Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE
|
|
|
- Ruby Charles
- 6 years ago
- Views:
Transcription
, National")

1 Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract No. N01-AI-50022, entitled HIV Clinical Research Support (CRS) Services.
2 Agenda 8:00a 8:10a Introduction and Course Objectives 8:10a 9:00a Section I - Overview of US FDA and the Inspection Process - Inspection Preparation and Readiness - Using FDA Resources 9:00a 9:10a Break 9:10a 10:20a Section II - OCSO Trend Analyses Overview - Quality (QM) and the Clinical Quality Plan (CQMP) - Roles and Responsibilities - Key Quality Indicators 10:20a 10:30a Break 10:30a 11:50a - QM Implementation and Documentation - Problems and Trends Found Now What? 11:50a 12:00p Conclusion
3 Objectives At the conclusion of this presentation, VOICE site staff will be able to: Explain the FDA inspection process Identify FDA inspection documents and outline the purpose and flow of each Identify areas for improvement from the site-specific trend analysis that may require attention prior to an FDA inspection Identify resources available to the site and how to use them in preparation for an FDA inspection 3
4 Objectives Continued Describe the relationship between quality management activities and FDA inspection preparedness Implement quality management techniques that will assist with identification of areas for improvement in preparation for a possible FDA inspection
 and FDA Inspection")
5 Overview of the United States Food and Drug Administration (FDA) and FDA Inspection Process
6 FDA US Government Executive Branch Other Departments Department of Health and Human Services Food and Drug Administration Other Agencies The FDA is an agency within the Department of Health and Human Services (DHHS) of the US Government. 6

7 FDA s Mission Protecting Public Health Assuring safety, efficacy, and security of human drugs, biological products, and medical devices, as well as veterinary drugs, cosmetics, products that emit radiation, and the US food supply. Advancing Public Health 7
 Center for")
 Center for Devices & Radiological Health (CDRH) Center for Biologics Evaluation Research (CBER) Center for")
8 FDA Today Office of the Commissioner Office of Special Medical Programs Office of External Affairs Office of Foods Office of the Chief Scientist Office of International Programs Office of Administration Office of Equal Employment Office of Regulatory Affairs (ORA) Center for Food Safety & Applied Nutrition (CFSAN) Center for Veterinary Medicine (CVM) National Center for Toxicological Research (NCTR) Center for Drug Evaluation and Research (CDER) Center for Devices & Radiological Health (CDRH) Center for Biologics Evaluation Research (CBER) Center for Tobacco Products

9 Bioresearch Monitoring (BIMO) Overview Bioresearch Monitoring (BIMO) Program An FDA program designed to monitor all aspects of the conduct and reporting of FDA-regulated research by conducting on-site inspections and data audits Inspections are conducted domestically and internationally Over 1000 inspections are conducted annually Routine and directed inspections /ucm htm 9
10 Bioresearch Monitoring (BIMO) Program Non-Clinical Laboratories Clinical Investigator Sites Principal Investigators BIMO conducts on-site inspections of: Institutional Review Boards/Ethics Committees (IRB/EC) Sponsors Contract Research Organizations (CROs) 10
 *Based on inspection start date DSI database")
![[4/01/2011] IRB/RDRC include some CBER/CDRH related](/docs-images/77/75758860/images/11-6.jpg "Inspections http://www.fda.")
11 Bioresearch Monitoring Program Inspections* (CDER, FY ) *Based on inspection start date DSI database [4/01/2011] IRB/RDRC include some CBER/CDRH related Inspections 11

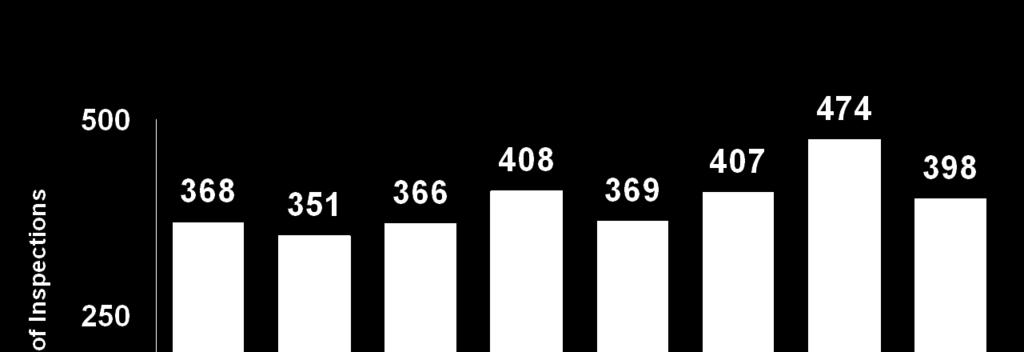
12 Clinical Investigator Inspections (CDER, FY ) 12


13 Routine vs. Directed Inspections Routine Inspection Large volume of work Efficacy too good Toxicity too low Out-of-range laboratory results Pivotal study Submitted for new drug application Site has singular importance Directed Inspection Past FDA inspection history Whistleblower reports Complaints FDA/Sponsor raised issues High protocol noncompliance Large volume of clinical trials Work outside of specialty Inconsistent safety reporting Unusually high enrollment High number of subjects with disease
14 Purpose of Inspections Ensure protection of human subjects Verify data Quality and integrity Verify compliance with regulations and GCP guidance Verify control of study product Facilitate sound decision making Safety and efficacy
15 Regulations Title 21 Code of Federal Regulations (CFR) Part 11 Part 50 Part 54 FINANCIAL DISCLOSURE BY CLINICAL INVESTIGATORS Part 56 Part 312
16 International Inspections 40%-65% of clinical studies investigating FDAregulated products are conducted outside of the US In 2008, 80% of marketing applications received by the FDA contained data from international clinical studies 78% of the participants involved in the studies supporting these applications were enrolled at international sites 54% of the clinical sites conducting these studies were located outside the US Challenges to FDA s Ability to Monitor and Inspect Foreign Clinical Trials, DHHS, June


")

17 International Clinical Investigator Inspections (CDER, FY ) 17
18 What Do You Think? Why might a VOICE site be inspected by the FDA?
19 VOICE-Specific FDA Inspections Large volume of work (>5000 participants) High enrollment Pivotal study for tenofovir gel and supplemental marketing application for Truvada to the FDA International sites International inspections are generally assigned when the studies covered are part of a marketing application to FDA and provide data critical to decision-making on product approval. FDA Compliance Program Guidance Manual Manual/ucm htm


