SRM UNIVERSITY FACULTY OF ENGINEERING & TECHNOLOGY SCHOOL OF BIOENGINEERING DEPARTMENT OF BIOINFORMATICS BI0213- COMPUTATIONAL BIOLOGY LABORATORY
|
|
|
- Julia Jocelyn King
- 6 years ago
- Views:
Transcription
1 SRM UNIVERSITY FACULTY OF ENGINEERING & TECHNOLOGY SCHOOL OF BIOENGINEERING DEPARTMENT OF BIOINFORMATICS BI0213- COMPUTATIONAL BIOLOGY LABORATORY LAB MANUAL Semester: III Code: BI0213 Lab: Computational Biology Laboratory Staff Handling: Mrs. J. B. Sheema Ex no: 1 & 2 Knowledge of different biological database & Sequence retrieval from biological database Aim: To study & to analyze the in formations from the various biological databases available on the World Wide Web. Description: Biological datas are highly complex and interrelated. Vast amount of biological information needs to be stored organized and indexed so that the information can be retrieved and
2 used. There are five major types of databases namely nucleotide databases, protein databases, protein structure databases, metabolic pathway databases and the bibliographic databases. Procedure: 1. Open web browser and type the web address of the required database. 2. Explore the database and analyze the various information available in the database. 3. Use the tools provided by the databases. 4. Save the output into a separate folder. A. Nucleotide Databases 1. NCBI: National Center for Biotechnology Information The challenge is in finding new approaches to deal with the volume and complexity of biological datas and in providing researchers with better access to analysis and computing tools to advance understanding of genetic legacy and its role in health and disease. The late Senator Claude Pepper recognized the importance of computerized information processing methods for the conduct of biomedical research and sponsored legislation that established the National Center for Biotechnology Information (NCBI) on November 4, 1988, as a division of the National Library of Medicine (NLM) at the National Institutes of Health (NIH). NLM was chosen for its experience in creating and maintaining biomedical databases, and because as part of NIH, it could establish an intramural research program in computational molecular biology. The collective research components of NIH make up the largest biomedical research facility in the world. As a national resource for molecular biology information, NCBI's mission is to develop new information technologies to aid in the understanding of fundamental molecular and genetic processes that control health and disease. More specifically, the NCBI has been charged with creating automated systems for storing and analyzing knowledge about molecular biology, biochemistry, and genetics; facilitating the use of such databases and software by the research and
3 medical community; coordinating efforts to gather biotechnology information both nationally and internationally; and performing research into advanced methods of computer-based information processing for analyzing the structure and function of biologically important molecules. To carry out its diverse responsibilities, NCBI: conducts research on fundamental biomedical problems at the molecular level using mathematical and computational methods maintains collaborations with several NIH institutes, academia, industry, and other governmental agencies. fosters scientific communication by sponsoring meetings, workshops, and lecture series. supports training on basic and applied research in computational biology for postdoctoral fellows through the NIH Intramural Research Program. engages members of the international scientific community in informatics research and training through the Scientific Visitors Program. develops, distributes, supports, and coordinates access to a variety of databases and software for the scientific and medical communities develops and promotes standards for databases, data deposition and exchange, and biological nomenclature.

4 The home page of NCBI can be seen as follows:
5 Output: The file format of the particular protein lysin can be shown follows :( GenPept format)
")
6 The file format of the particular protein lysin can be shown follows:(fasta format)
7 2. DDBJ DDBJ (DNA Data Bank of Japan) began DNA data bank activities in earnest in 1986 at the National Institute of Genetics (NIG) with the endorsement of the Ministry of Education, Science, Sport and Culture. From the beginning, DDBJ has been functioning as one of the International DNA Databases, including EBI (European Bioinformatics Institute; responsible for the EMBL database) in Europe and NCBI (National Center for Biotechnology Information; responsible for GenBank database) in the USA as the two other members. Consequently, we have been collaborating with the two data banks through exchanging data and information on Internet and by regularly holding two meetings, the International DNA Data Banks Advisory Meeting and the International DNA Data Banks Collaborative Meeting. DDBJ is the sole DNA data bank in Japan, which is officially certified to collect DNA sequences from researchers and to issue the internationally recognized accession number to data submitters. We collect data mainly from Japanese researchers, but of course accept data and issue the accession number to researchers in any other countries. Since we exchange the collected data with EMBL/EBI and GenBank/NCBI on a daily basis, the three data banks share virtually the same data at any given time. We also provided worldwide many tools for data retrieval and analysis developed by at DDBJ and others.

8 The home page of DDBJ can be viewed as follows:
9 3. EMBL The European Bioinformatics Institute (EMBL-EBI), which is part of the European Molecular Biology Laboratory (EMBL), is one of the few places in the world that has the resources and expertise to fulfil this important task. The EMBL-EBI lies in the 55 acres of landscaped parkland in rural Cambridgeshire that make up the Wellcome Trust Genome Campus. The Campus also houses the Wellcome Trust Sanger Institute, making it one of the world's largest concentrations of expertise in genomics and bioinformatics. We play a vital role in achieving EMBL's mission of providing a top-quality research environment that also develops new technologies, and provides services and training to Europe's molecular life scientists. Like the other EMBL sites, we have an extremely cosmopolitan staff base, and alumni who have moved on to successful careers all over the world. Mission: To provide freely available data and bioinformatics services to all facts of the scientific community in ways that promote scientific progress To contribute to the advancement of biology through basic investigator-driven research in bioinformatics To provide advanced bioinformatics training to scientists at all levels, from PhD students to independent investigators To help disseminate cutting-edge technologies to industry

10 The home page of EMBL can be viewed as follows:
11 B. Protein Databases 1. SWISSPROT UniProtKB/Swiss-Prot is a manually annotated protein knowledgebase established in 1986 and maintained since 2003 by the UniProt Consortium, collaboration between the Swiss Institute of Bioinformatics (SIB) and the Department of Bioinformatics and Structural Biology of the Geneva University, the European Bioinformatics Institute (EBI) and the Georgetown University Medical Center's Protein Information Resource (PIR). The UniProt Knowledgebase consists of sequence entries. Sequence entries are composed of different line-types, each with their own format. For standardization purposes the format of the UniProt Knowledgebase follows as closely as possible that of the EMBL Nucleotide Sequence Database. The UniProtKB/Swiss-Prot database distinguishes itself from other protein sequence databases by three distinct criteria: 1. Annotation 2. Minimal redundancy 3. Integration with other databases

12 The home page can be viewed as follows:
13 2. PIR: PROTEIN INFORMATION RESOURCES The Protein Information Resource (PIR), located at Georgetown University Medical Center (GUMC), is an integrated public bioinformatics resource to support genomic and proteomic research, and scientific studies (Wu et al., 2003). PIR was established in 1984 by the National Biomedical Research Foundation (NBRF) as a resource to assist researchers in the identification and interpretation of protein sequence information. For four decades, PIR has provided many protein databases and analysis tools freely accessible to the scientific community, including the Protein Sequence Database (PSD), the first international database (see PIR-International), which grew out of Atlas of Protein Sequence and Structure. In 2002, PIR along with its international partners, EBI (European Bioinformatics Institute) and SIB (Swiss Institute of Bioinformatics), were awarded a grant from NIH to create UniProt, a single worldwide database of protein sequence and function, by unifying the PIR-PSD, Swiss- Prot, and TrEMBL databases. PIR offers a wide variety of resources mainly oriented to assist the propagation and standardization of protein annotation.

14 The home page can be seen as follows:
. The server functions in collaboration with the European Institute of Bioinformatics.")
15 3. ExPASy: The ExPASy (Expert Protein Analysis System) is a proteomics server of the Swiss Institute of Bioinformatics (SIB) which analyses protein sequences and structures and twodimensional gel electrophoresis (2-D Page electrophoresis). The server functions in collaboration with the European Institute of Bioinformatics. ExPASy also produces the protein sequence knowledgebase, UniProtKB/Swiss-prot, and its computer annotated supplement, UniProtKB/TrEmbl. The home page can be seen as follows:
16 C. Structure Databases 1. SCOP The SCOP database aims to provide a detailed and comprehensive description of the structural and evolutionary relationships between all proteins whose structure is known, including all entries in the Protein Data Bank (PDB). It is available as a set of tightly linked hypertext documents which make the large database comprehensible and accessible. In addition, the hypertext pages offer a panoply of representations of proteins, including links to PDB entries, sequences, references, images and interactive display systems. World Wide Web URL is the entry point to the database (MRC site). Existing automatic sequence and structure comparison tools cannot identify all structural and evolutionary relationships between proteins. The SCOP classification of proteins has been constructed manually by visual inspection and comparison of structures, but with the assistance of tools to make the task manageable and help provide generality. The job is made more challenging--and theoretically daunting--by the fact that the entities being organized are not homogeneous: sometimes it makes more sense to organize by individual domains, and other times by whole multi-domain proteins. Classification: Proteins are classified to reflect both structural and evolutionary relatedness. Many levels exist in the hierarchy, but the principal levels are family, superfamily and fold, described below. The exact position of boundaries between these levels are to some degree subjective. Our evolutionary classification is generally conservative: where any doubt about relatedness exists, we made new divisions at the family and superfamily levels. Thus, some researchers may prefer to focus on the higher levels of the classification tree, where proteins with structural similarity are clustered. The different major levels in the hierarchy are: 1. Family: Clear evolutionarily relationship Proteins clustered together into families are clearly evolutionarily related. Generally, this
17 means that pairwise residue identities between the proteins are 30% and greater. However, in some cases similar functions and structures provide definitive evidence of common descent in the absense of high sequence identity; for example, many globins form a family though some members have sequence identities of only 15%. 2. Superfamily: Probable common evolutionary origin Proteins that have low sequence identities, but whose structural and functional features suggest that a common evolutionary origin is probable are placed together in superfamilies. For example, actin, the ATPase domain of the heat shock protein, and hexakinase together form a superfamily. 3. Fold: Major structural similarity Proteins are defined as having a common fold if they have the same major secondary structures in the same arrangement and with the same topological connections. Different proteins with the same fold often have peripheral elements of secondary structure and turn regions that differ in size and conformation. In some cases, these differing peripheral regions may comprise half the structure. Proteins placed together in the same fold category may not have a common evolutionary origin: the structural similarities could arise just from the physics and chemistry of proteins favoring certain packing arrangements and chain topologies.

18 The home page can be seen as follows:
19 2. CATH: The CATH database is a hierarchical domain classification of protein structures in the Protein Data Bank (PDB, Berman et al. 2003). Only crystal structures solved to resolution better than 4.0 angstroms are considered, together with NMR structures. All non-proteins, models, and structures with greater than 30% "C-alpha only" are excluded from CATH. This filtering of the PDB is performed using the SIFT protocol (Michie et al., 1996). Protein structures are classified using a combination of automated and manual procedures. There are four major levels in this hierarchy: Class, Architecture, Topology (fold family) and Homologous superfamily (Orengo et al., 1997). Each level is described below, together with the methods used for defining domain boundaries and assigning structures to a specific family. The CATH Hierarchy and Classification Procedures: Automated Procedures If a given domain has sufficiently high sequence and structural similarity (ie. 35% sequence identity, SSAP score >= 80) with a domain that has been previously classified in CATH, the classification is automatically inherited from the other domain. Otherwise, the domain is classified manually, based upon an analysis of the results derived primarily from a range of comparison algorithms CATHEDRAL, HMMs, SSAP scores and relevant literature. Manual and Automated Procedures Combined Class, C-level Class is determined according to the secondary structure composition and packing within the structure. Three major classes are recognised; mainly-alpha, mainly-beta and alpha-beta. This last class (alpha-beta) includes both alternating alpha/beta structures and alpha+beta structures, as originally defined by Levitt and Chothia (1976). A fourth class is also identified which contains protein domains which have low secondary structure content.
20 Architecture, A-level This describes the overall shape of the domain structure as determined by the orientations of the secondary structures but ignores the connectivity between the secondary structures. It is currently assigned manually using a simple description of the secondary structure arrangement e.g. barrel or 3-layer sandwich. Reference is made to the literature for well-known architectures (e.g the betapropellor or alpha four helix bundle). Topology (Fold family), T-level Structures are grouped into fold groups at this level depending on both the overall shape and connectivity of the secondary structures. This is done using the structure comparison algorithm SSAP (Taylor & Orengo, 1989) and CATHEDRAL (Harrison et al. 2002, 2003). Parameters for clustering domains into the same fold family have been determined by empirical trials throughout the databank (Orengo et al. 1992; Orengo et al. 1993; Harrison et al. 2002, 2003). Structures which have a SSAP score of 70 and where at least 60% of the larger protein matches the smaller protein are assigned to the same T level or fold group. Some fold fgroups are very highly populated (Orengo et al. 1994); Orengo & Thornton, 2005) particularly within the mainly-beta 2- layer sandwich architectures and the alpha-beta 3-layer sandwich architectures. Homologous Superfamily, H-level This level groups together protein domains which are thought to share a common ancestor and can therefore be described as homologous. Similarities are identified either by high sequence identity or structure comparison using SSAP. Structures are clustered into the same homologous superfamily if they satisfy one of the following criteria:

21 The home page can be seen as follows:
22 3. PDB: The Protein Data Bank (PDB) is a repository for 3-D structural data of proteins and nucleic acids. This data, typically obtained by X-ray crystallography or NMR spectroscopy, is submitted by biologists and biochemists from around the world, is released into the public domain, and can be accessed for free. Founded in 1971 by Brookhaven National Laboratory, management of the Protein Data Bank was transferred in 1998 to members of the Research Collaboratory for Structural Bioinformatics (RCSB).The PDB is a key resource in structural biology and is critical to more recent work in structural genomics. Countless derived databases and projects have been developed to integrate and classify the PDB in terms of protein structure, protein function and protein evolution. When the PDB was originally founded it contained just 7 protein structures. Since then it has undergone an approximate exponential growth in the number of structures, which does not show any sign of falling off. Note that the database stores information about the exact location of all atoms in a large biomolecule (although, usually without the hydrogen atoms, as their positions are more of a statistical estimate); if one is only interested in sequence data, i.e. the list of amino acids making up a particular protein or the list of nucleotides making up a particular nucleic acid, the much larger databases from Swiss-Prot and the International Nucleotide Sequence Database Collaboration should be used.

