for the Attestation of Conformity to EN1337 Revision 00 Approved by BT3 on 20050525, by Board of directors on 20050610 Operating Procedure for the Attestation of Conformity
POSITION PAPER Moving from the MDD to the MDR
 A summary of Key Changes regarding Sterile Packaging and considerations on recommended changes to standards Introduction EN ISO 11607 specifies the requirements and test methods for materials, preformed
A summary of Key Changes regarding Sterile Packaging and considerations on recommended changes to standards Introduction EN ISO 11607 specifies the requirements and test methods for materials, preformed
INDUSTRY S VIEW ON THE FUTURE OF IN VITRO DIAGNOSTIC (IVD) LEGISLATION IN EUROPE. May 2013 SPECIAL REPRINT. By Jesús Rueda Rodríguez, EDMA
 LEGISLATION IN EUROPE. May 2013 SPECIAL REPRINT. By Jesús Rueda Rodríguez, EDMA") May 2013 SPECIAL REPRINT INDUSTRY S VIEW ON THE FUTURE OF IN VITRO DIAGNOSTIC (IVD) LEGISLATION IN EUROPE By Jesús Rueda Rodríguez, EDMA Reproduced with the kind permission of Global Regulatory Press from
May 2013 SPECIAL REPRINT INDUSTRY S VIEW ON THE FUTURE OF IN VITRO DIAGNOSTIC (IVD) LEGISLATION IN EUROPE By Jesús Rueda Rodríguez, EDMA Reproduced with the kind permission of Global Regulatory Press from
Copyright GCI, LLC Remediation of Legacy Medical Devices
 Copyright GCI, LLC. 2016 Remediation of Legacy Medical Devices Copyright GCI, LLC. 2016 What Drives Legacy Remediation? Proactive Recertification / CE Mark Audit Readiness M&A / Due Diligence Reactive
Copyright GCI, LLC. 2016 Remediation of Legacy Medical Devices Copyright GCI, LLC. 2016 What Drives Legacy Remediation? Proactive Recertification / CE Mark Audit Readiness M&A / Due Diligence Reactive
Surveillance and Medical Devices
 Surveillance and Medical Devices Nicole Pratt Senior Research Fellow Medicine and Device Centre for Research Excellence Contact: Prof Libby Roughead UniSA, GPO Box 2471, Adelaide SA 5001 Libby.roughead@unisa.edu.au
Surveillance and Medical Devices Nicole Pratt Senior Research Fellow Medicine and Device Centre for Research Excellence Contact: Prof Libby Roughead UniSA, GPO Box 2471, Adelaide SA 5001 Libby.roughead@unisa.edu.au
Dangers of Over-Regulation (or Under- Regulation) of Genetic Testing at EU level. David Barton
 of Genetic Testing at EU level. David Barton") Dangers of Over-Regulation (or Under- Regulation) of Genetic Testing at EU level David Barton Department of Clinical Genetics Our Lady s Children s Hospital Dublin Background Diagnostic tests, known as
Dangers of Over-Regulation (or Under- Regulation) of Genetic Testing at EU level David Barton Department of Clinical Genetics Our Lady s Children s Hospital Dublin Background Diagnostic tests, known as
New Pharmacovigilance legislation. Post-authorisation safety studies. ENCePP Plenary meeting. 3 May 2012
 New Pharmacovigilance legislation Post-authorisation safety studies ENCePP Plenary meeting 3 May 2012 Presented by: Annalisa Rubino, PhV and Risk Management, EMA An agency of the European Union Why? Public
New Pharmacovigilance legislation Post-authorisation safety studies ENCePP Plenary meeting 3 May 2012 Presented by: Annalisa Rubino, PhV and Risk Management, EMA An agency of the European Union Why? Public
Regulatory and ethical requirements in medical device studies. Finland
 Regulatory and ethical in medical device studies Finland SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Regulatory and ethical in medical device studies Finland SECTIONS A.Type of research SECTIONS A.Type of research We have differentiated 8 types of research: Medical device alone with CE mark use within
Flexible, robust solutions from BSI. An In Vitro Diagnostic Notified Body. Expertise and experience. IVD regulatory solutions
 Flexible, robust solutions from BSI An In Vitro Diagnostic Notified Body Expertise and experience IVD regulatory solutions Updated May 2017 Your guide to the In Vitro Diagnostic Directive The purpose of
Flexible, robust solutions from BSI An In Vitro Diagnostic Notified Body Expertise and experience IVD regulatory solutions Updated May 2017 Your guide to the In Vitro Diagnostic Directive The purpose of
Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations
 Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations Paul Brooks Vice President Healthcare, Americas Presentation to:
Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations Paul Brooks Vice President Healthcare, Americas Presentation to:
TUV SUD BABT PRODUCTION QUALITY CERTIFICATION SCHEME
 TUV SUD BABT PRODUCTION QUALITY CERTIFICATION SCHEME Copyright TUV SUD BABT 2017 A Certification Body of: Page 1 of 33 Contents AMENDMENT RECORD 3 0. INTRODUCTION 3 1. PRE-CONDITIONS TO SUPPORT PRODUCTION
TUV SUD BABT PRODUCTION QUALITY CERTIFICATION SCHEME Copyright TUV SUD BABT 2017 A Certification Body of: Page 1 of 33 Contents AMENDMENT RECORD 3 0. INTRODUCTION 3 1. PRE-CONDITIONS TO SUPPORT PRODUCTION
By: Clay Anselmo COO, DLLS, Founder Reglera and Terry J. Dagnon M.S., Vice President Regulatory Affairs, DLSS
 AN INTRODUCTION TO THE WORLDWIDE REGULATORY FRAMEWORK FOR MEDICAL DEVICES By: Clay Anselmo COO, DLLS, Founder Reglera and Terry J. Dagnon M.S., Vice President Regulatory Affairs, DLSS COURSE OUTLINE /
AN INTRODUCTION TO THE WORLDWIDE REGULATORY FRAMEWORK FOR MEDICAL DEVICES By: Clay Anselmo COO, DLLS, Founder Reglera and Terry J. Dagnon M.S., Vice President Regulatory Affairs, DLSS COURSE OUTLINE /
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS
 FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
Guideline on good pharmacovigilance practices (GVP)
") 22 June 2012 EMA/816573/2011 Guideline on good pharmacovigilance practices (GVP) Module II Pharmacovigilance system master file Draft finalised by the Agency in collaboration with Member States and submitted
22 June 2012 EMA/816573/2011 Guideline on good pharmacovigilance practices (GVP) Module II Pharmacovigilance system master file Draft finalised by the Agency in collaboration with Member States and submitted
Fourth Stakeholders forum on the implementation of the new Pharmacovigilance legislation
 Fourth Stakeholders forum on the implementation of the new Pharmacovigilance legislation Module II - Pharmacovigilance system master file Presented by: Joanna Harper Inspections, Enforcement & Standards
Fourth Stakeholders forum on the implementation of the new Pharmacovigilance legislation Module II - Pharmacovigilance system master file Presented by: Joanna Harper Inspections, Enforcement & Standards
National TFS Office, Dublin City Council [NTFSO] Guidance for Completing Notification Document (Annex 1A) & Movement Document (Annex 1B)
![National TFS Office, Dublin City Council [NTFSO] Guidance for Completing Notification Document (Annex 1A) & Movement Document (Annex 1B)](/thumbs/75/72492936.jpg "National TFS Office, Dublin City Council [NTFSO] Guidance for Completing Notification Document (Annex 1A) & Movement Document (Annex 1B)") National TFS Office, Dublin City Council [NTFSO] Guidance for Completing Notification Document (Annex 1A) & Movement Document (Annex 1B) Introduction A planned shipment subject to the procedure of prior
National TFS Office, Dublin City Council [NTFSO] Guidance for Completing Notification Document (Annex 1A) & Movement Document (Annex 1B) Introduction A planned shipment subject to the procedure of prior
The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective
 The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective Almath Spooner, Irish Medicines Board Pharmaceutical Society of Ireland National Pharmacy Summit, November 2008. Presentation Topics
The Role of the Pharmacist in Pharmacovigilance A Regulatory Perspective Almath Spooner, Irish Medicines Board Pharmaceutical Society of Ireland National Pharmacy Summit, November 2008. Presentation Topics
CAMD Implementation Taskforce
 CAMD Implementation Taskforce Medical Devices Regulation/In-vitro Diagnostics Regulation (MDR/IVDR) Roadmap Overview and proposed terms of reference The Competent Authorities for Medical Devices (CAMD)
CAMD Implementation Taskforce Medical Devices Regulation/In-vitro Diagnostics Regulation (MDR/IVDR) Roadmap Overview and proposed terms of reference The Competent Authorities for Medical Devices (CAMD)
Serious Adverse Event Reporting During European Device Clinical Investigations
 regulations and standards Serious Adverse Event Reporting During European Device Clinical Investigations In December 2010, two new European guidelines on medical device clinical studies were published.
regulations and standards Serious Adverse Event Reporting During European Device Clinical Investigations In December 2010, two new European guidelines on medical device clinical studies were published.