20 Overview of the FDA Inspection Process Three Distinct Phases of an FDA Inspection: Phase 1 Before Phase 2 During Phase 3 After
21 FDA Inspection Process Role Site Task Before During After Implement inspection preparation activities Perform assigned roles and responsibilities and provide information to the Inspector Review observations identified during the inspection and respond to the FDA in writing (if applicable) Inspector Review all information regarding the upcoming inspection Evaluate site practices and procedures to determine compliance with applicable regulations May or may not issue FDA Form 483 Document inspectional observations in the final report and forward to FDA management FDA Determine the site to be inspected Act as a resource to the on-site Inspector by providing guidance and clarification Review the final report from the Inspector and determine appropriate actions and/or consequences
 Guidelines, E6 Section 4.")
22 Investigator Responsibilities The Principal Investigator is ultimately responsible for all study-related activities at his or her site, regardless of who has been delegated the various study-related activities References discussing Investigator Responsibilities: International Conference on Harmonisation (ICH) Guidelines, E6 Section 4.0 FDA 21 CFR 312 (Subpart D) FDA Guidance for Industry Investigator Responsibilities Protecting the Rights, Safety, and Welfare of Study Subjects (October 2009) ces/ucm pdf Institutional, local, state, and national regulations




23 Key Documents Form FDA 1572 Statement of Investigator The statement completed by the Study Investigator that he/she will abide by the federal guidelines set forth in the Code of Federal Regulations for the use of investigational products in the research setting Form FDA 482 Notice of Inspection Form FDA 483 Inspectional Observations

24 Key Documents Establishment Inspection Report The report written by the FDA Inspector that describes the observational findings of the inspection FDA Warning Letter Close-Out Letter A letter that may be issued when, based on the FDA s evaluation, the firm has taken corrective action to address the violations contained in the Warning Letter
25 Inspection Preparedness and Readiness
26 Inspection Preparedness and Readiness What is it? To be inspection ready at all times! Why now? So issues are identified and corrected long before the site gets an audit notification Where to begin? With an effective quality management plan! Future Steps FDA Inspection Checklist FDA Inspection Preparation Team
27 Using FDA Resources

28
29 29
30 FDA Website Guidance Documents Information Sheet Guidance For IRBs, Clinical Investigators, and Sponsors: FDA Inspection of Clinical Investigators CM pdf Database of FDA Form 483 Findings Warning Letters ult.htm Clinical Trials Section ltrials/default.htm
31 Warning Letter Findings for investigators You failed to ensure that the investigation was conducted according to the investigational plan [21 CFR ] 5 investigators You failed to personally conduct or supervise the clinical investigation [21 CFR ] 5 investigators You failed to prepare and maintain adequate and accurate case histories that record all observations and other data pertinent to the investigation on each individual administered the investigational drug or employed as a control in the investigation [21 CFR (b)]
32 Warning Letter Findings for investigators You failed to report promptly to the IRB all changes in the research activity [21 CFR ] 2 investigators You failed to obtain informed consent of each subject in accordance with the provisions of 21 CFR part 50 [21 CFR ] 2 investigators You failed to prepare and maintain adequate and accurate case histories that record all observations and other data pertinent to the investigation on each individual administered the investigational drug or employed as a control in the investigation [21 CFR (b)] 1 investigator You failed to maintain adequate records of the disposition of the drug, including dates, quantity, and use by subjects [21 CFR (a)]
33 Questions?
34 Section II: OCSO Trend Analyses and Quality This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract No. N01-AI-50022, entitled HIV Clinical Research Support (CRS) Services.
35 Objectives for Section II Identify areas for improvement from the site-specific trend analysis that may require attention prior to an FDA inspection Describe the relationship between quality management activities and FDA inspection preparedness Identify resources available to the site and how to use them in preparation for an FDA inspection Implement quality management techniques that will assist with identification of areas for improvement in preparation for possible FDA inspection 35
36 OCSO Trend Analysis Overview To identify trends from findings reported in the PPD CSSM Site Monitoring Reports. Including Clinical, Regulatory, Pharmacy and Lab To distribute information obtained from the trend analysis for possible corrective and preventive actions, as well as evaluation and modification of current CQMP processes and site-specific SOPs 36

37 VOICE Enrolled versus Monitored

38 Total Sample Size Monitored = 16% Total Enrollment 5027 Monitored Not monitored What about the other 84%?
 676 Failure to maintain adequate or accurate case histories 171 Failure to complete assessments or procedures as required by the protocol Failure to")
 4 Failure to follow the Inclusion/Exclusion criteria or timelines for enrollment as required by the")
39 Monitoring Trends As of September 14, 2011 Monitoring Trends Findings Warning Letter Examples Procedural Inadequate Source Documentation (A72) Missed Tests/Study Procedures (A14 [inadeq] = 46, A24 [adeq] = 125) 676 Failure to maintain adequate or accurate case histories 171 Failure to complete assessments or procedures as required by the protocol Failure to repeat labs as required by the protocol Unreported Adverse Events (A62) 23 Failure to conduct the studies or ensure they were conducted according to the investigational plans, and to protect the rights, safety, and welfare of subjects Enrollment Violations (A2) 4 Failure to follow the Inclusion/Exclusion criteria or timelines for enrollment as required by the protocol 16%
40 Site Reported Protocol Deviations As of September 12, 2011 MTN-003 Protocol Deviations Findings Warning Letter Examples Omitted Study Procedures 83 Failure to complete assessments or procedures as required by the protocol Missed Repeat Labs 32 Failure to complete assessments or procedures as required by the protocol Study Product Errors Study Product Hold (17) Study Product Dispensing (14) 31 Failure to adequately document study drug accountability or failure to hold the study drug as required by the protocol Enrollment Deviations 26 Failure to follow the Inclusion/Exclusion criteria or timelines for enrollment as required by the protocol
41 Objective Check Identify areas for improvement from the sitespecific trend analysis that may require attention prior to an FDA inspection 41





42 Big Picture

43 How could one define Quality in the clinic research setting? The reliability and confidence of the data collected for the purpose of answering a scientific medical question while complying with ethical principles, clinical research trial requirements, and the laws and regulations governing human subject clinical research.