23 The home page can be seen as follows:
24 4. MMDB: NCBI's structure database is called MMDB (Molecular Modeling Data Base), and it is a subset of three-dimensional structures obtained from the Protein Data Bank (PDB), excluding theoretical models. MMDB is a database of ASN.1-formatted records. It was designed for flexibility, and as such, is capable of archiving conventional structural data as well as future descriptions of bio-molecules, such as those generated by electron microscopy (surface models). Protein sequences from MMDB are extracted and available in the Entrez protein sequence database. They are linked to the 3-D -structures, therefore it is possible to determine whether a protein sequence in Entrez has homologs amongst known structures by examining its Related Sequences or Protein Neighbors and checking whether this set has any Structure Links.

25 The home page can be seen as follows:
26 D. Metabolic Pathway Databases 1. KEGG: Kyoto Encyclopedia of Genes & Genomes KEGG is a "biological systems" database integrating both molecular building block information and higher-level systemic information. Molecular building blocks are distinguished between genetic building blocks (KEGG GENES) and chemical building blocks (KEGG LIGAND), while the systemic information is represented as molecular wiring diagrams (KEGG PATHWAY) and hierarchies and relationships among biological objects (KEGG BRITE). KEGG PATHWAY - Manually drawn pathway maps representing our knowledge on the molecular interaction and reaction networks for metabolism, other cellular processes, and human diseases. KEGG BRITE - Functional hierarchies and binary relations of KEGG objects, including genes and proteins, compounds and reactions, drugs and diseases, and cells and organisms. KEGG GENES - Gene catalogs of all complete genomes and some partial genomes with ortholog annotation (KO assignment), enabling KEGG PATHWAY mapping and BRITE mapping. KEGG LIGAND - A composite database of chemical substances and reactions representing our knowledge on the chemical repertoire of biological systems and environments.

27 The home page can be seen as follows:
28 2. BRENDA: BRaunschweig ENzyme DAtabase BRENDA is the main collection of enzyme functional data available to the scientific community. It is available free of charge for academic, non-profit users via the internet ( and as an in-house database for commercial users (requests to our distributor Biobase). Recent progress on enzyme immobilisation, enzyme production, enzyme inhibition, coenzyme regeneration and enzyme engineering has opened up fascinating new fields for the potential application of enzymes in a large range of different areas. The enzymes are classified according to the Enzyme Commission list of enzymes. Some 3500 "different" enzymes are covered. Frequently enzymes with very different properties are included under the same EC number. Naturally it is not possible to cover all the numerous literature references for each enzyme (for some enzymes up to 40000) if the data representation is to be concise as is intended. The data collection is being developed into a metabolic network information system with links to Enzyme expression and regulation information. BRENDA is maintained and developed at the institute of Biochemistry at the University of Cologne. Data on enzyme function are extracted directly from the primary literature by scientists holding a degree in Biology or Chemistry. Formal and consistency checks are done by computer programs, each data set on a classified enzyme is checked manually by at least one biologist and one chemist.

29 The home page can be seen as follows:
30 3. METACYC: MetaCyc is a database of non-redundant experimentally elucidated metabolic pathways. It stores predominantly qualitative information rather than quantitative data, although we have recently began capturing quantitative data such as enzyme kinetics data. "MetaCyc" is pronounced "met-a-sike". It rhymes with "encyclopedia". A unique property of MetaCyc is that it is curated from the scientific experimental literature according to an extensive process such that: More than 900 different organisms are represented The majority of pathways occur in microorganisms and plants More than 900 metabolic pathways are stored, with more than 6,000 enzymatic reactions and more than 12,000 associated literature citations MetaCyc stores all enzyme-catalyzed reactions that have been assigned EC numbers by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) MetaCyc also stores hundreds of additional enzyme-catalyzed reactions that have not yet been assigned an EC number MetaCyc stores pathways involved in: Primary metabolism and Secondary metabolism MetaCyc also stores compounds, proteins, protein complexes and genes associated with these pathways. MetaCyc is extensively linked to other biological databases containing protein and nucleic-acid sequence data, bibliographic data and protein structures.unlike EcoCyc, MetaCyc provides little genomic data. MetaCyc does contain objects for the genes that encode many enzymes within the DB, but MetaCyc contains no sequence data. It does contain links to external sequence databases.

31 The home page can be seen as follows:
32 4. ECOCYC: EcoCyc is a bioinformatics database for the bacterium Escherichia coli K-12. The EcoCyc project performs literature-based curation of the E. coli genome, and of E. coli transcriptional regulation, transporters, and metabolic pathways. EcoCyc is a regularly updated electronic encyclopedia that contains written summaries of E. coli genes, distilled from more than 15,000 scientific articles. EcoCyc is also a computable description of the genome and cellular networks of E. coli that supports data mining and computational analyses. Objects in the EcoCyc database describe each E. coli gene and gene product. Database objects also describe molecular interactions, including metabolic pathways, transport events, and the regulation of gene expression.

33 The home page can be seen as follows:
34 E. Bibliographic Databases 1. PUBMED: Aim: To search books and articles related to PubMed. Description : 1. PubMed is a free search engine offering access to the MEDLINE database of citations. 2. Abstracts of biomedical research articles will be available in Pubmed. 3. The core subject is medicine, and PubMed covers fields related to medicin such as nursing and other allied health disciplines. 4. It also provides very full coverage of the related biomedical sciences, such as biochemistry and cell biology. 5. It is offered by the United States National Library of Medicine as part of theentrez information retrieval system. 6. The inclusion of an article in PubMed does not endorse that article's contents 7. Pubmed website : Procedure: 1. Access Pubmed homepage. 2. Give the book or article name in the search option and start the search. 3. After the search results will be displayed on the page.

35 The home page can be viewed as follows:

36 Searching a protein from pubmed:
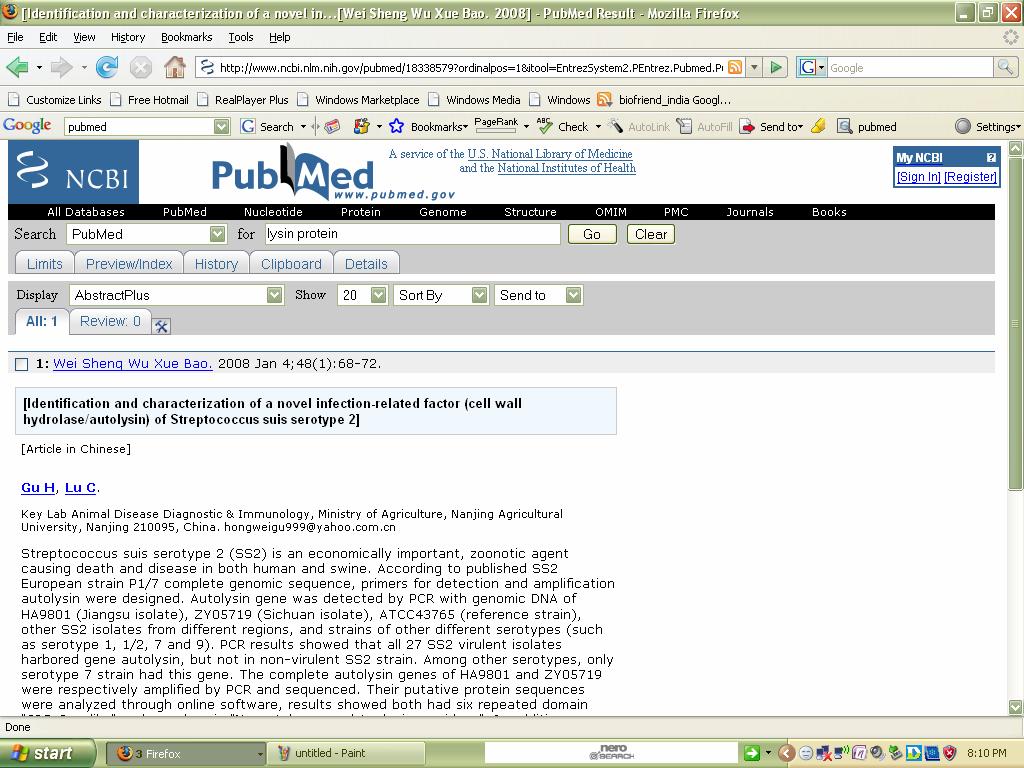
37
38 Result: Thus desired information can be taken from Pubmed. 2. OMIM (Online Mendelian Inheritance In Man): Aim: To find OMIM record for cancer. Description: 1. This database is a catalog of human genes and genetic disorders. 2. It has over entries. 3. This database is authored by DR.Victor A.Mc.Kusick and his colleagues at JohnsHopkins. 4. The database contains textual information and gives references. 5. It also contains copies to link Medline and sequence records in Entrez system and links related to resources at NCBI 6. Advanced search options are accessible via the Limits, Preview/Index History, and Clipboard options in the grey bar beneath the text box. Procedure: 1. Access the OMIM webpage. 2. Then give the desired search term in the search options. 3. It will search for that particular disease given and it will provide information. 4. Such that the desired information is displayed in the screen about the particular Disease.

39 The home page can be viewed as follows:
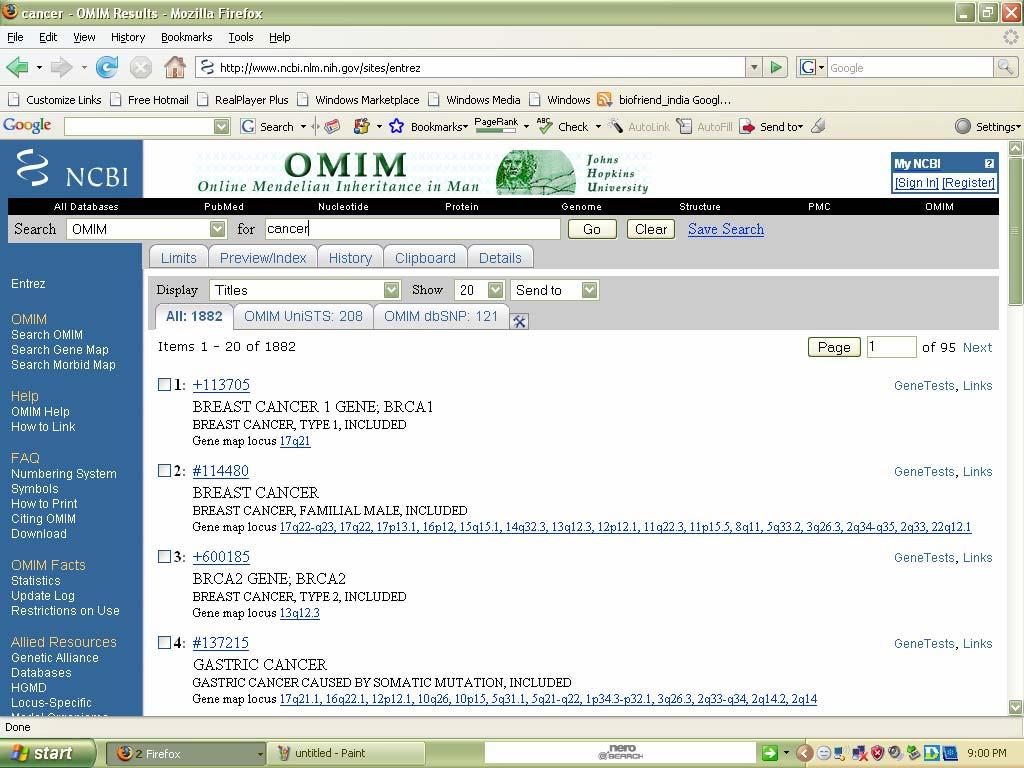
40
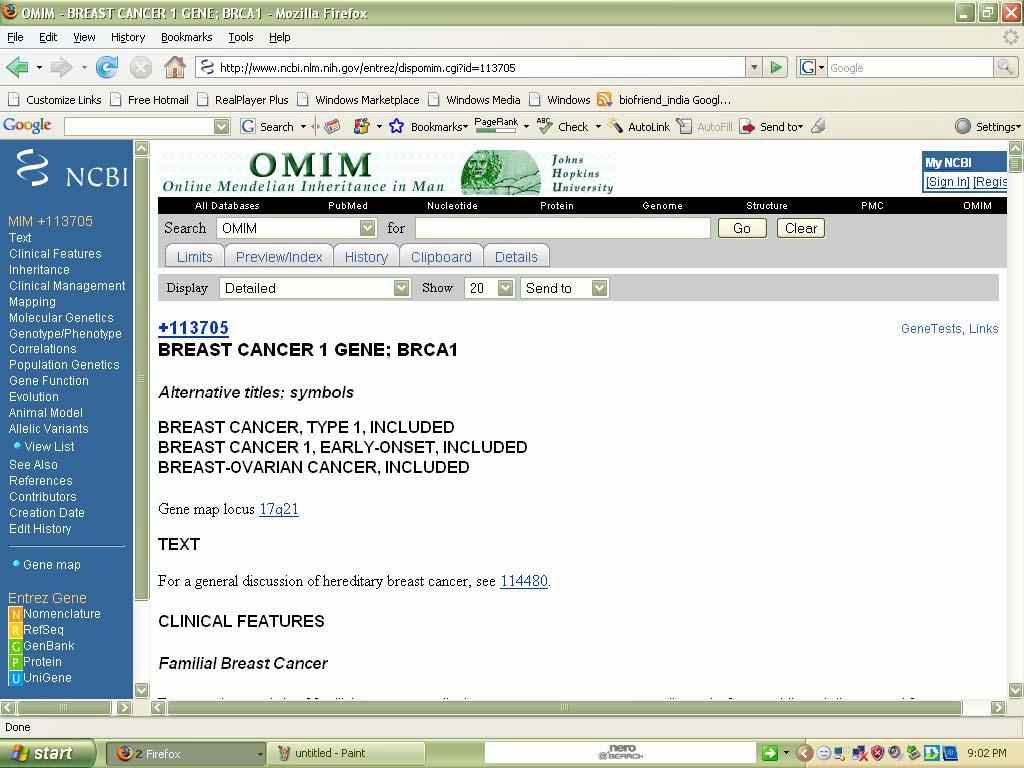
41 Result:
42 Thus information for a particular disease is successfully taken from OMIM. Ex no: 3 Date: Gene Prediction Tool (GenScan) Aim: To predict the gene regions in the given genomic DNA sequence by using a Gene Prediction Tool like GenScan. Procedure: We can utilize the file transfer protocol feature of NCBI to obtain complete genome sequences of any of the sequenced organisms freely available. To do this we enter the FTP site of the NCBI. It is available at OR we can go for genome option instead of all databases in ncbi for a particular organism. To use the FTP feature and gain access to all the utilities available at NCBI enter ftp.ncbi.nih.gov. We access the genomes directory of all the available options. A list of organisms is now available. Observation: GenScan server is maintained at the MIT. This server provides access to the program Genscan for predicting the locations and exon-intron structures of genes in genomic sequences from a variety of organisms. This server can accept sequences up to 1 million base pairs (1 Mbp) in length. The accuracy of GENSCAN is somewhat sensitive to exon length. GenScan is by far the most comprehensive and sophisticated Gene prediction tool available for free.