Mircea Ciuca, MD Global Head Medical & Clinical Drug Safety
 Mircea Ciuca, MD Global Head Medical & Clinical Drug Safety Disclaimer The views and opinions expressed in this presentation are solely those of the presenter and do not necessarily reflect those of Vifor,
Mircea Ciuca, MD Global Head Medical & Clinical Drug Safety Disclaimer The views and opinions expressed in this presentation are solely those of the presenter and do not necessarily reflect those of Vifor,
Accessories and other parts for Active Implantable Medical Devices
 Notified Bodies Medical Chapter: 2.1 Scope, field of application, explanation of terms Text: Key words: accessories, spare parts, labelling 1 Introduction and purpose With the application of the provisions
Notified Bodies Medical Chapter: 2.1 Scope, field of application, explanation of terms Text: Key words: accessories, spare parts, labelling 1 Introduction and purpose With the application of the provisions
MEDICAL DEVICES : Guidance document
 EUROPEAN COMMISSION DG ENTERPRISE Directorate G Unit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES : Guidance document MEDDEV 2. 1/2 rev 2 26 April 1994 GUIDELINES RELATING TO THE
EUROPEAN COMMISSION DG ENTERPRISE Directorate G Unit 4 - Pressure Equipment, Medical Devices, Metrology MEDICAL DEVICES : Guidance document MEDDEV 2. 1/2 rev 2 26 April 1994 GUIDELINES RELATING TO THE
Understanding Clinical Equivalence
 Understanding Clinical Equivalence BSI 2014 Medical Device Mini-Roadshow Ibim Tariah Ph.D Technical Director, Healthcare Solutions 1 Understanding Clinical Equivalence Review Requirements from Directives
Understanding Clinical Equivalence BSI 2014 Medical Device Mini-Roadshow Ibim Tariah Ph.D Technical Director, Healthcare Solutions 1 Understanding Clinical Equivalence Review Requirements from Directives
FDA s Final Rule on Medical Device Data Systems Signals Active Regulation of Data Communication Technologies
 ADVISORY February 2011 FDA s Final Rule on Medical Device Data Systems Signals Active Regulation of Data Communication Technologies Contacts On February 15, 2011, the U.S. Food and Drug Administration
ADVISORY February 2011 FDA s Final Rule on Medical Device Data Systems Signals Active Regulation of Data Communication Technologies Contacts On February 15, 2011, the U.S. Food and Drug Administration
Product Safety and Market Surveillance Package
 Product Safety and Market Surveillance Package Prague, 25 September 2013 European Commission The Package: Communication on More Product Safety and Better Market Surveillance in the Single Market Proposal
Product Safety and Market Surveillance Package Prague, 25 September 2013 European Commission The Package: Communication on More Product Safety and Better Market Surveillance in the Single Market Proposal
LABORATORY TRAINING LOGBOOK
 REGISTRATION TRAINING PORTFOLIO FOR THE IBMS CERTIFICATE OF COMPETENCE LABORATORY TRAINING LOGBOOK Version 4.1 www.ibms.org Trainee record details Registration Training Portfolio Case No: Surname: First
REGISTRATION TRAINING PORTFOLIO FOR THE IBMS CERTIFICATE OF COMPETENCE LABORATORY TRAINING LOGBOOK Version 4.1 www.ibms.org Trainee record details Registration Training Portfolio Case No: Surname: First
International Standards and EU regulation of medical device software an update
 International Standards and EU regulation of medical device software an update Sherman Eagles Partner, SoftwareCPR seagles@softwarecpr.com 612 865 0107 1 Who am I? 18 years at Medtronic, retired 2008 Last
International Standards and EU regulation of medical device software an update Sherman Eagles Partner, SoftwareCPR seagles@softwarecpr.com 612 865 0107 1 Who am I? 18 years at Medtronic, retired 2008 Last
Conformity Assessments Revised Toy Safety Directive 2009/48/EC
 Toy Safety Update Conformity Assessments Revised Toy Safety Directive 2009/48/EC Contents 1. Obligations of Economic Operators 2. Introduction 3. Overview 4. How to use this guide 5. The Conformity Assessment
Toy Safety Update Conformity Assessments Revised Toy Safety Directive 2009/48/EC Contents 1. Obligations of Economic Operators 2. Introduction 3. Overview 4. How to use this guide 5. The Conformity Assessment
THE EUROPEAN PARLIAMENT AND THE COUNCIL OF THE EUROPEAN UNION,