44 Protocol 44



45 45
46 Ethical Principles Regulatory Informed Consent Eligibility Laws and Regulations ICH GCP/GLP Data Study Procedures Sponsor Requirements Laboratory Evaluations Clinical Study Product 46
47 NIAID/DAIDS Ethical Principles Laws and Regulations ICH GCP/GLP Data Regulatory Informed Consent Eligibility Study Procedures CORE FHI SCHARP SDMC Network Laboratory IRB/EC Sponsor Requirements Laboratory Evaluations Clinical Study Product PPD CSSM 47
48 Quality Plan(s) NIAID/DAIDS Ethical Principles Laws and Regulations ICH GCP/GLP Data Regulatory Informed Consent Eligibility Study Procedures CORE FHI SCHARP SDMC Network Laboratory IRB/EC Sponsor Requirements Laboratory Evaluations Clinical Study Product PPD CSSM 48
49 Quality Plan(s) Quality NIAID/DAIDS Ethical Principles Laws and Regulations ICH GCP/GLP Data Regulatory Informed Consent Eligibility Study Procedures MTN/CORE FHI SCHARP SDMC Network Laboratory IRB/EC Sponsor Requirements Laboratory Evaluations Clinical Study Product PPD CSSM FDA Inspection Preparedness 49
50 Quality Quality Control Quality Assurance Quality Plan(s) 50 50
51 Basic Elements of a Quality Plan Responsibilities Quality Control Quality Assurance Key Quality Indicators Tools Evaluation Reporting 51
52 Benefits to Quality To ensure participant safety Verify accuracy of data; reduce error rates Identify areas in need of corrective action Assure compliance with study requirements Prepared for an external audit, monitoring visit, or FDA Inspection FDA Inspection Preparedness 52
53 Objective Check Describe the relationship between quality management activities and FDA inspection preparedness 53
54 Roles and Responsibilities

55 Informed Consent Regulatory Eligibility Data Study Procedures Laboratory Evaluations Study Product Investigator of Record Clinical An investigator is responsible for ensuring that an investigation is conducted according to the signed investigator statement [FDA Form 1572], the investigational plan, and applicable regulations; for protecting the rights, safety, and welfare of the subjects under the investigator s care; and for the control of the drugs under investigation. 21 CFR
56 Structure Investigator of Record Informed Consent Regulatory Eligibility Data Study Procedures Clinical Research Site Laboratory Evaluations Clinical Study Product Laboratory Pharmacy 56
57 Study Team Investigator of Record Informed Consent Regulatory Eligibility Data Study Procedures Clinical Research Coordinator Clinical Research Site Laboratory Evaluations Clinical Study Product Laboratory Laboratory Director Pharmacy Pharmacist of Record 57


58 Quality Informed Consent Investigator of Record Regulatory Eligibility QMP Data Study Procedures QMP Clinical Research Site Laboratory Evaluations Clinical Study Product Laboratory QMP Pharmacy 58
59 References Clinical Pharmacy DAIDS Policy: Requirements for Clinical Quality Plans MTN-003 SSP Manual: Section 16.1 Site Quality Plans Pharmacy Guidelines and Instructions for DAIDS Clinical Trials Networks: Responsibilities of the Pharmacist of Record Quality Laboratory DAIDS Guidelines for Good Clinical Laboratory Practice Standards: Quality HIV Prevention Trials Network (HPTN)-MTN Laboratory Manual: MTN Laboratory Quality Assessment and Quality Control Program 59
60 Cross-functional Communication Example During a periodic pharmacy quality assurance activity, the reviewer identifies multiple prescriptions written and signed by an unauthorized prescriber in the clinic. These prescriptions were subsequently filled by pharmacy staff. Problem 1: Unauthorized Prescriber - Clinic Problem 2: Filling prescriptions from unauthorized prescriber - Pharmacy Problem 3: Communication Clinic and Pharmacy 60
61 Key Quality Indicators

62 Key Quality Indicators Defined Performance areas and activities that are vital to compliance with accepted standards of performance. Data Regulatory Informed Consent Eligibility Study Procedures Measurable Laboratory Evaluations Study Product Clinical Standard/Threshold 62

63 Informed Consent Form and Processes IRB/EC Approval Informed Consent Inclusion Exclusion Criteria Regulatory Eligibility DataFax Submission Data Study Procedures Missed Tests/ Procedures Hematology Tests Laboratory Evaluations Clinical Study Product Study Product Accountability AE s, SAE s, EAE s 63

64 Key Quality Indicators Activity Based on the OCSO Trend Analysis, what are some additional KQIs that might be considered? Informed Consent Regulatory Eligibility Data Study Procedures Laboratory Evaluations Study Product 64 Clinical
65 10:00am Break (30 Minutes) 65
66 Quality Implementation and Documentation
67 MTN-003
68 Quality Quality Control Quality Assurance 68
69 Quality Control Sample Size 100% Records Quality Control Activities Documentation What will be reviewed? Consider Key Quality Indicators. What is the step-by-step process for completing QC activities? How will errors be documented for analysis? Frequency Real-time, daily Error Correction Communicate How will errors be corrected? How will the errors, corrective actions, and preventive actions be communicated? 69

70 Quality Assurance Sample Size Variable Records Quality Assurance Activities Documentation What will be reviewed? Consider Key Quality Indicators. What is the step-by-step process for completing QA activities? How will errors be documented for analysis? Frequency Periodic Error Correction How will errors be corrected? RETROACTIVE REVIEW Communicate How will the errors, corrective actions, and preventive actions be communicated? 70
71 Internal Quality Reporting Structure Quality Control Log Quality Quality Assurance Quality Assurance Quality Assurance Quality Tools Assurance Tools Assurance Tools Tools Tools Data Gathering Summary Report Communication Annual Summary Report 71
72 Tools! 72

73 MTN-003 Study Implementation Materials 73






74 Quality Assurance Tools Quality Control Tools SCHARP DM Quality Report Trending MTN CORE-FHI Assessment Reports PPD Monitoring Reports SCHARP QC Reports Quality Summary Report Tool Communicate
75 Objective Check Identify resources available to the site and how to use them in preparation for an FDA inspection 75

76 Trending Activity The monitoring visit has just concluded, and the monitor has left you, the Quality Manager, with completed copies of the Monitor Record Review Tools for each participant record reviewed. The Monitor Record Review Tools document the observations noted by the monitor for each participant record reviewed during the visit. As the Quality Manager, you will now compare observations from these monitoring tools, the Quality Chart Review Tools, and the Quality Control Log in an effort to identify observational trends. 76
77 Trending Activity Matrix Monitor (3) Quality Control (10) Quality Assurance (6)
78 Problems and Trends Found Now What?
79 Define Problem Evaluate Solution Implement Solution Problem- Solving Model Analyze Data Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 79
80 Define Problem Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 80
81 Analyze Data Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 81
82 Identify Solutions Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 82
83 Select Best Solution Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 83
84 Develop Action Plan Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 84
85 Implement Solution Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 85
86 Evaluate Solution Define Problem Evaluate Solution Analyze Data Implement Solution Problem- Solving Model Root Cause Analysis Identify Solutions Develop Action Plan Select Best Solution 86
87 Quality Plan Evaluation Immediate Periodic Annual 87
88 Objective Check Implement quality management techniques that will assist with identification of areas for improvement in preparation for possible FDA inspection 88