43 The home page of GENSCAN consists the following:
44 GenScan however has a few limitations as discussed below. Gene Number. The number of genes predicted in a sequence is usually approximately correct but it may also happen that, for instance, a predicted gene splices together exons from two real genes, or vice versa. Organism. The program was designed primarily to predict genes in human/vertebrate genomic sequences: accuracy may be lower for non-vertebrates. In particular, the vertebrate version of the program performs fairly well on Drosophila sequences and the maize and Arabidopsis versions perform fairly well on their respective organisms, but the program version for C. elegans sequences is still in the experimental stages. Other organisms have not been systematically tested. For prokaryotic or yeast sequences, the programs Glimmer, developed by Salzberg and colleagues, and/or GeneMark, developed by Borodovsky and McIninch, are recommended. Biases In Test Set. The Burset/Guigó test set is undoubtedly biased toward short genes with relatively simple exon/intron structure: as a consequence, the statistics given in the tables on the accuracy page may not be completely representative of the performance of the programs on typical genomic DNA sequences. Exon/Feature Type. As a rule, internal exons are predicted more accurately than initial or terminal exons, and exons are predicted more accurately than polyadenylation or promoter signals. Predicted promoters, in particular, are not reliable. If promoters are of primary interest, you may wish to try Martin Reese's NNPP program. Plant Splice Signals. If identification of splice sites in plant pre-mrnas is of primary interest, the SplicePredictor program developed by Brendel and colleagues is recommended.
45 Exon probabilities: 1) Very high probability GENSCAN exons (e.g., P > 0.99) are almost always exactly correct and thus can be used with confidence to design PCR primers for cdna amplification or for other purposes. 2) Moderate- to high-probability GENSCAN exons (e.g., 0.50 < P < 0.99) are exactly correct most of the time, with the likelihood of exact correctness only slightly lower on average than the stated probability. 3) Low-probability GENSCAN exons (P < 0.50) are not reliable and should be treated with caution. For certain purposes it might be desirable to ignore such exons. Under the probabilistic model of gene structural and compositional properties used by GENSCAN, each possible "parse" (gene structure description) which is compatible with the sequence is assigned a probability. The default output of the program is simply the "optimal" (highest probability) parse of the sequence. The exons in this optimal parse are referred to as "optimal exons" and the translation products of the corresponding "optimal genes" are printed as GENSCAN predicted peptides. The presence of certain types of repetitive elements in a sequence may sometimes distort the results of GENSCAN. In particular, L1 elements are often predicted as genes.
46 Genome:
47 Output: Input:
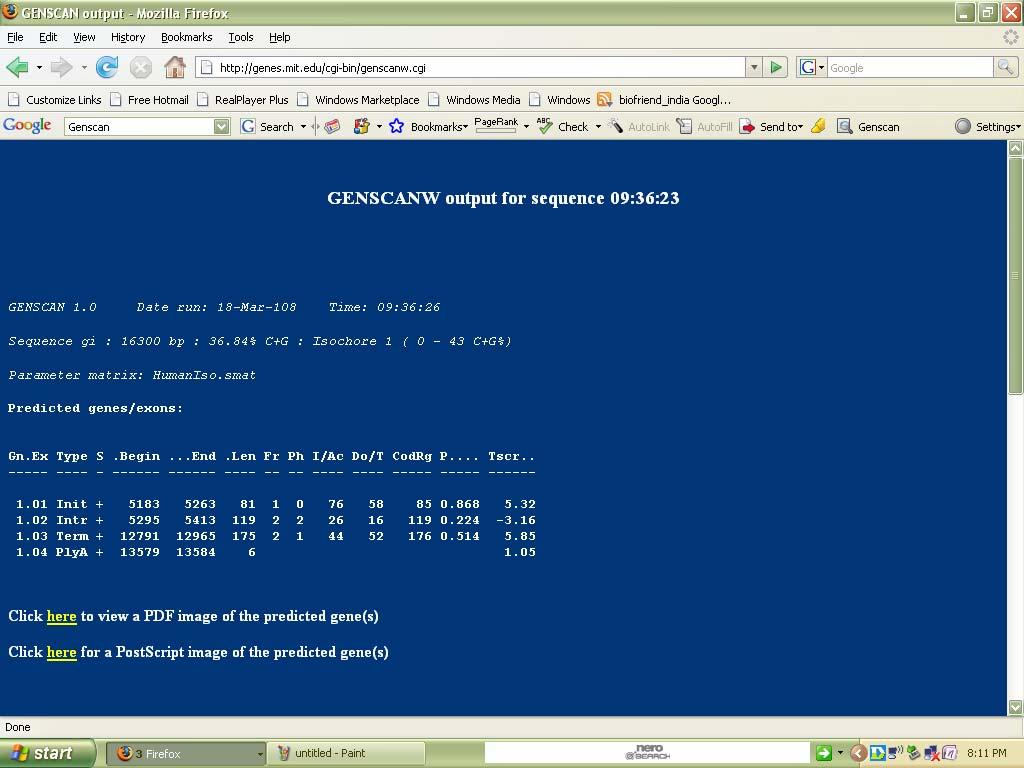
48
49
50 Gene Regions: to to to to Inference: Thus we can predict the gene and exons present in a genomic DNA sequence. The Exon type is also provided. The strand considered. The frame out of 6- frame translation considered. The 3 splice and 5 splice regions. and various other information can be interpreted via the output by GenScan. The accuracy depends upon the probability value. The higher it is the closer is the result to the best fit. Thus bioinformaticians can predict the gene regions to some degree of accuracy using GenScan.
51 Result: Thus we can predict the gene regions in the given genomic DNA sequence by using a Gene Prediction Tool like GenScan. Category Service Organism Gene Identification FGENEH integrated method Human Gene Identification GENE ID Vertebrates Gene Identification GENE MARK Many Gene Identification GENLANG Integrated method Dicots,Drosophilla, Vertebrates Gene Identification GENPARSER Integrated method Human
52 Gene Identification GENSCAN Integrated method Vertebrates & Plants Gene Identification GRAIL Human Gene Identification ECOPARSE E.coli Gene Identification GENEFINDER Any Gene Identification ORF FINDER Any Gene Identification PROCRUSTES many Gene Identification CBS-DENMARK Human & Arabidopsis Gene Identification ORPHEUS Bacterial
53 Ex.No: Date: Analysis of protein sequence using Expasy Characterisation Tools (Primary Structure Prediction) a. Prot Param tool Introduction: Protparam is a tool which allows the computation of various physical and chemical parameters for a given protein stored in Swiss-Prot or TrEMBL or for a user entered sequence. The computed parameters include the molecular weight, theoretical pi, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index and grand average of hydropathicity Procedure: 1. Go to expasy home from internet explorer and select expasy tools in the home page. 2. Select protparam from the expasy tools page and enter a Swiss-Prot/TrEMBL accession number (AC) in the space provided or you can paste your own sequence in the box provided. 3. Then click the compute parameters button provided.
54 The home page can be viewed as follows:

55 Protein: (GenPept form):
56 Input: GenPept format:
57 Output:
58
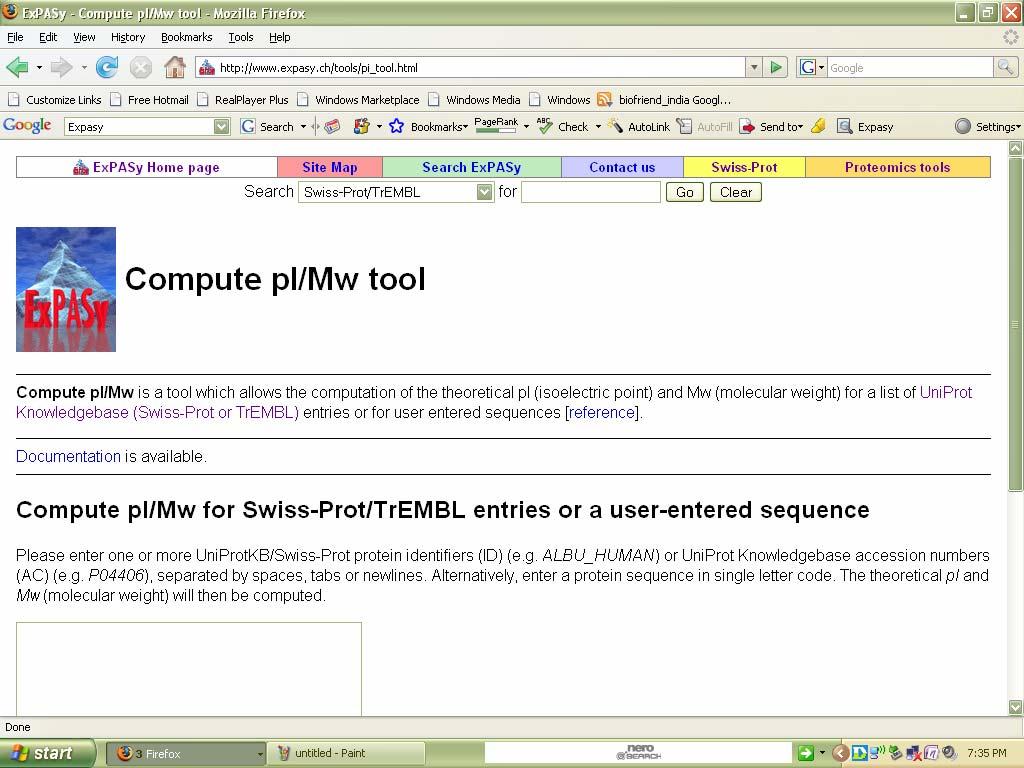
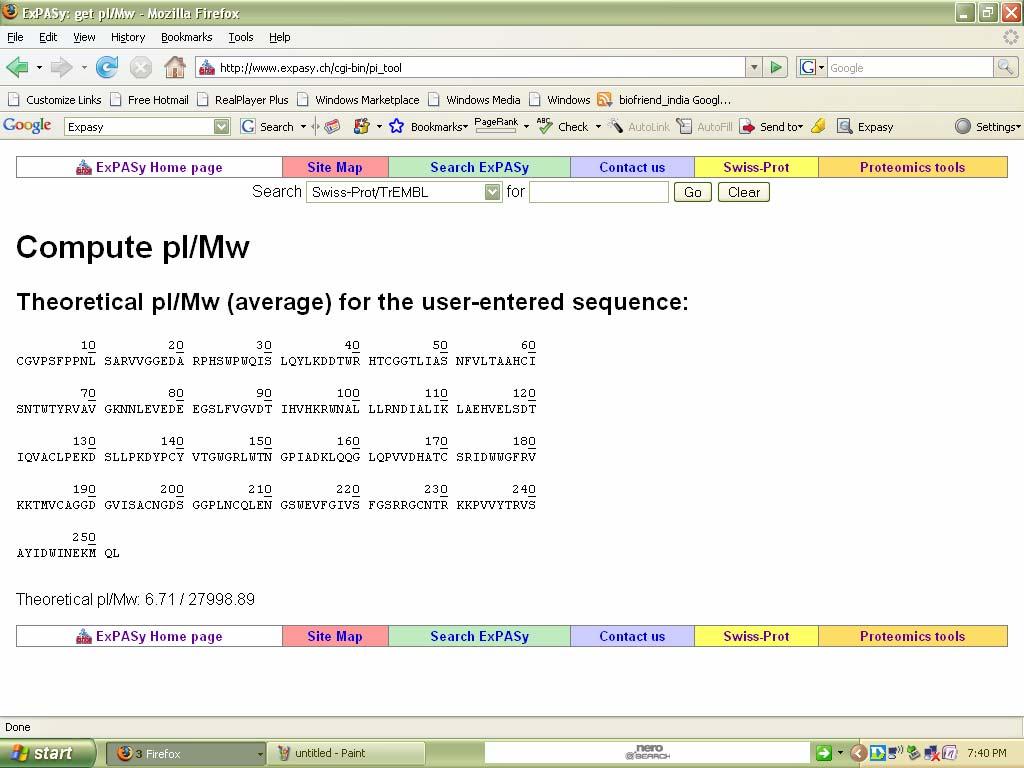
59 Compute pi/mw Introduction: Compute pi/mw: it is a tool which allows the computation of the theoretical pi (isoelectric point) and Mw (molecular weight) for a list of UniProt Knowledgebase (Swiss-Prot or TrEMBL) entries or for user entered sequences. Procedure: 1. Select compute pi/mw tool from expasy tools page that gets linked from the expasy home. 2. Enter a protein sequence in single letter code. The theoretical pi(isoelectric point) and Mw (molecular weight) will then be computed. 3.Then click compute parameters.
60 The home page can be viewed as follows
61 Protein:(GenPept form):
62 Input : fasta format:
63 Output:
64 Result: The above tools have been executed successfully Ex no: Date: Pattern & Profile Search PROSCAN Introduction: PROSCAN is a tool which is used to find the pattern &profile of the given protein.we can find the idendities in between the sequences. It has been developed by IBCP contribution to PBIL in Lyon, France. Procedure: 1.Go to Expasy homepage & select Expasy tools. 2. Select PROSCAN (pattern & profile tools) &enter the sequence in fasta format. 3. Then click submit.
65 The home page of PROSCAN can be viewed as follows:
66 Protein:(GenPept form):
67 Input: genpept format: Output:
68 Result: The above tool has been executed successfully.
69 Topological Prediction TMHMM Introduction: TMHMM is an Expasy tool which is used to predict the topology of a given Protein. Procedure: 1. Go to expasy home from internet explorer and select expasy tools in the home page. 2. Select TMHMM from the expasy tools page and you can paste your own protein sequence in FASTA format in the box provided in that page. 3. Then click the submit button provided there.
70 The home page of TMHMM can be viewed as following:

71 Protein: (GenPept form):
72 Input : FASTA format: Output:
73 Result:
74 No Helix was found in the given protein sequence. Ex no: Date: Secondary Structure prediction Tool Aim: To predict the secondary structure of a given protein sequence using GOR(IV) tool of expasy secondary structure prediction tools. GOR: GOR IV is the fourth version of GOR secondary structure prediction methods based on the information theory. There is no defined decision constant. GOR IV uses all possible pair frequencies within the window of 17 amino acid residues. Procedure: 1. Go to expasy home from your internet explorer and select expasy tools in the home page. 2. Select GOR (IV) from the expasy tools page and you can paste your own protein sequence in the box provided in that page. 3. Then click the submit button provided there.
75 The home page can be viewed as follows:
76

77 Protein: Input: GenPept format Output:
78 Result:
79 tool. The secondary structure of given protein sequence is determined using GOR (IV) Expasy Ex no: Date: Translate Tools a. Translate Introduction: Translate is a tool which allows the translation of a nucleotide (DNA/RNA) sequence to a protein sequence. Procedure: 1. Select translate tool from expasy tools page. 2. Here paste your DNA sequence(query) in the box provided. 3. Now click translate sequence button to obtain the result.
80 The home page can be viewed as follows:
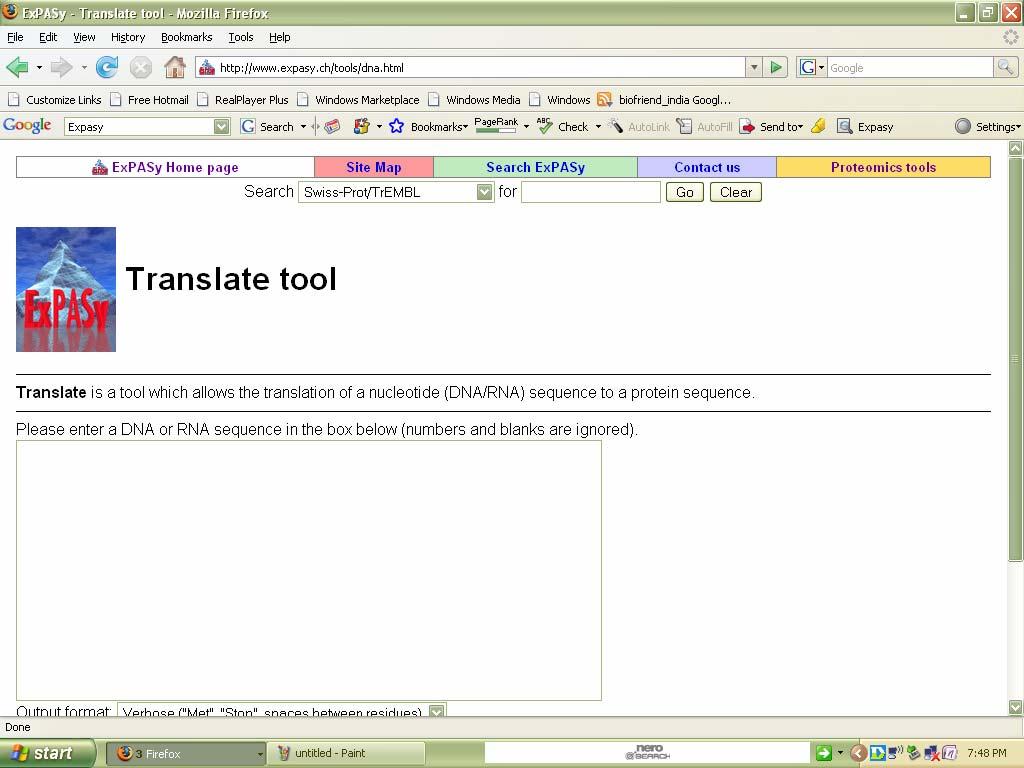
81
82 Nucleotide Sequence: Input : genpept format:
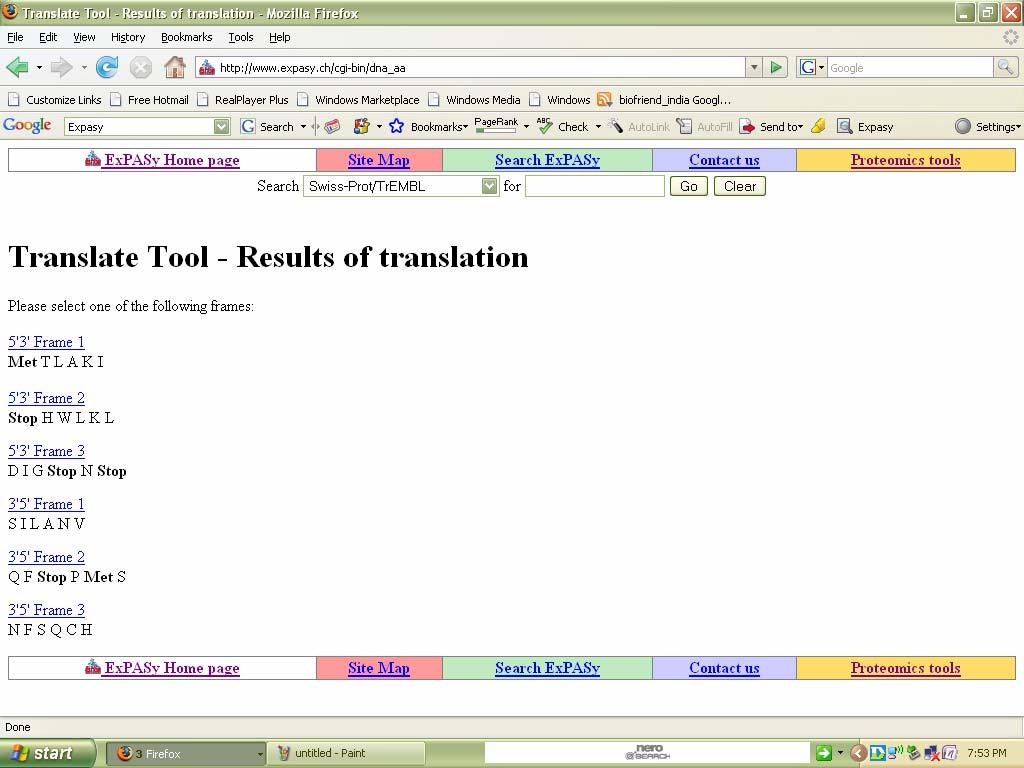
83 Output:
84 Result: Thus the given C-RNA sequence is translated into six frames protein sequences. Ex no: 5&6 Date: Sequence similarity searching of nucleotide sequences & Sequence similarity searching of protein sequences Similarity Search Tools
85 BLAST Aim: To study the BLAST similarity search tool. Algorithm: 1. Go to NCBI home page and click BLAST. 2. Then select the type of BLAST search you want. 3. After that paste your sequence in the box. 4. click format or BLAST to perform search operation. 5. You ll get results displayed based on the e-value and score. Introduction: The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families. Types of BLAST searches: Nucleotide Quickly search for highly similar sequences (megablast) Quickly search for divergent sequences (discontiguous megablast) Nucleotide-nucleotide BLAST (blastn) Search for short, nearly exact matches Search trace archives with megablast or discontiguous megablast
86 Protein Protein-protein BLAST (blastp) Position-specific iterated and pattern-hit initiated BLAST (PSI- and PHI-BLAST) Search for short, nearly exact matches Search the conserved domain database (rpsblast) Protein homology by domain architecture (cdart) Translated Translated query vs. protein database (blastx) Protein query vs. translated database (tblastn) Translated query vs. translated database (tblastx) Genomes Human, mouse, rat, chimp, cow, pig, dog, sheep, cat Chicken, puffer fish, zebrafish Fly, honey bee, other insects Microbes, environmental samples Plants, nematodes, Fungi, protozoa, other eukaryotes Special Search for gene expression data (GEO BLAST) Align two sequences (bl2seq)
87 Screen for vector contamination (VecScreen) Immunoglobin BLAST (IgBlast) Expectation value: The Expect value (E) is a parameter that describes the number of hits one can "expect" to see just by chance when searching a database of a particular size. It decreases exponentially with the Score (S) that is assigned to a match between two sequences. Essentially, the E value describes the random background noise that exists for matches between sequences. For example, an E value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance. This means that the lower the E- value, or the closer it is to "0" the more "significant" the match is. However, keep in mind that searches with short sequences, can be virtually identical and have relatively high EValue. This is because the calculation of the E-value also takes into account the length of the Query sequence. This is because shorter sequences have a high probability of occurring in the database purely by chance. For more details please see the calculations in the BLAST Course. The Expect value can also be used as a convenient way to create a significance threshold for reporting results. You can change the Expect value threshold on most main BLAST search pages. When the Expect value is increased from the default value of 10, a larger list with more lowscoring hits can be reported.
88 The home page can be seen as follows:
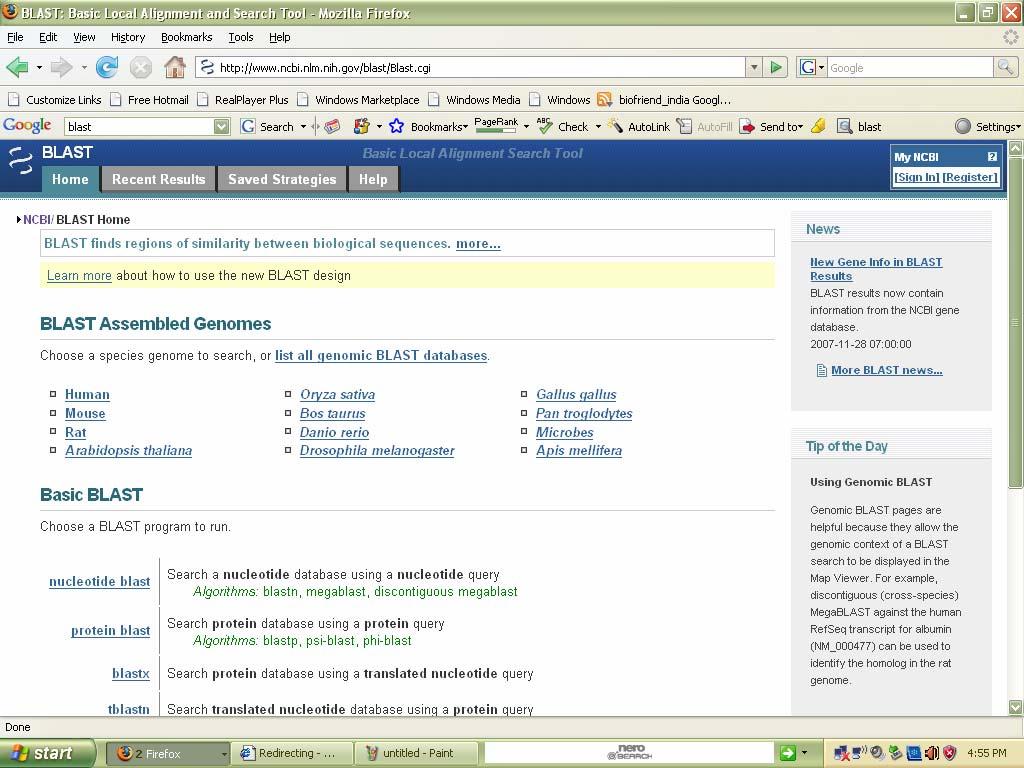
89
90 1.Blast Protein: GenPept format: Input: fasta format