 Regulation (EC) No 1071/2009 of the European Parliament and of the Council of 21 October 2009 establishing common rules concerning the conditions to be complied with to pursue the occupation of road transport
Regulation (EC) No 1071/2009 of the European Parliament and of the Council of 21 October 2009 establishing common rules concerning the conditions to be complied with to pursue the occupation of road transport
FINAL DOCUMENT. Global Harmonization Task Force (revision of GHTF/SG1/N29:2005)
") GHTF/SG1/N071:2012 FINAL DOCUMENT Global Harmonization Task Force (revision of GHTF/SG1/N29:2005) Title: Definition of the Terms Medical Device and In Vitro Diagnostic (IVD) Medical Device Authoring Group:
GHTF/SG1/N071:2012 FINAL DOCUMENT Global Harmonization Task Force (revision of GHTF/SG1/N29:2005) Title: Definition of the Terms Medical Device and In Vitro Diagnostic (IVD) Medical Device Authoring Group:
UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS
 UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS Executive Summary European Medicines Agency (EMA) regulations for licensing of medical devices include the use of authorized
UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS Executive Summary European Medicines Agency (EMA) regulations for licensing of medical devices include the use of authorized
Clarifying digital health and software regulation: FDA releases three new guidance documents
 Clarifying digital health and software regulation: FDA releases three new guidance documents December 15, 2017 On December 7, 2017, the Food and Drug Administration (FDA or the Agency) released three guidance
Clarifying digital health and software regulation: FDA releases three new guidance documents December 15, 2017 On December 7, 2017, the Food and Drug Administration (FDA or the Agency) released three guidance
CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT
 TF N 548 Rev1 2012-03-29 CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT How to draft clauses on Assessment and Verification of the Constancy of Performance (AVCP) in harmonized standards for construction
TF N 548 Rev1 2012-03-29 CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT How to draft clauses on Assessment and Verification of the Constancy of Performance (AVCP) in harmonized standards for construction
The Risk Management + Design Controls Connection: What Device Makers Need to Know
 !!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
!!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC
 Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC Institute for Testing and Certification, Inc., Czech Republic 1. Introduction
Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC Institute for Testing and Certification, Inc., Czech Republic 1. Introduction
Your complimentary Clinica Medtech Intelligence content
 Your complimentary Clinica Medtech Intelligence content Clinica provides you with up to the minute coverage and opinion on company, product, market and regulatory developments as well as market size, growth
Your complimentary Clinica Medtech Intelligence content Clinica provides you with up to the minute coverage and opinion on company, product, market and regulatory developments as well as market size, growth
National laws. National Competent Authority / Authorities. Orders, ordinances (supervision)
") 1 The Regulatory World Only for products classified higher than class I: Notification of the body, Supervision National laws National Competent Authority / Authorities Orders, ordinances (supervision)
1 The Regulatory World Only for products classified higher than class I: Notification of the body, Supervision National laws National Competent Authority / Authorities Orders, ordinances (supervision)
Clinical evaluation report,
 Clinical Evaluation Reports from the medical writer s perspective! Gillian Pritchard Sylexis Limited, Dundee, Scotland, UK Correspondence to: Dr Gillian Pritchard Sylexis Limited 30/34 Reform Street Dundee,
Clinical Evaluation Reports from the medical writer s perspective! Gillian Pritchard Sylexis Limited, Dundee, Scotland, UK Correspondence to: Dr Gillian Pritchard Sylexis Limited 30/34 Reform Street Dundee,
Guideline on good pharmacovigilance practices (GVP)