89 Quality Plan(s) Investigator of Record Quality Informed Consent Regulatory Eligibility Data Study Procedures Clinical Research Site Laboratory Evaluations Clinical Study Product Laboratory Pharmacy FDA Inspection Preparedness 89
90 Questions?
91 Next Steps Donna Germuga, DAIDS OCSO
92 Next Steps Modify CQMP as needed Conduct retrospective reviews on KQIs Develop corrective action as needed Re-evaluate corrective action-is IT WORKING? DOCUMENT all QC/QA activities *If currently no significant trends or findings, then continue internal QA/QC activities and proactively identify and resolve any issues or trends in preparation for FDA audit. Good Work!!! 92
93 Next Steps Initiate FDA inspection checklist Establish FDA inspection prep team (clinical, lab, pharmacy, regulatory, QA/QC) Appoint one person to act as central Coordinator Delegate individual/s to oversee completion of each section Schedule regular meetings to discuss progress, findings Develop a systematic plan and timeline for completion 93

94 FDA Checklist Administrative Complete later- post FDA training II -2Q013 Sites will develop SOP

95 FDA Checklist Regulatory Ensure all documents in place Ensure documents are complete, accurate, organized and easily found Training Documentation

96 FDA Checklist Clinical Determine sample size (OCSO can assist) Recommendation minimum 10% of PIDs per quarter for critical KQIs. Customize this checklist as needed
97 Next Steps Access FDA inspection resources on-line Appoint one person to be the go to person and resource for other staff Contact your OCSO PO with any questions or concerns 97



98 Updated FDA Inspection Preparation Timeline DATA LOCK October 2012 Presentation of Results February 2013 FDA Inspection Prep Training Part I Site Revisions to CQMP Initiate FDA checklist PPD Special Assignments FDA Inspection Prep Training Part II FDA Inspection TODAY October 10, Q011-3Q012 2Q013 3Q013??