91 Output: The distribution of list can be seen as follows

92 Alignment table:
93 Alignment in BLAST(first three alignments):

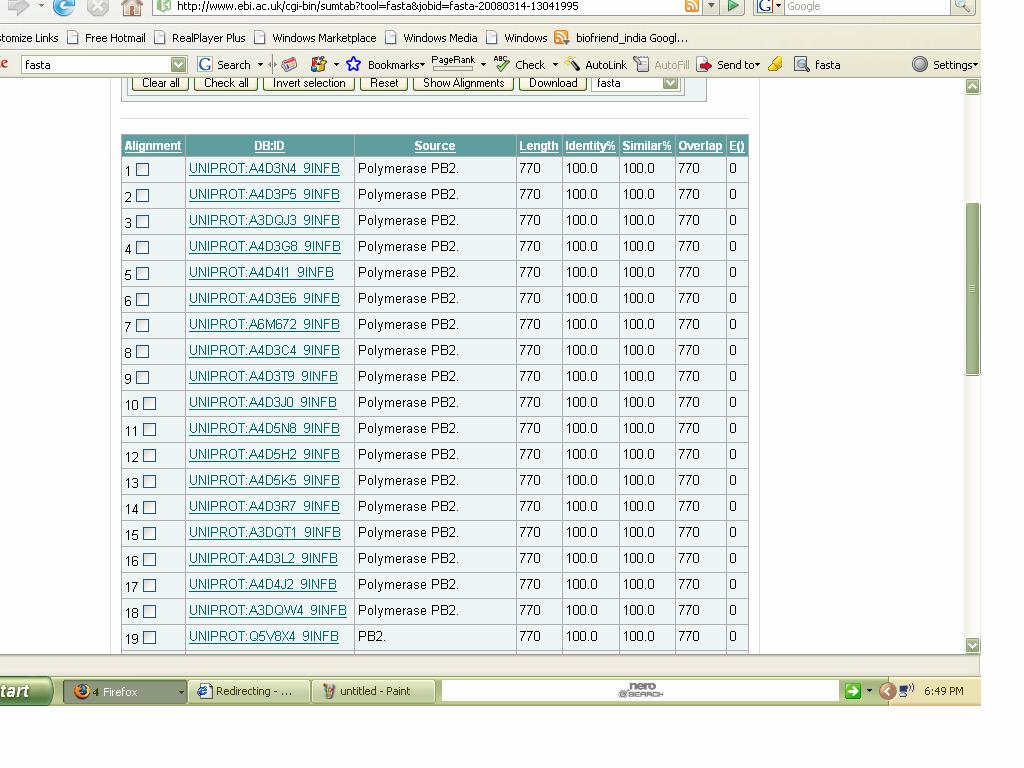
94
95 Hits Table: Hits Organism Score E- value Identity Gaps First Hit Heliotis sorenseni 321 7e % 0% Second Hit Heliotis sorenseni 292 4e-78 89% 0% Third Hit Heliotis sorenseni 292 5e-78 91% 0% 2. Blast Nucleotide: Nucleotide sequence (GenPept form):
96 Input (Fasta format): Output:

97

98 The distribution table can be observed as follows: Hits Table: Hits Organism Score E- value Identity Gaps First Hit Homo sapiens % 0% Second Hit Homo sapiens % 0% Third Hit Homo sapiens % 0% Result:
99 The BLAST tool has been studied with the above example. FASTA Introduction: FASTA stands for FAST-All, reflecting the fact that it can be used for a fast protein comparison or a fast nucleotide comparison. This program achieves a high level of sensitivity for similarity searching at high speed. This is achieved by performing optimised searches for local alignments using a substitution matrix. The high speed of this program is achieved by using the observed pattern of word hits to identify potential matches before attempting the more time consuming optimised search. The trade-off between speed and sensitivity is controlled by the ktup parameter, which specifies the size of the word. Increasing the ktup decreases the number of background hits. Not every word hit is investigated but instead initially looks for segment's containing several nearby hits. Types of FastA are: 1.Fast-A3 2.Fast-Y3 3.Fast-X3 4.Fast-F3 5.Fast-S3
100 FASTA HOME PAGE:

101 a. FASTA Nucleotide Aim: To perform local alignment for a given nucleotide sequence using FASTA. Procedure:
102 1.Open Google home page and click on FASTA link. 2.Choose the FASTA option given for Nucleotide. 3.Type the required sequence in FASTA format from NCBI. 4.Then click the format button and wait for a few seconds. 5.View the results and record the first 3 hits as observation. Nucleotide Sequence :(GenPept form) Input:
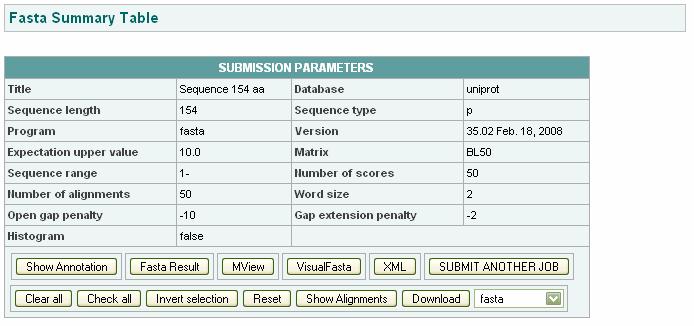
103 Fasta format: Observation: Fasta summary table:

104 Alignment table:
105

106 Alignments: Hits Table: Hits Organism Similarity E- value Identity Gaps First Hit Influenza B virus % 0% Second Hit Influenza B virus % 0%
107 Third Hit Influenza B virus % 0% 2. FASTA Protein: Aim: To perform local alignment for a given protein sequence using FASTA Procedure: 1.Open EMBL-EBI home page and click on FASTA link 2.Choose the FASTA option given for protein. 3.Type the required sequence in FastA format from NCBI. 4.Then click the format button and wait for a few seconds. 5.View the results and record the first 3 hits as observation.
108 Protein (GenPept form): Input : fasta format
109 Observation:
110 Alignment table:

111 Alignments:

112 Hits Table: Hits Organism Similarity E- value Identity Gaps First Hit Haliotis sorenseni e % 0% Second Hit Haliotis sorenseni % 0% Third Hit Haliotis sorenseni e % 0% Result: The local alignment for a given nucleotide and protein sequences were performed using FASTA and the observations was recorded.
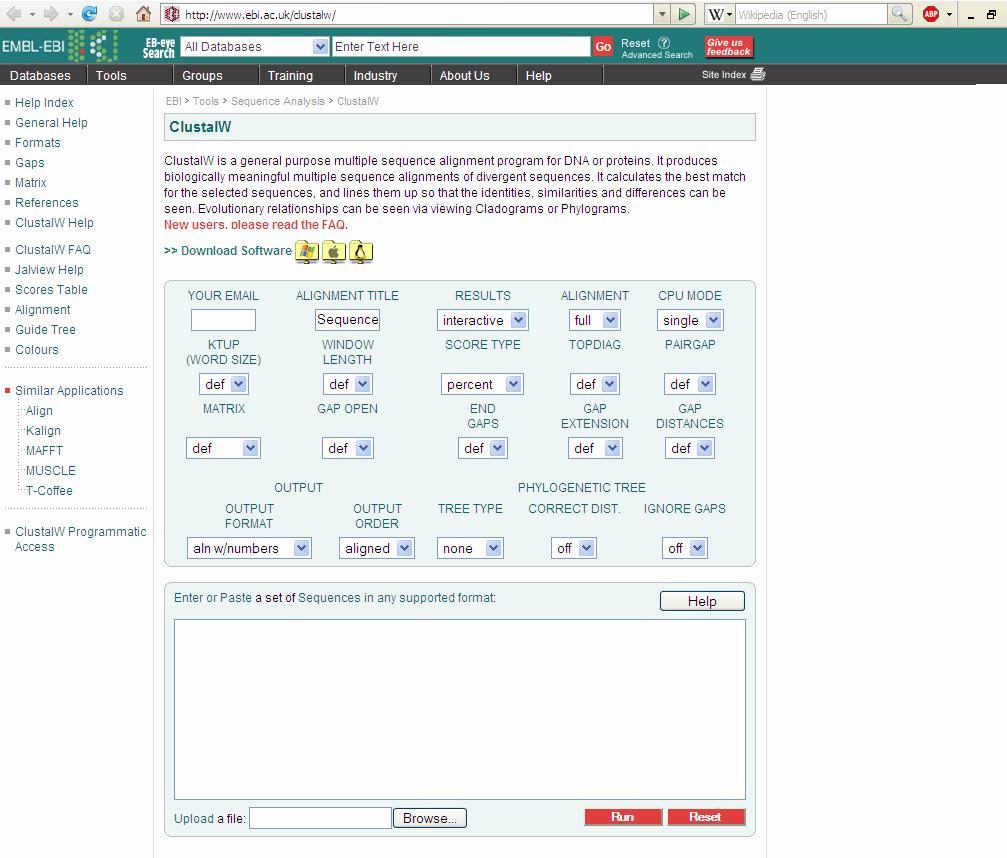
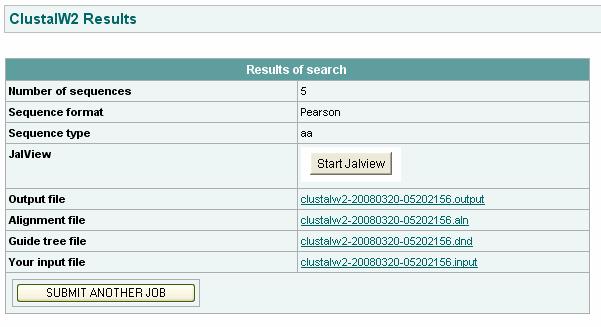
113 Ex no: 7 Date: Multiple sequence alignment ClustalW Aim: To find the evolutionary relationships between peptide sequences by performing Multiple Sequence Alignment using ClustalW. Given: 5 peptide sequences of different species to find their evolutionary relationship.the five peptide sequences in FASTA format is the input. Procedure: To perform Multiple Sequence Alignment: Step 1: Obtain peptide sequences of the given accession numbers from NCBI in FASTA format. Step 2: Enter these sequences into the ClustalW software in the given space. Step 3: We keep all the result modification options at default. Step 4: We Run ClustalW. We obtain the output. The alignment is done which can be interpreted. Step 5: A phylogram and a cladogram make interpretation of results easy.

114 Observation: Multiple alignments of protein sequences are important tools in studying sequences. The basic information they provide is identification of conserved sequence regions. This is very useful in designing experiments to test and modify the function of specific proteins, in predicting the function and structure of proteins, and in identifying new members of protein families. Sequences can be aligned across their entire length (global alignment) or only in certain regions (local alignment). This is true for pairwise and multiple alignments. Global alignments need to use gaps (representing insertions/deletions) while local alignments can avoid them, aligning regions between gaps. ClustalW is a fully automatic program for global multiple alignment of DNA and protein sequences. The alignment is progressive and considers the sequence redundancy. Trees can also be calculated from multiple alignments. The program has some adjustable parameters with reasonable defaults. Colour Codes: Consensus symbol: An alignment will display by default the following symbols denoting the degree of conservation observed in each column: "*" means that the residues or nucleotides in that column are identical in all sequences in the alignment.
115 ":" means that conserved substitutions have been observed, according to the COLOUR table above. "." means that semi-conserved substitutions are observed. Phylogenetic Tree: Phylogram: Phylogram is a branching diagram (tree) assumed to be an estimate of a phylogeny, branch lengths are proportional to the amount of inferred evolutionary change. Cladogram: A Cladogram is a branching diagram (tree) assumed to be an estimate of a phylogeny where the branches are of equal length, thus cladograms show common ancestry, but do not indicate the amount of evolutionary "time" separating taxa. The home page can be viewed as follows:
116 Input proteins:fasta id:
117 Output:

118 Cladogram: Phylogram: Result: Thus the evolutionary relationships between peptide sequences is found by performing Multiple Sequence Alignment using ClustalW.
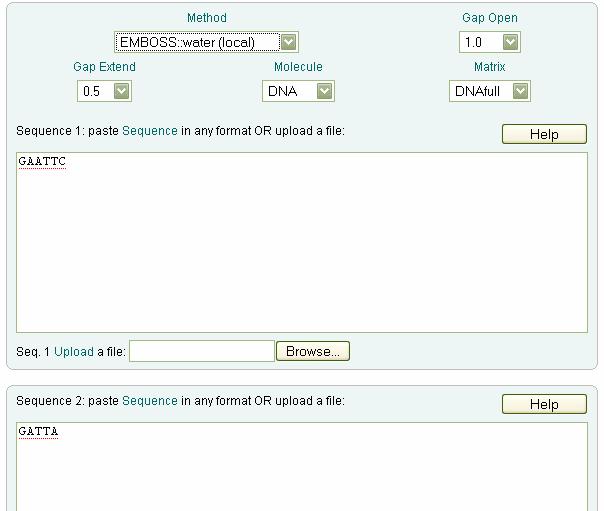
119 Ex no: 8&9 Date: Dynamic programming method- local alignment & Dynamic programming method- global alignment Dynamic programming method- local alignment Dynamic programming method- global alignment Global and local alignment of sequences Aim: To perform Global and Local alignment for the given sequences.
120 Dynamic Programming: In dynamic programming technique,first we solve small sub problems on then using the solutions of small problems major main problem will be solved. We use dynamic programming for both local and global alignment of two sequences A. Global Alignment Global alignment: Global alignment considers similarity across the full extend of the sequences. Global alignment aligns two sequences with maximum score by introducing gap wherever possible. In 1970Needleman and Wunch proposed a maximum-match pathway that can be obtained computationally by applying a straight forward algorithm. Procedure: Step1: First we have to build scoring matrix based on the given base condition for the given sequences Step2: We have to fill the path graph for the two sequences. The formula for filing the path graph is F(i,j)=max[f(i-1,j-1)+s(x i,y j ), f(i,j-1)+d, f(i-1,j)+d] Where F(i,j) the entry in the i th row and j th column of the path graph
121 s(x i,y j )--the score residues being aligned according to the base condition. f(i-1,j-1)+s(x i,y j ) represents a diagonal move on the path graph f(i,j-1)+d represents horizontal move in which the corresponding residue is aligned with gap. d is the penalty for gap given in base condition. f(i-1,j)+d represents a vertical move on the path graph in which the corresponding residue is aligned with gap. Step3: Backtracking should be done to optimal alignment Example of global alignment: Given sequences Seq1: GAATTC Seq2: GATTA Base Condition: GAP = -2 MATCH = 2 MISMATCH = -1
122 SCORING MATRIX - G A A T T C G A T T A Path Graph y - G A A T T C
123 G A T T A x Optimal Alignment: G A A T T C. G _ A T T A
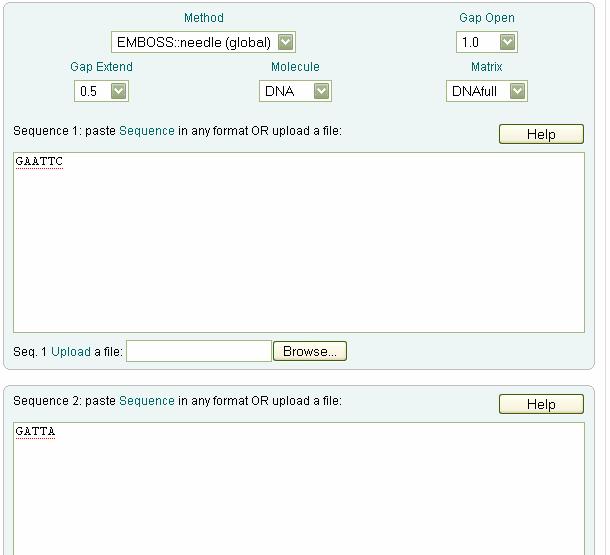
124 Output:

125 B. Local Alignment
126 Local alignment: Local alignment is used for aligning two sequences of almost same length. it is used to find similar regions in the given two sequences. Here gaps are not allowed. In 1981 Smith-waterman described a method for finding common regions of similarity Procedure: Step1: First we have to build scoring matrix based on the given base condition for the given sequences. Since gaps and mismatches are not allowed usually we will change all the negative values to zero Step2: We have to fill the path graph for the two sequences. The formula for filing the path graph is F(i,j)=max[f(i-1,j-1)+s(x i,y j ), f(i,j-1)+d, f(i-1,j)+d] Where F(i,j) the entry in the i th row and j th column of the path graph s(x i, y j )--the score residues being aligned according to the base condition. f(i-1,j-1)+s(x i,y j ) represents a diagonal move on the path graph f(i,j-1)+d represents horizontal move in which the corresponding residue is aligned with gap. d is the penalty for gap given in base condition. f(i-1,j)+d represents a vertical move on the path graph in which the corresponding residue is aligned with gap. Step3: Backtracking should be done to find optimal alignment. We should backtrack from maximum score found in the path graph
127 Example of local alignment: Given sequences are Seq1: GAATTC Seq2: GATTA Base Condition: MATCH = 2 MISMATCH = -1 GAP = -2 PATH GRAPH - G A A T T C G A T
128 T A
129 Output:

130 Result: The sequences were aligned using Global and Local Alignment.
Two Mark question and Answers
 1. Define Bioinformatics Two Mark question and Answers Bioinformatics is the field of science in which biology, computer science, and information technology merge into a single discipline. There are three
1. Define Bioinformatics Two Mark question and Answers Bioinformatics is the field of science in which biology, computer science, and information technology merge into a single discipline. There are three
Sequence Databases and database scanning
 Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Sequence Databases and database scanning Marjolein Thunnissen Lund, 2012 Types of databases: Primary sequence databases (proteins and nucleic acids). Composite protein sequence databases. Secondary databases.
Protein Bioinformatics Part I: Access to information
 Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Protein Bioinformatics Part I: Access to information 260.655 April 6, 2006 Jonathan Pevsner, Ph.D. pevsner@kennedykrieger.org Outline [1] Proteins at NCBI RefSeq accession numbers Cn3D to visualize structures
Bioinformatics Tools. Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine
 Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
Types of Databases - By Scope
 Biological Databases Bioinformatics Workshop 2009 Chi-Cheng Lin, Ph.D. Department of Computer Science Winona State University clin@winona.edu Biological Databases Data Domains - By Scope - By Level of
Biological Databases Bioinformatics Workshop 2009 Chi-Cheng Lin, Ph.D. Department of Computer Science Winona State University clin@winona.edu Biological Databases Data Domains - By Scope - By Level of
ELE4120 Bioinformatics. Tutorial 5
 ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
ELE4120 Bioinformatics Tutorial 5 1 1. Database Content GenBank RefSeq TPA UniProt 2. Database Searches 2 Databases A common situation for alignment is to search through a database to retrieve the similar
Introduction to Bioinformatics CPSC 265. What is bioinformatics? Textbooks
 Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
Introduction to Bioinformatics CPSC 265 Thanks to Jonathan Pevsner, Ph.D. Textbooks Johnathan Pevsner, who I stole most of these slides from (thanks!) has written a textbook, Bioinformatics and Functional
I nternet Resources for Bioinformatics Data and Tools
 ~i;;;;;;;'s :.. ~,;;%.: ;!,;s163 ~. s :s163:: ~s ;'.:'. 3;3 ~,: S;I:;~.3;3'/////, IS~I'//. i: ~s '/, Z I;~;I; :;;; :;I~Z;I~,;'//.;;;;;I'/,;:, :;:;/,;'L;;;~;'~;~,::,:, Z'LZ:..;;',;';4...;,;',~/,~:...;/,;:'.::.
~i;;;;;;;'s :.. ~,;;%.: ;!,;s163 ~. s :s163:: ~s ;'.:'. 3;3 ~,: S;I:;~.3;3'/////, IS~I'//. i: ~s '/, Z I;~;I; :;;; :;I~Z;I~,;'//.;;;;;I'/,;:, :;:;/,;'L;;;~;'~;~,::,:, Z'LZ:..;;',;';4...;,;',~/,~:...;/,;:'.::.
Sequence Based Function Annotation
 Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Sequence Based Function Annotation 1. Given a sequence, how to predict its biological
Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Sequence Based Function Annotation 1. Given a sequence, how to predict its biological
Protein Sequence Analysis. BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl)
") Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
G4120: Introduction to Computational Biology
 G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Lecture 3 February 13, 2003 Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics
G4120: Introduction to Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Lecture 3 February 13, 2003 Copyright 2003 Oliver Jovanovic, All Rights Reserved. Bioinformatics
Introduction to BIOINFORMATICS
 Introduction to BIOINFORMATICS Antonella Lisa CABGen Centro di Analisi Bioinformatica per la Genomica Tel. 0382-546361 E-mail: lisa@igm.cnr.it http://www.igm.cnr.it/pagine-personali/lisa-antonella/ What
Introduction to BIOINFORMATICS Antonella Lisa CABGen Centro di Analisi Bioinformatica per la Genomica Tel. 0382-546361 E-mail: lisa@igm.cnr.it http://www.igm.cnr.it/pagine-personali/lisa-antonella/ What
ONLINE BIOINFORMATICS RESOURCES
 Dedan Githae Email: d.githae@cgiar.org BecA-ILRI Hub; Nairobi, Kenya 16 May, 2014 ONLINE BIOINFORMATICS RESOURCES Introduction to Molecular Biology and Bioinformatics (IMBB) 2014 The larger picture.. Lower
Dedan Githae Email: d.githae@cgiar.org BecA-ILRI Hub; Nairobi, Kenya 16 May, 2014 ONLINE BIOINFORMATICS RESOURCES Introduction to Molecular Biology and Bioinformatics (IMBB) 2014 The larger picture.. Lower
NCBI web resources I: databases and Entrez
 NCBI web resources I: databases and Entrez Yanbin Yin Most materials are downloaded from ftp://ftp.ncbi.nih.gov/pub/education/ 1 Homework assignment 1 Two parts: Extract the gene IDs reported in table
NCBI web resources I: databases and Entrez Yanbin Yin Most materials are downloaded from ftp://ftp.ncbi.nih.gov/pub/education/ 1 Homework assignment 1 Two parts: Extract the gene IDs reported in table
Outline. Evolution. Adaptive convergence. Common similarity problems. Chapter 7: Similarity searches on sequence databases
 Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Textbook Reading Guidelines
 Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Compiled by Mr. Nitin Swamy Asst. Prof. Department of Biotechnology
 Bioinformatics Model Answers Compiled by Mr. Nitin Swamy Asst. Prof. Department of Biotechnology Page 1 of 15 Previous years questions asked. 1. Describe the software used in bioinformatics 2. Name four
Bioinformatics Model Answers Compiled by Mr. Nitin Swamy Asst. Prof. Department of Biotechnology Page 1 of 15 Previous years questions asked. 1. Describe the software used in bioinformatics 2. Name four
FACULTY OF BIOCHEMISTRY AND MOLECULAR MEDICINE
 FACULTY OF BIOCHEMISTRY AND MOLECULAR MEDICINE BIOMOLECULES COURSE: COMPUTER PRACTICAL 1 Author of the exercise: Prof. Lloyd Ruddock Edited by Dr. Leila Tajedin 2017-2018 Assistant: Leila Tajedin (leila.tajedin@oulu.fi)
FACULTY OF BIOCHEMISTRY AND MOLECULAR MEDICINE BIOMOLECULES COURSE: COMPUTER PRACTICAL 1 Author of the exercise: Prof. Lloyd Ruddock Edited by Dr. Leila Tajedin 2017-2018 Assistant: Leila Tajedin (leila.tajedin@oulu.fi)
Why learn sequence database searching? Searching Molecular Databases with BLAST
 Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
Why learn sequence database searching? Searching Molecular Databases with BLAST What have I cloned? Is this really!my gene"? Basic Local Alignment Search Tool How BLAST works Interpreting search results
Bioinformatics for Proteomics. Ann Loraine
 Bioinformatics for Proteomics Ann Loraine aloraine@uab.edu What is bioinformatics? The science of collecting, processing, organizing, storing, analyzing, and mining biological information, especially data
Bioinformatics for Proteomics Ann Loraine aloraine@uab.edu What is bioinformatics? The science of collecting, processing, organizing, storing, analyzing, and mining biological information, especially data
EECS 730 Introduction to Bioinformatics Sequence Alignment. Luke Huan Electrical Engineering and Computer Science
 EECS 730 Introduction to Bioinformatics Sequence Alignment Luke Huan Electrical Engineering and Computer Science http://people.eecs.ku.edu/~jhuan/ Database What is database An organized set of data Can
EECS 730 Introduction to Bioinformatics Sequence Alignment Luke Huan Electrical Engineering and Computer Science http://people.eecs.ku.edu/~jhuan/ Database What is database An organized set of data Can
Genomic Annotation Lab Exercise By Jacob Jipp and Marian Kaehler Luther College, Department of Biology Genomics Education Partnership 2010
 Genomic Annotation Lab Exercise By Jacob Jipp and Marian Kaehler Luther College, Department of Biology Genomics Education Partnership 2010 Genomics is a new and expanding field with an increasing impact
Genomic Annotation Lab Exercise By Jacob Jipp and Marian Kaehler Luther College, Department of Biology Genomics Education Partnership 2010 Genomics is a new and expanding field with an increasing impact
FUNCTIONAL BIOINFORMATICS
 Molecular Biology-2018 1 FUNCTIONAL BIOINFORMATICS PREDICTING THE FUNCTION OF AN UNKNOWN PROTEIN Suppose you have found the amino acid sequence of an unknown protein and wish to find its potential function.
Molecular Biology-2018 1 FUNCTIONAL BIOINFORMATICS PREDICTING THE FUNCTION OF AN UNKNOWN PROTEIN Suppose you have found the amino acid sequence of an unknown protein and wish to find its potential function.
Just the Facts: A Basic Introduction to the Science Underlying NCBI Resources
 National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools News About NCBI Site Map
National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools News About NCBI Site Map
Bioinformatics Prof. M. Michael Gromiha Department of Biotechnology Indian Institute of Technology, Madras. Lecture - 5a Protein sequence databases
 Bioinformatics Prof. M. Michael Gromiha Department of Biotechnology Indian Institute of Technology, Madras Lecture - 5a Protein sequence databases In this lecture, we will mainly discuss on Protein Sequence
Bioinformatics Prof. M. Michael Gromiha Department of Biotechnology Indian Institute of Technology, Madras Lecture - 5a Protein sequence databases In this lecture, we will mainly discuss on Protein Sequence
Bioinformatics & Protein Structural Analysis. Bioinformatics & Protein Structural Analysis. Learning Objective. Proteomics
 The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
Basic Bioinformatics: Homology, Sequence Alignment,
 Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Introduction to Bioinformatics
 Introduction to Bioinformatics Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com
Introduction to Bioinformatics Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com
G4120: Introduction to Computational Biology
 ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
Biotechnology Explorer
 Biotechnology Explorer C. elegans Behavior Kit Bioinformatics Supplement explorer.bio-rad.com Catalog #166-5120EDU This kit contains temperature-sensitive reagents. Open immediately and see individual
Biotechnology Explorer C. elegans Behavior Kit Bioinformatics Supplement explorer.bio-rad.com Catalog #166-5120EDU This kit contains temperature-sensitive reagents. Open immediately and see individual
Gene Identification in silico
 Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Genome Informatics. Systems Biology and the Omics Cascade (Course 2143) Day 3, June 11 th, Kiyoko F. Aoki-Kinoshita
 Day 3, June 11 th, Kiyoko F. Aoki-Kinoshita") Genome Informatics Systems Biology and the Omics Cascade (Course 2143) Day 3, June 11 th, 2008 Kiyoko F. Aoki-Kinoshita Introduction Genome informatics covers the computer- based modeling and data processing
Genome Informatics Systems Biology and the Omics Cascade (Course 2143) Day 3, June 11 th, 2008 Kiyoko F. Aoki-Kinoshita Introduction Genome informatics covers the computer- based modeling and data processing
ab initio and Evidence-Based Gene Finding
 ab initio and Evidence-Based Gene Finding A basic introduction to annotation Outline What is annotation? ab initio gene finding Genome databases on the web Basics of the UCSC browser Evidence-based gene
ab initio and Evidence-Based Gene Finding A basic introduction to annotation Outline What is annotation? ab initio gene finding Genome databases on the web Basics of the UCSC browser Evidence-based gene
Following text taken from Suresh Kumar. Bioinformatics Web - Comprehensive educational resource on Bioinformatics. 6th May.2005
 Bioinformatics is the recording, annotation, storage, analysis, and searching/retrieval of nucleic acid sequence (genes and RNAs), protein sequence and structural information. This includes databases of
Bioinformatics is the recording, annotation, storage, analysis, and searching/retrieval of nucleic acid sequence (genes and RNAs), protein sequence and structural information. This includes databases of
Product Applications for the Sequence Analysis Collection
 Product Applications for the Sequence Analysis Collection Pipeline Pilot Contents Introduction... 1 Pipeline Pilot and Bioinformatics... 2 Sequence Searching with Profile HMM...2 Integrating Data in a
Product Applications for the Sequence Analysis Collection Pipeline Pilot Contents Introduction... 1 Pipeline Pilot and Bioinformatics... 2 Sequence Searching with Profile HMM...2 Integrating Data in a
Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH. BIOL 7210 A Computational Genomics 2/18/2015
 Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH BIOL 7210 A Computational Genomics 2/18/2015 The $1,000 genome is here! http://www.illumina.com/systems/hiseq-x-sequencing-system.ilmn Bioinformatics bottleneck
Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH BIOL 7210 A Computational Genomics 2/18/2015 The $1,000 genome is here! http://www.illumina.com/systems/hiseq-x-sequencing-system.ilmn Bioinformatics bottleneck
Lecture 7 Motif Databases and Gene Finding
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 7 Motif Databases and Gene Finding Motif Databases & Gene Finding Motifs Recap Motif Databases TRANSFAC
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il Lecture 7 Motif Databases and Gene Finding Motif Databases & Gene Finding Motifs Recap Motif Databases TRANSFAC
A New Database of Genetic and. Molecular Pathways. Minoru Kanehisa. sequencing projects have been. Mbp) and for several bacteria including
 and for several bacteria including") Toward Pathway Engineering: A New Database of Genetic and Molecular Pathways Minoru Kanehisa Institute for Chemical Research, Kyoto University From Genome Sequences to Functions The Human Genome Project
Toward Pathway Engineering: A New Database of Genetic and Molecular Pathways Minoru Kanehisa Institute for Chemical Research, Kyoto University From Genome Sequences to Functions The Human Genome Project
Klinisk kemisk diagnostik BIOINFORMATICS
 Klinisk kemisk diagnostik - 2017 BIOINFORMATICS What is bioinformatics? Bioinformatics: Research, development, or application of computational tools and approaches for expanding the use of biological,
Klinisk kemisk diagnostik - 2017 BIOINFORMATICS What is bioinformatics? Bioinformatics: Research, development, or application of computational tools and approaches for expanding the use of biological,
Genomic region (ENCODE) Gene definitions
 Gene definitions") DNA From genes to proteins Bioinformatics Methods RNA PROMOTER ELEMENTS TRANSCRIPTION Iosif Vaisman mrna SPLICE SITES SPLICING Email: ivaisman@gmu.edu START CODON STOP CODON TRANSLATION PROTEIN From genes
DNA From genes to proteins Bioinformatics Methods RNA PROMOTER ELEMENTS TRANSCRIPTION Iosif Vaisman mrna SPLICE SITES SPLICING Email: ivaisman@gmu.edu START CODON STOP CODON TRANSLATION PROTEIN From genes
Sequence Based Function Annotation. Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University
 Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
Sequence Based Function Annotation Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Usage scenarios for sequence based function annotation Function prediction of newly cloned
COMPUTER RESOURCES II:
 COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
G4120: Introduction to Computational Biology
 ICB Fall 2009 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Copyright 2009 Oliver Jovanovic, All Rights Reserved. Analysis of Protein
ICB Fall 2009 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology & Immunology Copyright 2009 Oliver Jovanovic, All Rights Reserved. Analysis of Protein
Computational Biology and Bioinformatics
 Computational Biology and Bioinformatics Computational biology Development of algorithms to solve problems in biology Bioinformatics Application of computational biology to the analysis and management
Computational Biology and Bioinformatics Computational biology Development of algorithms to solve problems in biology Bioinformatics Application of computational biology to the analysis and management
This place covers: Methods or systems for genetic or protein-related data processing in computational molecular biology.
 G16B BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY Methods or systems for genetic
G16B BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY Methods or systems for genetic
Will discuss proteins in view of Sequence (I,II) Structure (III) Function (IV) proteins in practice
 Structure (III) Function (IV) proteins in practice") Will discuss proteins in view of Sequence (I,II) Structure (III) Function (IV) proteins in practice integration - web system (V) 1 Touring the Protein Space (outline) 1. Protein Sequence - how rich? How
Will discuss proteins in view of Sequence (I,II) Structure (III) Function (IV) proteins in practice integration - web system (V) 1 Touring the Protein Space (outline) 1. Protein Sequence - how rich? How
The University of California, Santa Cruz (UCSC) Genome Browser
 Genome Browser") The University of California, Santa Cruz (UCSC) Genome Browser There are hundreds of available userselected tracks in categories such as mapping and sequencing, phenotype and disease associations, genes,
The University of California, Santa Cruz (UCSC) Genome Browser There are hundreds of available userselected tracks in categories such as mapping and sequencing, phenotype and disease associations, genes,
This practical aims to walk you through the process of text searching DNA and protein databases for sequence entries.
 PRACTICAL 1: BLAST and Sequence Alignment The EBI and NCBI websites, two of the most widely used life science web portals are introduced along with some of the principal databases: the NCBI Protein database,
PRACTICAL 1: BLAST and Sequence Alignment The EBI and NCBI websites, two of the most widely used life science web portals are introduced along with some of the principal databases: the NCBI Protein database,
BLAST. Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences.
 BLAST Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences. An example could be aligning an mrna sequence to genomic DNA. Proteins are frequently composed of
BLAST Basic Local Alignment Search Tool. Optimized for finding local alignments between two sequences. An example could be aligning an mrna sequence to genomic DNA. Proteins are frequently composed of
Kyoto Encyclopedia of Genes and Genomes (KEGG)
") NPTEL Biotechnology -Systems Biology Kyoto Encyclopedia of Genes and Genomes (KEGG) Dr. M. Vijayalakshmi School of Chemical and Biotechnology SASTRA University Joint Initiative of IITs and IISc Funded
NPTEL Biotechnology -Systems Biology Kyoto Encyclopedia of Genes and Genomes (KEGG) Dr. M. Vijayalakshmi School of Chemical and Biotechnology SASTRA University Joint Initiative of IITs and IISc Funded
Textbook Reading Guidelines
 Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: January 16, 2013 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: January 16, 2013 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
BME 110 Midterm Examination
 BME 110 Midterm Examination May 10, 2011 Name: (please print) Directions: Please circle one answer for each question, unless the question specifies "circle all correct answers". You can use any resource
BME 110 Midterm Examination May 10, 2011 Name: (please print) Directions: Please circle one answer for each question, unless the question specifies "circle all correct answers". You can use any resource
Why Use BLAST? David Form - August 15,
 Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
Wolbachia Workshop 2017 Bioinformatics BLAST Basic Local Alignment Search Tool Finding Model Organisms for Study of Disease Can yeast be used as a model organism to study cystic fibrosis? BLAST Why Use
Web-based Bioinformatics Applications in Proteomics