") 1 2 3 19 June 2012 EMA/119871/2012 4 5 Guideline on good pharmacovigilance practices (GVP) Module III Pharmacovigilance inspections Draft finalised by the Agency in collaboration with Member States and
1 2 3 19 June 2012 EMA/119871/2012 4 5 Guideline on good pharmacovigilance practices (GVP) Module III Pharmacovigilance inspections Draft finalised by the Agency in collaboration with Member States and
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL GUIDELINES ON MEDICAL DEVICES
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Competitiveness in pharmaceuticals, medical devices, cosmetics MEDDEV. 2.11/1 rev.1 April 2005 GUIDELINES ON MEDICAL DEVICES
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Competitiveness in pharmaceuticals, medical devices, cosmetics MEDDEV. 2.11/1 rev.1 April 2005 GUIDELINES ON MEDICAL DEVICES
AAMI Quality Systems White Paper: Comparison of 21 CFR Part 820 to ISO 13485:2016 1
 AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
INSTRUCTIONS FOR CERTIFICATION OF FACTORY PRODUCTION CONTROL
 . August 2017 were approved by Manager of Product Certification Bureau of the Polish Register of Shipping on 25 August 2017 Copyright by, 2017. GDAŃSK, AUGUST 2017 1/12 CONTENTS 1. Factory Production Control
. August 2017 were approved by Manager of Product Certification Bureau of the Polish Register of Shipping on 25 August 2017 Copyright by, 2017. GDAŃSK, AUGUST 2017 1/12 CONTENTS 1. Factory Production Control
The rules governing medicinal products in the European Union. Presentation and content of the dossier Edition
 The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
Guideline on good pharmacovigilance practices (GVP)
") 9 October 2017 EMA/813938/2011 Rev 3* Guideline on good pharmacovigilance practices (GVP) Module VIII Post-authorisation safety studies (Rev 3) Date for coming into effect of first version 2 July 2012
9 October 2017 EMA/813938/2011 Rev 3* Guideline on good pharmacovigilance practices (GVP) Module VIII Post-authorisation safety studies (Rev 3) Date for coming into effect of first version 2 July 2012
TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE MANAGEMENT Q12
 INTERNATIONAL CONCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE
INTERNATIONAL CONCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE
IMQ REGULATION FOR THE CERTIFICATION OF MEDICAL DEVICES CE Marking Directive 93/42/EEC
 Title Reference Review and date of entry into force Approved by IMQ REGULATION FOR THE CERTIFICATION OF MEDICAL DEVICES CE Marking Directive 93/42/EEC Reg. IMQ/ON/MDD_E Rev. 1 dated 12/05/2017 IMQ S.p.A.
Title Reference Review and date of entry into force Approved by IMQ REGULATION FOR THE CERTIFICATION OF MEDICAL DEVICES CE Marking Directive 93/42/EEC Reg. IMQ/ON/MDD_E Rev. 1 dated 12/05/2017 IMQ S.p.A.
ISO INTERNATIONAL STANDARD. Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process
 INTERNATIONAL STANDARD ISO 10993-1 Fourth edition 2009-10-15 Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process Évaluation biologique des dispositifs
INTERNATIONAL STANDARD ISO 10993-1 Fourth edition 2009-10-15 Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process Évaluation biologique des dispositifs
Guidance on Independent Assessment. Rail Industry Guidance Note. Published by: RSSB Block 2 Angel Square 1 Torrens Street London EC1V 1NY
 GN Published by: Block 2 Angel Square 1 Torrens Street London EC1V 1NY Copyright 2014 Rail Safety and Standards Board Limited GE/GN8645 Issue One: June 2014 Rail Industry Guidance Note Issue record Issue
GN Published by: Block 2 Angel Square 1 Torrens Street London EC1V 1NY Copyright 2014 Rail Safety and Standards Board Limited GE/GN8645 Issue One: June 2014 Rail Industry Guidance Note Issue record Issue
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
Writing an Assessment Report
 Safeguarding public health Writing an Assessment Report Name: Malcolm Dash Date: Programme Why we need Assessment Reports Writing style Deficiency points Potential Serious Risk to Public Health Targeted
Safeguarding public health Writing an Assessment Report Name: Malcolm Dash Date: Programme Why we need Assessment Reports Writing style Deficiency points Potential Serious Risk to Public Health Targeted
(Non-legislative acts) REGULATIONS