99 Remember we re all in this together...
100 Questions?
In consultation with the Behavioral Research Working Group (BRWG), the Statistical and Data Management Center (SDMC), the Network Laboratory (NL),
, the Statistical and Data Management Center (SDMC), the Network Laboratory (NL),") 1 2 In consultation with the Behavioral Research Working Group (BRWG), the Statistical and Data Management Center (SDMC), the Network Laboratory (NL), and the U.S. National Institute for Allergy and Infectious
1 2 In consultation with the Behavioral Research Working Group (BRWG), the Statistical and Data Management Center (SDMC), the Network Laboratory (NL), and the U.S. National Institute for Allergy and Infectious
Introduction to Clinical Research
 Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
Quality Assurance QA STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits
 Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
Guidance for Industry - Computerized Systems Used in Clinical Trials
 Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
1 The Clinical Research Coordinator (CRC)... 1
... 1") TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
GCP Refresher and GCP/GCDMP Trends. in the CTN. Denise King, MS, RD, CCRA & Lauren Yesko, BS. Presented by:
 GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
Standard Operating Procedures (SOPs)
") Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Good Clinical Practice
 Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
TOP 10 INVESTIGATOR RESPONSIBILITIES WHEN CONDUCTING HUMAN SUBJECTS RESEARCH
 Investigators Responsibility #1: Design and Implement Ethical Research Consistent with the Three Ethical Principles Delineated in The Belmont Report. The Belmont Report: Three Basic Ethical Principles:
Investigators Responsibility #1: Design and Implement Ethical Research Consistent with the Three Ethical Principles Delineated in The Belmont Report. The Belmont Report: Three Basic Ethical Principles:
Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework
 1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
Clinical Trials and the Code of Federal Regulations. Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014
 Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Human Research Audit Program. Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance
 Human Research Audit Program Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance Monday, September 11, 2017 Objectives 1. Understand the purpose of the audit program 2. Identify
Human Research Audit Program Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance Monday, September 11, 2017 Objectives 1. Understand the purpose of the audit program 2. Identify
1.4 Applicable Regulatory Requirement(s) Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.
 Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.") 1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
OCTC 2012 CRO Selection
 OCTC 2012 CRO Selection Colin Macaulay Viron Therapeutics Inc. 15 Nov 2012 Viron Therapeutics Inc. Virtual Biotech Company (6) Phase 2a (48 pt) clinical trial in acute coronary syndrome (ACS) completed
OCTC 2012 CRO Selection Colin Macaulay Viron Therapeutics Inc. 15 Nov 2012 Viron Therapeutics Inc. Virtual Biotech Company (6) Phase 2a (48 pt) clinical trial in acute coronary syndrome (ACS) completed
Food and Drug Administration (FDA) 101
 101") Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
Inspections: an academic perspective
 Inspections: an academic perspective Patricia Henley Quality and Governance Manager Head, Research Governance & Integrity Office London School of Hygiene & Tropical Medicine Email: patricia.henley@lshtm.ac.uk
Inspections: an academic perspective Patricia Henley Quality and Governance Manager Head, Research Governance & Integrity Office London School of Hygiene & Tropical Medicine Email: patricia.henley@lshtm.ac.uk
The Role of Chemists in the FDA Drug Approval Process
 The Role of Chemists in the FDA Drug Approval Process 231 st ACS National Meeting Atlanta, GA M. Scott Furness, Ph.D. March 26, 2006 Introduction Presentation Outline FDA Organization CDER Organization
The Role of Chemists in the FDA Drug Approval Process 231 st ACS National Meeting Atlanta, GA M. Scott Furness, Ph.D. March 26, 2006 Introduction Presentation Outline FDA Organization CDER Organization
Ethics Committees/IRBs Today: Challenges for Efficiency and Quality
 Marjorie A. Speers, Ph.D. President and CEO Ethics Committees/IRBs Today: Challenges for Efficiency and Quality Copyright 2013 AAHRPP All rights reserved Goal Clinical Research Globally Access to patients
Marjorie A. Speers, Ph.D. President and CEO Ethics Committees/IRBs Today: Challenges for Efficiency and Quality Copyright 2013 AAHRPP All rights reserved Goal Clinical Research Globally Access to patients
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval
 Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
Investigator Manual. Human Subjects Protection Program
 Human Subjects Protection Program HRP-103, Revised June 1, 2015 HRP-103 06/01/2015 2 of 33 Table of Contents Scope... 3 What is the purpose of this manual?... 3 What is Human Research?... 3 What is the
Human Subjects Protection Program HRP-103, Revised June 1, 2015 HRP-103 06/01/2015 2 of 33 Table of Contents Scope... 3 What is the purpose of this manual?... 3 What is Human Research?... 3 What is the
Chapter 1. Pharmaceutical Industry Overview
 Chapter 1 Pharmaceutical Industry Overview 1.1 Introduction 2 1.2 Regulations 2 1.2.1 Health Insurance Portability and Accountability Act 2 1.2.2 The Code of Federal Regulations 3 1.2.3 Guidance for Industry
Chapter 1 Pharmaceutical Industry Overview 1.1 Introduction 2 1.2 Regulations 2 1.2.1 Health Insurance Portability and Accountability Act 2 1.2.2 The Code of Federal Regulations 3 1.2.3 Guidance for Industry
Preparing for Close-Out of Studies and Sites
 Preparing for Close-Out of Studies and Sites Presented by: Christie Thomas, MPH, CCRA Matthew Wright, BS, CCRP Dikla Blumberg, PhD CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
Preparing for Close-Out of Studies and Sites Presented by: Christie Thomas, MPH, CCRA Matthew Wright, BS, CCRP Dikla Blumberg, PhD CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
Stephanie Gentilin, CCRA
 Elements of Clinical Trial Quality Assurance Stephanie Gentilin, CCRA Regulatory Coordinator SCTR SUCCESS Center QA Monitor NIDA Monitor s Responsibilities ICH E6 Section 5.18 Overall Purpose of Monitoring
Elements of Clinical Trial Quality Assurance Stephanie Gentilin, CCRA Regulatory Coordinator SCTR SUCCESS Center QA Monitor NIDA Monitor s Responsibilities ICH E6 Section 5.18 Overall Purpose of Monitoring
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order
 Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
Multi-Site Coordination Process. Drafted by: Ester Dimayuga Page 1 of 18
 Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Source And Regulatory Documentation for DMID Clinical Studies
 Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
FDA Audit Preparation
 Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
GCP Basics - refresher
 p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
Center for Drug Evaluation and Research. CDER Small Business and Industry Assistance. (CDER SBIA) and New Drug Review.
 and New Drug Review.") Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
Study Files and Filing
 Study Files and Filing The current version of all Hillingdon Hospital R&D Guidance Documents and Standard Operating Procedures are available from the R&D Intranet and Internet sites: www.ths.nhs.uk/departments/research/research.htm
Study Files and Filing The current version of all Hillingdon Hospital R&D Guidance Documents and Standard Operating Procedures are available from the R&D Intranet and Internet sites: www.ths.nhs.uk/departments/research/research.htm
Applicability of US Regulations to Canadian Research
 Applicability of US Regulations to Canadian Research Mary Kate Needler Capital District Health Authority Halifax, Nova Scotia, Canada N2 Stakeholder Meeting August 18, 2014 MK s Conventions: 1. US Regulation