 Web-based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu January 30, 2009 NCBI (National Center for Biotechnology Information) http://www.ncbi.nlm.nih.gov/ 1 Pubmed
Web-based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu January 30, 2009 NCBI (National Center for Biotechnology Information) http://www.ncbi.nlm.nih.gov/ 1 Pubmed
BLAST. compared with database sequences Sequences with many matches to high- scoring words are used for final alignments
 BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
Data Retrieval from GenBank
 Data Retrieval from GenBank Peter J. Myler Bioinformatics of Intracellular Pathogens JNU, Feb 7-0, 2009 http://www.ncbi.nlm.nih.gov (January, 2007) http://ncbi.nlm.nih.gov/sitemap/resourceguide.html Accessing
Data Retrieval from GenBank Peter J. Myler Bioinformatics of Intracellular Pathogens JNU, Feb 7-0, 2009 http://www.ncbi.nlm.nih.gov (January, 2007) http://ncbi.nlm.nih.gov/sitemap/resourceguide.html Accessing
Data Mining for Biological Data Analysis
 Data Mining for Biological Data Analysis Data Mining and Text Mining (UIC 583 @ Politecnico di Milano) References Data Mining Course by Gregory-Platesky Shapiro available at www.kdnuggets.com Jiawei Han
Data Mining for Biological Data Analysis Data Mining and Text Mining (UIC 583 @ Politecnico di Milano) References Data Mining Course by Gregory-Platesky Shapiro available at www.kdnuggets.com Jiawei Han
Chapter 2: Access to Information
 Chapter 2: Access to Information Outline Introduction to biological databases Centralized databases store DNA sequences Contents of DNA, RNA, and protein databases Central bioinformatics resources: NCBI
Chapter 2: Access to Information Outline Introduction to biological databases Centralized databases store DNA sequences Contents of DNA, RNA, and protein databases Central bioinformatics resources: NCBI
Dina El-Khishin (Ph.D.) Bioinformatics Research Facility. Deputy Director of AGERI & Head of the Genomics, Proteomics &
 Bioinformatics Research Facility. Deputy Director of AGERI & Head of the Genomics, Proteomics &") Dina El-Khishin (Ph.D.) Deputy Director of AGERI & Head of the Genomics, Proteomics & Bioinformatics Research Facility Agricultural Genetic Engineering Research Institute (AGERI) Giza EGYPT Bioinformatics
Dina El-Khishin (Ph.D.) Deputy Director of AGERI & Head of the Genomics, Proteomics & Bioinformatics Research Facility Agricultural Genetic Engineering Research Institute (AGERI) Giza EGYPT Bioinformatics
KEGG: Kyoto Encyclopedia of Genes and Genomes
 1999 Oxford University Press Nucleic Acids Research, 1999, Vol. 27, No. 1 29 34 KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono
1999 Oxford University Press Nucleic Acids Research, 1999, Vol. 27, No. 1 29 34 KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono
The Gene Ontology Annotation (GOA) project application of GO in SWISS-PROT, TrEMBL and InterPro
 project application of GO in SWISS-PROT, TrEMBL and InterPro") Comparative and Functional Genomics Comp Funct Genom 2003; 4: 71 74. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/cfg.235 Conference Review The Gene Ontology Annotation
Comparative and Functional Genomics Comp Funct Genom 2003; 4: 71 74. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/cfg.235 Conference Review The Gene Ontology Annotation
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools
 CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
CAP 5510: Introduction to Bioinformatics : Bioinformatics Tools ECS 254A / EC 2474; Phone x3748; Email: giri@cis.fiu.edu My Homepage: http://www.cs.fiu.edu/~giri http://www.cs.fiu.edu/~giri/teach/bioinfs15.html
user s guide Question 3
 Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
CHAPTER 21 LECTURE SLIDES
 CHAPTER 21 LECTURE SLIDES Prepared by Brenda Leady University of Toledo To run the animations you must be in Slideshow View. Use the buttons on the animation to play, pause, and turn audio/text on or off.
CHAPTER 21 LECTURE SLIDES Prepared by Brenda Leady University of Toledo To run the animations you must be in Slideshow View. Use the buttons on the animation to play, pause, and turn audio/text on or off.
BIMM 143: Introduction to Bioinformatics (Winter 2018)
") BIMM 143: Introduction to Bioinformatics (Winter 2018) Course Instructor: Dr. Barry J. Grant ( bjgrant@ucsd.edu ) Course Website: https://bioboot.github.io/bimm143_w18/ DRAFT: 2017-12-02 (20:48:10 PST
BIMM 143: Introduction to Bioinformatics (Winter 2018) Course Instructor: Dr. Barry J. Grant ( bjgrant@ucsd.edu ) Course Website: https://bioboot.github.io/bimm143_w18/ DRAFT: 2017-12-02 (20:48:10 PST
Comparative Bioinformatics. BSCI348S Fall 2003 Midterm 1
 BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
BSCI348S Fall 2003 Midterm 1 Multiple Choice: select the single best answer to the question or completion of the phrase. (5 points each) 1. The field of bioinformatics a. uses biomimetic algorithms to
Annotation Walkthrough Workshop BIO 173/273 Genomics and Bioinformatics Spring 2013 Developed by Justin R. DiAngelo at Hofstra University
 Annotation Walkthrough Workshop NAME: BIO 173/273 Genomics and Bioinformatics Spring 2013 Developed by Justin R. DiAngelo at Hofstra University A Simple Annotation Exercise Adapted from: Alexis Nagengast,
Annotation Walkthrough Workshop NAME: BIO 173/273 Genomics and Bioinformatics Spring 2013 Developed by Justin R. DiAngelo at Hofstra University A Simple Annotation Exercise Adapted from: Alexis Nagengast,
Exercise I, Sequence Analysis
 Exercise I, Sequence Analysis atgcacttgagcagggaagaaatccacaaggactcaccagtctcctggtctgcagagaagacagaatcaacatgagcacagcaggaaaa gtaatcaaatgcaaagcagctgtgctatgggagttaaagaaacccttttccattgaggaggtggaggttgcacctcctaaggcccatgaagt
Exercise I, Sequence Analysis atgcacttgagcagggaagaaatccacaaggactcaccagtctcctggtctgcagagaagacagaatcaacatgagcacagcaggaaaa gtaatcaaatgcaaagcagctgtgctatgggagttaaagaaacccttttccattgaggaggtggaggttgcacctcctaaggcccatgaagt
Annotation of contig27 in the Muller F Element of D. elegans. Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans.
 David Wang Bio 434W 4/27/15 Annotation of contig27 in the Muller F Element of D. elegans Abstract Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans. Genscan predicted six
David Wang Bio 434W 4/27/15 Annotation of contig27 in the Muller F Element of D. elegans Abstract Contig27 is a 60,000 bp region located in the Muller F element of the D. elegans. Genscan predicted six
Bioinformatics Databases
 Bioinformatics Databases Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com Agenda
Bioinformatics Databases Dr. Taysir Hassan Abdel Hamid Lecturer, Information Systems Department Faculty of Computer and Information Assiut University taysirhs@aun.edu.eg taysir_soliman@hotmail.com Agenda
Bioinformatics for Cell Biologists
 Bioinformatics for Cell Biologists 15 19 March 2010 Developmental Biology and Regnerative Medicine (DBRM) Schedule Monday, March 15 09.00 11.00 Introduction to course and Bioinformatics (L1) D224 Helena
Bioinformatics for Cell Biologists 15 19 March 2010 Developmental Biology and Regnerative Medicine (DBRM) Schedule Monday, March 15 09.00 11.00 Introduction to course and Bioinformatics (L1) D224 Helena
Annotation Practice Activity [Based on materials from the GEP Summer 2010 Workshop] Special thanks to Chris Shaffer for document review Parts A-G
![Annotation Practice Activity [Based on materials from the GEP Summer 2010 Workshop] Special thanks to Chris Shaffer for document review Parts A-G](/thumbs/95/123686269.jpg "Annotation Practice Activity [Based on materials from the GEP Summer 2010 Workshop] Special thanks to Chris Shaffer for document review Parts A-G") Annotation Practice Activity [Based on materials from the GEP Summer 2010 Workshop] Special thanks to Chris Shaffer for document review Parts A-G Introduction: A genome is the total genetic content of
Annotation Practice Activity [Based on materials from the GEP Summer 2010 Workshop] Special thanks to Chris Shaffer for document review Parts A-G Introduction: A genome is the total genetic content of
Biological databases an introduction
 Biological databases an introduction By Dr. Erik Bongcam-Rudloff SLU 2017 Biological Databases Sequence Databases Genome Databases Structure Databases Sequence Databases The sequence databases are the
Biological databases an introduction By Dr. Erik Bongcam-Rudloff SLU 2017 Biological Databases Sequence Databases Genome Databases Structure Databases Sequence Databases The sequence databases are the
Comparative Genomics. Page 1. REMINDER: BMI 214 Industry Night. We ve already done some comparative genomics. Loose Definition. Human vs.
 Page 1 REMINDER: BMI 214 Industry Night Comparative Genomics Russ B. Altman BMI 214 CS 274 Location: Here (Thornton 102), on TV too. Time: 7:30-9:00 PM (May 21, 2002) Speakers: Francisco De La Vega, Applied
Page 1 REMINDER: BMI 214 Industry Night Comparative Genomics Russ B. Altman BMI 214 CS 274 Location: Here (Thornton 102), on TV too. Time: 7:30-9:00 PM (May 21, 2002) Speakers: Francisco De La Vega, Applied
Computational methods in bioinformatics: Lecture 1
 Computational methods in bioinformatics: Lecture 1 Graham J.L. Kemp 2 November 2015 What is biology? Ecosystem Rain forest, desert, fresh water lake, digestive tract of an animal Community All species
Computational methods in bioinformatics: Lecture 1 Graham J.L. Kemp 2 November 2015 What is biology? Ecosystem Rain forest, desert, fresh water lake, digestive tract of an animal Community All species
Making Sense of DNA and Protein Sequences. Lily Wang, PhD Department of Biostatistics Vanderbilt University
 Making Sense of DNA and Protein Sequences Lily Wang, PhD Department of Biostatistics Vanderbilt University 1 Outline Biological background Major biological sequence databanks Basic concepts in sequence
Making Sense of DNA and Protein Sequences Lily Wang, PhD Department of Biostatistics Vanderbilt University 1 Outline Biological background Major biological sequence databanks Basic concepts in sequence
NCBI Molecular Biology Resources
 NCBI Molecular Biology Resources Part 2: Using NCBI BLAST December 2009 Using BLAST Basics of using NCBI BLAST Using the new Interface Improved organism and filter options New Services Primer BLAST Align
NCBI Molecular Biology Resources Part 2: Using NCBI BLAST December 2009 Using BLAST Basics of using NCBI BLAST Using the new Interface Improved organism and filter options New Services Primer BLAST Align
VALLIAMMAI ENGINEERING COLLEGE
 VALLIAMMAI ENGINEERING COLLEGE SRM Nagar, Kattankulathur 603 203 DEPARTMENT OF COMPUTER SCIENCE AND ENGINEERING QUESTION BANK VII SEMESTER BM6005 BIO INFORMATICS Regulation 2013 Academic Year 2018-19 Prepared
VALLIAMMAI ENGINEERING COLLEGE SRM Nagar, Kattankulathur 603 203 DEPARTMENT OF COMPUTER SCIENCE AND ENGINEERING QUESTION BANK VII SEMESTER BM6005 BIO INFORMATICS Regulation 2013 Academic Year 2018-19 Prepared
21.5 The "Omics" Revolution Has Created a New Era of Biological Research
 21.5 The "Omics" Revolution Has Created a New Era of Biological Research 1 Section 21.5 Areas of biological research having an "omics" connection are continually developing These include proteomics, metabolomics,
21.5 The "Omics" Revolution Has Created a New Era of Biological Research 1 Section 21.5 Areas of biological research having an "omics" connection are continually developing These include proteomics, metabolomics,
Identifying Regulatory Regions using Multiple Sequence Alignments
 Identifying Regulatory Regions using Multiple Sequence Alignments Prerequisites: BLAST Exercise: Detecting and Interpreting Genetic Homology. Resources: ClustalW is available at http://www.ebi.ac.uk/tools/clustalw2/index.html
Identifying Regulatory Regions using Multiple Sequence Alignments Prerequisites: BLAST Exercise: Detecting and Interpreting Genetic Homology. Resources: ClustalW is available at http://www.ebi.ac.uk/tools/clustalw2/index.html
From AP investigative Laboratory Manual 1
 Comparing DNA Sequences to Understand Evolutionary Relationships. How can bioinformatics be used as a tool to determine evolutionary relationships and to better understand genetic diseases? BACKGROUND
Comparing DNA Sequences to Understand Evolutionary Relationships. How can bioinformatics be used as a tool to determine evolutionary relationships and to better understand genetic diseases? BACKGROUND
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. GEP goals: Evidence Based Annotation. Evidence for Gene Models 12/26/2018
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Applied Bioinformatics
 Applied Bioinformatics Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Course overview What is bioinformatics Data driven science: the creation and advancement
Applied Bioinformatics Bing Zhang Department of Biomedical Informatics Vanderbilt University bing.zhang@vanderbilt.edu Course overview What is bioinformatics Data driven science: the creation and advancement
Big picture and history
 Big picture and history (and Computational Biology) CS-5700 / BIO-5323 Outline 1 2 3 4 Outline 1 2 3 4 First to be databased were proteins The development of protein- s (Sanger and Tuppy 1951) led to the
Big picture and history (and Computational Biology) CS-5700 / BIO-5323 Outline 1 2 3 4 Outline 1 2 3 4 First to be databased were proteins The development of protein- s (Sanger and Tuppy 1951) led to the
CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU
 CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU !2 Sequence Alignment! Global: Needleman-Wunsch-Sellers (1970).! Local: Smith-Waterman (1981) Useful when commonality
CAP 5510/CGS 5166: Bioinformatics & Bioinformatic Tools GIRI NARASIMHAN, SCIS, FIU !2 Sequence Alignment! Global: Needleman-Wunsch-Sellers (1970).! Local: Smith-Waterman (1981) Useful when commonality
Tutorial for Stop codon reassignment in the wild
 Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Tutorial for Stop codon reassignment in the wild Learning Objectives This tutorial has two learning objectives: 1. Finding evidence of stop codon reassignment on DNA fragments. 2. Detecting and confirming
Web based Bioinformatics Applications in Proteomics. Genbank
 Web based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu February 9, 2010 Genbank Primary nucleic acid sequence database Maintained by NCBI National Center for Biotechnology
Web based Bioinformatics Applications in Proteomics Chiquito Crasto ccrasto@genetics.uab.edu February 9, 2010 Genbank Primary nucleic acid sequence database Maintained by NCBI National Center for Biotechnology
Challenging algorithms in bioinformatics
 Challenging algorithms in bioinformatics 11 October 2018 Torbjørn Rognes Department of Informatics, UiO torognes@ifi.uio.no What is bioinformatics? Definition: Bioinformatics is the development and use
Challenging algorithms in bioinformatics 11 October 2018 Torbjørn Rognes Department of Informatics, UiO torognes@ifi.uio.no What is bioinformatics? Definition: Bioinformatics is the development and use
Collect, analyze and synthesize. Annotation. Annotation for D. virilis. Evidence Based Annotation. GEP goals: Evidence for Gene Models 08/22/2017
 Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Annotation Annotation for D. virilis Chris Shaffer July 2012 l Big Picture of annotation and then one practical example l This technique may not be the best with other projects (e.g. corn, bacteria) l
Introduction to Bioinformatics for Medical Research. Gideon Greenspan TA: Oleg Rokhlenko. Lecture 1
 Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il TA: Oleg Rokhlenko Lecture 1 Introduction to Bioinformatics Introduction to Bioinformatics What is Bioinformatics?
Introduction to Bioinformatics for Medical Research Gideon Greenspan gdg@cs.technion.ac.il TA: Oleg Rokhlenko Lecture 1 Introduction to Bioinformatics Introduction to Bioinformatics What is Bioinformatics?
Protein structure. Wednesday, October 4, 2006
 Protein structure Wednesday, October 4, 2006 Introduction to Bioinformatics Johns Hopkins School of Public Health 260.602.01 J. Pevsner pevsner@jhmi.edu Copyright notice Many of the images in this powerpoint
Protein structure Wednesday, October 4, 2006 Introduction to Bioinformatics Johns Hopkins School of Public Health 260.602.01 J. Pevsner pevsner@jhmi.edu Copyright notice Many of the images in this powerpoint
B I O I N F O R M A T I C S
 B I O I N F O R M A T I C S Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be SUPPLEMENTARY CHAPTER: DATA BASES AND MINING 1 What
B I O I N F O R M A T I C S Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be SUPPLEMENTARY CHAPTER: DATA BASES AND MINING 1 What
TIGR THE INSTITUTE FOR GENOMIC RESEARCH
 Introduction to Genome Annotation: Overview of What You Will Learn This Week C. Robin Buell May 21, 2007 Types of Annotation Structural Annotation: Defining genes, boundaries, sequence motifs e.g. ORF,
Introduction to Genome Annotation: Overview of What You Will Learn This Week C. Robin Buell May 21, 2007 Types of Annotation Structural Annotation: Defining genes, boundaries, sequence motifs e.g. ORF,
BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology
 BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology Jeremy Buhler March 15, 2004 In this lab, we ll annotate an interesting piece of the D. melanogaster genome. Along the way, you ll get
BIO4342 Lab Exercise: Detecting and Interpreting Genetic Homology Jeremy Buhler March 15, 2004 In this lab, we ll annotate an interesting piece of the D. melanogaster genome. Along the way, you ll get
RESEARCH METHODOLOGY, BIOSTATISTICS AND IPR
 MB 401: RESEARCH METHODOLOGY, BIOSTATISTICS AND IPR Objectives: The overall aim of the course is to deepen knowledge regarding basic concepts of Biostatistics, the research process in occupational therapy
MB 401: RESEARCH METHODOLOGY, BIOSTATISTICS AND IPR Objectives: The overall aim of the course is to deepen knowledge regarding basic concepts of Biostatistics, the research process in occupational therapy
user s guide Question 1
 Question 1 How does one find a gene of interest and determine that gene s structure? Once the gene has been located on the map, how does one easily examine other genes in that same region? doi:10.1038/ng966
Question 1 How does one find a gene of interest and determine that gene s structure? Once the gene has been located on the map, how does one easily examine other genes in that same region? doi:10.1038/ng966
Since 2002 a merger and collaboration of three databases: Swiss-Prot & TrEMBL
 Since 2002 a merger and collaboration of three databases: Swiss-Prot & TrEMBL PIR-PSD Funded mainly by NIH (US) to be the highest quality, most thoroughly annotated protein sequence database o A high quality
Since 2002 a merger and collaboration of three databases: Swiss-Prot & TrEMBL PIR-PSD Funded mainly by NIH (US) to be the highest quality, most thoroughly annotated protein sequence database o A high quality
BIO 152 Principles of Biology III: Molecules & Cells Acquiring information from NCBI (PubMed/Bookshelf/OMIM)
") BIO 152 Principles of Biology III: Molecules & Cells Acquiring information from NCBI (PubMed/Bookshelf/OMIM) Note: This material is adapted from Web-based Bioinformatics Tutorials: Exploring Genomes by
BIO 152 Principles of Biology III: Molecules & Cells Acquiring information from NCBI (PubMed/Bookshelf/OMIM) Note: This material is adapted from Web-based Bioinformatics Tutorials: Exploring Genomes by
Genetics and Bioinformatics
 Genetics and Bioinformatics Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be Lecture 1: Setting the pace 1 Bioinformatics what s
Genetics and Bioinformatics Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be Lecture 1: Setting the pace 1 Bioinformatics what s