 REGULATIONS") 11.12.2010 Official Journal of the European Union L 327/13 II (Non-legislative acts) REGULATIONS COMMISSION REGULATION (EU) No 1169/2010 of 10 December 2010 on a common safety method for assessing conformity
11.12.2010 Official Journal of the European Union L 327/13 II (Non-legislative acts) REGULATIONS COMMISSION REGULATION (EU) No 1169/2010 of 10 December 2010 on a common safety method for assessing conformity
-Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009
 No.1394/2007 -Regulation (EC) No. 668/2009") Introduction to Advanced Therapy Medicinal Products Regulation -Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009 -Directive 2009/120/EC Dr. Maura O Donovan F.R.C.O.G. MA MD M.R.C.P.I. CAT member
Introduction to Advanced Therapy Medicinal Products Regulation -Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009 -Directive 2009/120/EC Dr. Maura O Donovan F.R.C.O.G. MA MD M.R.C.P.I. CAT member
LEARNING, TRAINING, AND PERFORMANCE
 9 Overview Learning, Training, and Performance Training is one of the most important investments a firm can make as it produces products that meet GMP expectations. Training is not done just for the sake
9 Overview Learning, Training, and Performance Training is one of the most important investments a firm can make as it produces products that meet GMP expectations. Training is not done just for the sake
Market surveillance of medical devices
 Market surveillance of medical devices A Joint Action to reinforce public health protection and medical devices monitoring by implementing joint manufacturer inspections and improving clinical process
Market surveillance of medical devices A Joint Action to reinforce public health protection and medical devices monitoring by implementing joint manufacturer inspections and improving clinical process
Expert Commentary on BS EN ISO 13485:2016, Medical devices Quality management systems Requirements for regulatory purposes
 Expert Commentary on BS EN ISO 13485:2016, Medical devices Quality management systems Requirements for regulatory purposes Author: Eamonn Hoxey, PhD, F.R.Pharm.S., Vice President, Medical Devices Quality
Expert Commentary on BS EN ISO 13485:2016, Medical devices Quality management systems Requirements for regulatory purposes Author: Eamonn Hoxey, PhD, F.R.Pharm.S., Vice President, Medical Devices Quality
COUNTRY OVERVIEW: THAILAND. August 2012 SPECIAL REPRINT. By Rarana Phanudulkitti
 August 2012 SPECIAL REPRINT COUNTRY OVERVIEW: THAILAND By Rarana Phanudulkitti Reproduced with the kind permission of Global Regulatory Press from the Journal of Medical Device Regulation, 2012, 9(3),
August 2012 SPECIAL REPRINT COUNTRY OVERVIEW: THAILAND By Rarana Phanudulkitti Reproduced with the kind permission of Global Regulatory Press from the Journal of Medical Device Regulation, 2012, 9(3),
VOLUME 4 Good manufacturing practices ANNEX 13 Manufacture of investigational medicinal products JULY 2003
 EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single market : management & legislation for consumer goods Pharmaceuticals : regulatory framework and market authorisations Brussels, F2/BL D(2003) Revision
EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single market : management & legislation for consumer goods Pharmaceuticals : regulatory framework and market authorisations Brussels, F2/BL D(2003) Revision
White Paper: Improvements to the Australian Regulatory System for Medical Devices. 23 May 2014
 White Paper: Improvements to the Australian Regulatory System for Medical Devices 23 May 2014 mtaa.org.au Medical technology for a healthier Australia 0 www.mtaa.org.au Level 12, 54 Miller St, North Sydney
White Paper: Improvements to the Australian Regulatory System for Medical Devices 23 May 2014 mtaa.org.au Medical technology for a healthier Australia 0 www.mtaa.org.au Level 12, 54 Miller St, North Sydney
Updated EU Medical Device Regula2ons: What s In, What s Out, and What s Hot?
 Updated EU Medical Device Regula2ons: What s In, What s Out, and What s Hot? Introduc2on EU Medical Device Overview Current Medical Device Direc2ves ü Governed by 3 directives (past 15-20 yrs): Polling
Updated EU Medical Device Regula2ons: What s In, What s Out, and What s Hot? Introduc2on EU Medical Device Overview Current Medical Device Direc2ves ü Governed by 3 directives (past 15-20 yrs): Polling