Applicability of US Regulations to Canadian Research Mary Kate Needler Capital District Health Authority Halifax, Nova Scotia, Canada N2 Stakeholder Meeting August 18, 2014 MK s Conventions: 1. US Regulation
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success. Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist
 with the FDA: Keys for Success. Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist") Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist Agenda How an investigator should prepare and submit
Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist Agenda How an investigator should prepare and submit
Investigator-Initiated INDs
 Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
5 th Training Workshop for Micro-, Small- and Medium- Sized Enterprises (SMEs)
") 5 th Training Workshop for Micro-, Small- and Medium- Sized Enterprises (SMEs) GCP Requirements and Compliance Dr. Katalina Mettke BfArM, Germany Agenda Specialities of Early Development Trials Examples
5 th Training Workshop for Micro-, Small- and Medium- Sized Enterprises (SMEs) GCP Requirements and Compliance Dr. Katalina Mettke BfArM, Germany Agenda Specialities of Early Development Trials Examples
The Clinical Investigator Site Audit Process-Driven Good Clinical Practice
 The Clinical Investigator Site Audit Process-Driven Good Clinical Practice Terry Winchell* GCP Innovative Dynamics, LLC, 7400 Kansas Ave., Kansas City, Kansas 66111-2639, USA Summary This article describes
The Clinical Investigator Site Audit Process-Driven Good Clinical Practice Terry Winchell* GCP Innovative Dynamics, LLC, 7400 Kansas Ave., Kansas City, Kansas 66111-2639, USA Summary This article describes
Human Research Protection Program Good Clinical Practice Guidance for Investigators Regulatory File Essential Documents
 Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
FDA Initiatives and Regulatory Trends for Life Sciences. Larry Spears President L. Spears Consulting
 FDA Initiatives and Regulatory Trends for Life Sciences Larry Spears President L. Spears Consulting Before We Begin If you experience technical problems, please contact GoToMeeting Technical Support at
FDA Initiatives and Regulatory Trends for Life Sciences Larry Spears President L. Spears Consulting Before We Begin If you experience technical problems, please contact GoToMeeting Technical Support at
The interface between Good Clinical Practice and Good Manufacturing Practice
 1 The interface between Good Clinical Practice and Good Manufacturing Practice your partner in compliance 1 The interface between GCP and GMP Generally, studies are designed and planned by physicians who
1 The interface between Good Clinical Practice and Good Manufacturing Practice your partner in compliance 1 The interface between GCP and GMP Generally, studies are designed and planned by physicians who
GxP Auditing, Remediation, and Quality System Resourcing
 GxP Auditing, Remediation, and Quality System Resourcing TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 6 GLP Auditing 7 Pharmacovigilance Auditing 7 Vendor/Supplier Auditing
GxP Auditing, Remediation, and Quality System Resourcing TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 6 GLP Auditing 7 Pharmacovigilance Auditing 7 Vendor/Supplier Auditing
Global Clinical Trials in Korea
 Global Clinical Trials in Korea In-Sook Park Department of Drug Evaluation Korea Food & Drug Administration Contents Regulatory changes relevant to Clinical Trials in Korea Current Status of Clinical Trials
Global Clinical Trials in Korea In-Sook Park Department of Drug Evaluation Korea Food & Drug Administration Contents Regulatory changes relevant to Clinical Trials in Korea Current Status of Clinical Trials
WCG ACADEMY COURSE OVERVIEW
 WCG ACADEMY COURSE OVERVIEW Investigator Course Title Documentation of Informed Consent Duration Ethical Principles Underlying Consent Documentation Long Form Short Form Consent Records Special Considerations
WCG ACADEMY COURSE OVERVIEW Investigator Course Title Documentation of Informed Consent Duration Ethical Principles Underlying Consent Documentation Long Form Short Form Consent Records Special Considerations
Quality Assurance in Clinical Trials Introduction
 Quality Assurance in Clinical Trials Introduction Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 2012 1 Delphine TEPPE-CROITROU - Catherine CORNU (Hospices Civils de Lyon)
Quality Assurance in Clinical Trials Introduction Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 2012 1 Delphine TEPPE-CROITROU - Catherine CORNU (Hospices Civils de Lyon)
GOOD CLINICAL PRACTICE (GCP) Series Catalog
 Series Catalog") GOOD CLINICAL PRACTICE (GCP) Series Catalog CITI Program s GCP series consists of three basic courses and three refresher courses. The basic courses include: GCP for Clinical Trials with Investigational
GOOD CLINICAL PRACTICE (GCP) Series Catalog CITI Program s GCP series consists of three basic courses and three refresher courses. The basic courses include: GCP for Clinical Trials with Investigational
Objectives. The Regulatory Binder = Investigator Site File= Trial Center File 8/16/2010. Essential Documents: Maintaining the Site's Regulatory Binder
 Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Quality Assurance in Clinical Trials
 Quality Assurance in Clinical Trials Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 5th, 2011 1 Introduction: quality in clinical research in human subjects Regulatory requirements:
Quality Assurance in Clinical Trials Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 5th, 2011 1 Introduction: quality in clinical research in human subjects Regulatory requirements:
How did it evolve? o Public disasters, serious fraud and abuse of human rights. o Trials of War criminals-nuremberg code 1949
 ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
CANCER CENTER SCIENTIFIC REVIEW COMMITTEE
 CANCER CENTER SCIENTIFIC REVIEW COMMITTEE The Clinical Scientific Review Committee (SRC) at The Medical College of Wisconsin Cancer Center plays a vital role in protocol review and monitoring to ensure
CANCER CENTER SCIENTIFIC REVIEW COMMITTEE The Clinical Scientific Review Committee (SRC) at The Medical College of Wisconsin Cancer Center plays a vital role in protocol review and monitoring to ensure
Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products
 1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
Source Documents and Regulatory Binders October 6, 2016
 Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010
 FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010 FDA Nanotechnology Task Force Carlos Peña, PhD Office of the Commissioner & Subhas Malghan, PhD Center for Devices and
FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010 FDA Nanotechnology Task Force Carlos Peña, PhD Office of the Commissioner & Subhas Malghan, PhD Center for Devices and
GxP Auditing, Remediation, and Staff Augmentation
 GxP Auditing, Remediation, and Staff Augmentation TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 5 GLP Auditing 6 Pharmacovigilance Auditing 6 Vendor/Supplier Auditing 7
GxP Auditing, Remediation, and Staff Augmentation TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 5 GLP Auditing 6 Pharmacovigilance Auditing 6 Vendor/Supplier Auditing 7
Medidée Services SA. Nano-Tera.ch. 05 February 2015 part 8. PMA, 510k, IDE. Pierre-Alain Sommer
 Nano-Tera.ch 05 February 2015 part 8 PMA, 510k, IDE Pierre-Alain Sommer Pierre-alain.sommer@medidee.com www.medidee.com Nano-Tera 2015 05.02.2015 USA/FDA Pre Market Approval System - PMA, Pre Market Notifcation
Nano-Tera.ch 05 February 2015 part 8 PMA, 510k, IDE Pierre-Alain Sommer Pierre-alain.sommer@medidee.com www.medidee.com Nano-Tera 2015 05.02.2015 USA/FDA Pre Market Approval System - PMA, Pre Market Notifcation
ICH GCP E6(R2): Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc.
: Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc.") ICH GCP E6(R2): Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc. 25 October 2017: University of Rochester Achieving High Quality Clinical Research
ICH GCP E6(R2): Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc. 25 October 2017: University of Rochester Achieving High Quality Clinical Research
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155
 MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
Guidance for Industry Part 11, Electronic Records; Electronic Signatures Scope and Application
 Guidance for Industry Part 11, Electronic Records; Electronic Signatures Scope and Application DRAFT GUIDANCE This guidance is being distributed for comment purposes only. Comments and suggestions regarding
Guidance for Industry Part 11, Electronic Records; Electronic Signatures Scope and Application DRAFT GUIDANCE This guidance is being distributed for comment purposes only. Comments and suggestions regarding
\\NAS1\George\Docs\SoCRA\CCRP communications\study guide management
 Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
PHARMA CONGRESS. Pharmacovigilance and Drug Safety: Assessing Future Regulatory and Compliance Developments. October 28, 2008
 PHARMA CONGRESS October 28, 2008 Pharmacovigilance and Drug Safety: Assessing Future Regulatory and Compliance Developments Beverly H. Lorell, MD Senior Medical & Policy Advisor King & Spalding LLP Assessing
PHARMA CONGRESS October 28, 2008 Pharmacovigilance and Drug Safety: Assessing Future Regulatory and Compliance Developments Beverly H. Lorell, MD Senior Medical & Policy Advisor King & Spalding LLP Assessing
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited.
. All rights reserved. Further dissemination without express written consent strictly prohibited.") Statutory Framework for Devices Medical Devices Investigational Use Application IDE (21 CFR 812) Abbreviated IDE Exempt Pre-Market Approval Applications 510(k) Pre-marketing Notification (21 CFR 807(e))
Statutory Framework for Devices Medical Devices Investigational Use Application IDE (21 CFR 812) Abbreviated IDE Exempt Pre-Market Approval Applications 510(k) Pre-marketing Notification (21 CFR 807(e))
STANDARD OPERATING PROCEDURE
 STANDARD OPERATING PROCEDURE Title Reference Number Archiving and the Destruction of Records SOP-RES-028 Version Number 2 Issue Date 08 th Dec 2015 Effective Date 22 nd January 2016 Review Date 22 nd January
STANDARD OPERATING PROCEDURE Title Reference Number Archiving and the Destruction of Records SOP-RES-028 Version Number 2 Issue Date 08 th Dec 2015 Effective Date 22 nd January 2016 Review Date 22 nd January
GW Policy on Human Research Protection Program
 GW Policy on Human Research Protection Program 1. PURPOSE 1.1. This policy establishes the [Organization] s Human Research Protection Program (HRPP) and its commitment to protect the rights and welfare
GW Policy on Human Research Protection Program 1. PURPOSE 1.1. This policy establishes the [Organization] s Human Research Protection Program (HRPP) and its commitment to protect the rights and welfare
elearning Catalog DIAglobal.org/eLearning
 elearning Catalog DIAglobal.org/eLearning Continuing education DIA s Continuing education program has been approved by a wide range of leading accrediting bodies that oversee the awarding of continuing
elearning Catalog DIAglobal.org/eLearning Continuing education DIA s Continuing education program has been approved by a wide range of leading accrediting bodies that oversee the awarding of continuing
The European Medicines Agency Inspections ANNEX IV TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA:
 The European Medicines Agency Inspections London, 20 September 2007 EMEA/INS/GCP/197221/2005 Procedure no.: INS/GCP/3/IV ANNEX IV TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA: SPONSOR
The European Medicines Agency Inspections London, 20 September 2007 EMEA/INS/GCP/197221/2005 Procedure no.: INS/GCP/3/IV ANNEX IV TO PROCEDURE FOR CONDUCTING GCP INSPECTIONS REQUESTED BY THE EMEA: SPONSOR
Expanded Access and the Individual Patient IND
 Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Strengthening GLP Compliance Through Internal Audits
 Strengthening GLP Compliance Through Internal Audits Henry Li*, Dominique Pifat, Gerald Klein and Steve Petteway Talecris Biotherapeutics, Research Triangle Park, NC, USA Summary The Quality Assurance
Strengthening GLP Compliance Through Internal Audits Henry Li*, Dominique Pifat, Gerald Klein and Steve Petteway Talecris Biotherapeutics, Research Triangle Park, NC, USA Summary The Quality Assurance
GxP Auditing, Remediation, and Staff Augmentation
 GxP Auditing, Remediation, and Staff Augmentation TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 6 GLP Auditing 7 Pharmacovigilance Auditing 7 Vendor/Supplier Auditing 8
GxP Auditing, Remediation, and Staff Augmentation TABLE OF CONTENTS 3 Introduction 4 GxP Auditing 4 GMP Auditing 5 GCP Auditing 6 GLP Auditing 7 Pharmacovigilance Auditing 7 Vendor/Supplier Auditing 8
Expanded Access. to Investigational Drugs & Biologics. for Treatment Use
 SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
Establishment of Clinical Trial Infrastructure
 Taiwan s Strategy in the Establishment of Clinical Trial Infrastructure Chei-Hsiang Chen, Ph. D. Director, Biotechnology and Pharmaceutical Industries Program Office, Ministry of Economic Affairs, Taiwan
Taiwan s Strategy in the Establishment of Clinical Trial Infrastructure Chei-Hsiang Chen, Ph. D. Director, Biotechnology and Pharmaceutical Industries Program Office, Ministry of Economic Affairs, Taiwan
National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services
 National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services The Division of Microbiology and Infectious Diseases (DMID), National Institute
National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services The Division of Microbiology and Infectious Diseases (DMID), National Institute
Human Research Protection Program Guidance for Human Research Determination
 Human Research Protection Program Guidance for Human Research Determination I.1.A The sole purpose of the Institutional Review Board (IRB), as defined in federal statutes, is the protection of human subjects
Human Research Protection Program Guidance for Human Research Determination I.1.A The sole purpose of the Institutional Review Board (IRB), as defined in federal statutes, is the protection of human subjects
Case Report from Audit Firm Inspection Results
 Case Report from Audit Firm Inspection Results July 2014 Certified Public Accountants and Auditing Oversight Board Table of Contents Expectations for Audit Firms... 1 Important Points for Users of this
Case Report from Audit Firm Inspection Results July 2014 Certified Public Accountants and Auditing Oversight Board Table of Contents Expectations for Audit Firms... 1 Important Points for Users of this
Trial oversight SOP for HEY-sponsored CTIMPs
 R&D Department Trial oversight SOP for HEY-sponsored CTIMPs Hull And East Yorkshire Hospitals NHS Trust 2010 All Rights Reserved No part of this document may be reproduced, stored in a retrieval system
R&D Department Trial oversight SOP for HEY-sponsored CTIMPs Hull And East Yorkshire Hospitals NHS Trust 2010 All Rights Reserved No part of this document may be reproduced, stored in a retrieval system
Good Clinical Practice
 Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
EU and FDA GMP Regulations: Overview and Comparison
 THE QUALITY ASSURANCE JOURNAL, VOL. 2, 55 60 (1997) EU and FDA GMP Regulations: Overview and Comparison The increasing emphasis on global supply of drug products, as well as starting materials and investigational
THE QUALITY ASSURANCE JOURNAL, VOL. 2, 55 60 (1997) EU and FDA GMP Regulations: Overview and Comparison The increasing emphasis on global supply of drug products, as well as starting materials and investigational
Globalization and the FDA The Alliance for a Stronger FDA Quarterly Member Meeting February 8, 2012
 Globalization and the FDA The Alliance for a Stronger FDA Quarterly Member Meeting February 8, 2012 Deborah M. Autor, Esq. Deputy Commissioner for Global Regulatory Operations and Policy U.S. Food and
Globalization and the FDA The Alliance for a Stronger FDA Quarterly Member Meeting February 8, 2012 Deborah M. Autor, Esq. Deputy Commissioner for Global Regulatory Operations and Policy U.S. Food and
Investigator Manual HRP-103
 HRP-103 Revised October 04, 2016 Table of Contents 1. Introduction... 3 A. Scope... 3 B. Purpose of this manual... 3 C. Where to get additional information and answers to questions... 3 E. Human Research
HRP-103 Revised October 04, 2016 Table of Contents 1. Introduction... 3 A. Scope... 3 B. Purpose of this manual... 3 C. Where to get additional information and answers to questions... 3 E. Human Research
R&D Manager Hillingdon Hospital. Revision History Effective Date Reason For Change. recommendations Version no:
 Research as a Participating site STANDARD OPERATING PROCEDURE FOR OVERSIGHT SOP No: P08/PF2 V2 Effective Date: 31 st July 2013 Supersedes: P08/PF2 Revision Date: 31 st March 2014 Author: Position: Approved
Research as a Participating site STANDARD OPERATING PROCEDURE FOR OVERSIGHT SOP No: P08/PF2 V2 Effective Date: 31 st July 2013 Supersedes: P08/PF2 Revision Date: 31 st March 2014 Author: Position: Approved
External IRB Review What Does it Mean for Your Institution
 External IRB Review What Does it Mean for Your Institution Wesley G Byerly, Pharm.D. Associate Vice President for Research Integrity and Regulatory Affairs University of Connecticut and UCONN Health HCCA
External IRB Review What Does it Mean for Your Institution Wesley G Byerly, Pharm.D. Associate Vice President for Research Integrity and Regulatory Affairs University of Connecticut and UCONN Health HCCA
Recommendations for Strengthening the Investigator Site Community
 Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
INVESTIGATIONAL DEVICE EXEMPTION APPLICATION. IDE Title (if title being used)
") INVESTIGATIONAL DEVICE EXEMPTION APPLICATION IDE Title (if title being used) Name of Sponsor Investigator, MD X Professor, Department Icahn School of Medicine at Mount Sinai Date of Submission Form version
INVESTIGATIONAL DEVICE EXEMPTION APPLICATION IDE Title (if title being used) Name of Sponsor Investigator, MD X Professor, Department Icahn School of Medicine at Mount Sinai Date of Submission Form version
Managing a Research Team. Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services
 Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Responding to an FDA 483
 Responding to an FDA 483 Jim Melancon VP, Associate General Counsel BioScrip, Inc. Marc Stranz, PharmD Healthcare Consultant Disclosure The speakers declare no conflicts of interest or financial interest
Responding to an FDA 483 Jim Melancon VP, Associate General Counsel BioScrip, Inc. Marc Stranz, PharmD Healthcare Consultant Disclosure The speakers declare no conflicts of interest or financial interest
Risk-Based Approach to SAS Program Validation
 Paper FC04 Risk-Based Approach to SAS Program Validation Keith C. Benze SEC Associates, Inc. 3900 Paramount Parkway, Suite 150 South Morrisville, NC 27560 ABSTRACT SAS is widely used throughout the FDA
Paper FC04 Risk-Based Approach to SAS Program Validation Keith C. Benze SEC Associates, Inc. 3900 Paramount Parkway, Suite 150 South Morrisville, NC 27560 ABSTRACT SAS is widely used throughout the FDA
Clinical Research: A Multifaceted Discipline
 Clinical Research: A Multifaceted Discipline Anuradha Kulkarni a, Arun Bhatt b* a Senior Associate - Medical & Regulatory Affairs, Clininvent Research Pvt Ltd, A-302, Everest Chambers, Marol Naka, Andheri
Clinical Research: A Multifaceted Discipline Anuradha Kulkarni a, Arun Bhatt b* a Senior Associate - Medical & Regulatory Affairs, Clininvent Research Pvt Ltd, A-302, Everest Chambers, Marol Naka, Andheri
MDSAP AUDIT PROCESS. A Manufacturer s Perspective. Connie Hoy EVP Regulatory Affairs Cynosure, Inc.
 MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
TITLE: Research Pharmacy Standard Policy SOP #: INV-100 Page: 1 of 5 Effective Date: 8/31/17
 SOP #: INV-100 Page: 1 of 5 1. POLICY STATEMENT: DF/HCC research pharmacies follow a standard set of policy requirements for clinical trials. 2. BACKGROUND: None 3. RESPONSIBLE PERSONNEL: 3.1. Research
SOP #: INV-100 Page: 1 of 5 1. POLICY STATEMENT: DF/HCC research pharmacies follow a standard set of policy requirements for clinical trials. 2. BACKGROUND: None 3. RESPONSIBLE PERSONNEL: 3.1. Research
THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE
 THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE Ali Mohamadi, M.D. Senior Director, Global Regulatory Patient Engagement and Policy BioMarin Pharmaceutical 30-July-2016 Goals of Today s Presentation
THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE Ali Mohamadi, M.D. Senior Director, Global Regulatory Patient Engagement and Policy BioMarin Pharmaceutical 30-July-2016 Goals of Today s Presentation
Preparing your Audit Response Communication with the Central Office
 Preparing your Audit Response Communication with the Central Office Sally Scherer, Audit Specialist CALGB Central Office CALGB Audit Prep Workshop, June 2006 Preparing Your Audit Response: Communication
Preparing your Audit Response Communication with the Central Office Sally Scherer, Audit Specialist CALGB Central Office CALGB Audit Prep Workshop, June 2006 Preparing Your Audit Response: Communication
Rules of Human Experimentation
 Rules of Human Experimentation Elaine Larson CUMC IRB Chair Associate Dean for Research, School of Nursing Professor of Epidemiology, Mailman School of Public Health Oversight for Human Research Office
Rules of Human Experimentation Elaine Larson CUMC IRB Chair Associate Dean for Research, School of Nursing Professor of Epidemiology, Mailman School of Public Health Oversight for Human Research Office
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS
 D i a g n o s t i c s a n d L a b o r a t o r y T e c h n o l o g y OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS Prequalification of Male Circumcision Devices PQMC_007
D i a g n o s t i c s a n d L a b o r a t o r y T e c h n o l o g y OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS Prequalification of Male Circumcision Devices PQMC_007
KINGSMANN CARE GROUP
 PHARMA CONSULTANTS KINGSMANN CARE GROUP KINGSMANN CONSULTANCY SERVICES Thank you for taking interest in Kingsmann Consultancy Services. Kingsmann Consultancy (KC) is a leading business development-consulting
PHARMA CONSULTANTS KINGSMANN CARE GROUP KINGSMANN CONSULTANCY SERVICES Thank you for taking interest in Kingsmann Consultancy Services. Kingsmann Consultancy (KC) is a leading business development-consulting
Harmonized Standard Operating Procedures for Academic Trials
 Harmonized Standard Operating Procedures for Academic Trials Heike Moenkemann*, Miriam Olderog*, Ursula Paulus** * Clinical Trials Center Cologne ** Clinical Trials Center Cologne and Head of QM working
Harmonized Standard Operating Procedures for Academic Trials Heike Moenkemann*, Miriam Olderog*, Ursula Paulus** * Clinical Trials Center Cologne ** Clinical Trials Center Cologne and Head of QM working
DRUG ORDERING AND MAINTENANCE
 AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter mainly apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter mainly apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
IMP Management and Accountability
 This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
Top Ten Investigator Responsibilities When Conducting Human Subjects Research
 Top Ten Investigator Responsibilities When Conducting Human Subjects Research Thanks to Ada Sue Selwitz, Univ. of Kentucky and PRIM&R (Public Responsibility in Medicine & Research) Investigator Responsibility
Top Ten Investigator Responsibilities When Conducting Human Subjects Research Thanks to Ada Sue Selwitz, Univ. of Kentucky and PRIM&R (Public Responsibility in Medicine & Research) Investigator Responsibility
The EFGCP Report on The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria
 Bulgaria") The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria Question 1: What laws or regulations apply to an application for conducting a clinical
The Procedure for the Ethical Review of Protocols for Clinical Research Projects in Europe (Update: April 2011) Bulgaria Question 1: What laws or regulations apply to an application for conducting a clinical