Genome Assembly: Background and Strategy
|
|
|
- Toby Dixon
- 6 years ago
- Views:
Transcription
1 Genome Assembly: Background and Strategy Monday, February 8, 2016 BIOL 7210: Genome Assembly Group Aroon Chande, Cheng Chen, Alicia Francis, Alli Gombolay, Namrata Kalsi, Ellie Kim, Tyrone Lee, Wilson Martin, Tannishtha Som, Peijue Zhang
2 Outline 1. Illumina Sequencing 2. Genome Pipeline a. Quality Control and Pre-processing b. Genome Pipeline Steps/Description c. Read assembly: de novo, reference-guided, hybrid d. Assembly improvement e. Assembly quality assessment 2
3 Objectives 1. Research classical and new tools to use 2. Evaluate and compare these available tools 3. Choose and combine best assemblers to utilize 4. Create wrapper for pipeline to increase efficiency 3
4 Illumina Sequencing 4
5 Illumina Sequencing "Sequence by synthesis" Sample DNA is sheared and ligated to primers Bridge amplification Sequencing reaction Each insert has a unique tag, barcode Barcode is unique for each DNA sample in run Barcode read step of synthesis reaction 5
6 Paired-end Sequencing Reads from both ends of the insert Insert is of unknown size Estimated average provided by sequencing facility PE help orient reads during assembly Spatial constraints for how far apart pairs can be placed 6

7 Project Data Haemophilus influenzae Sequencing data: Paired-end, 250bp reads From Illumina HiSeq2500 Already demultiplexed 7
8 Genome Pipeline 8
9 Genome Pipeline Quality Control and pre-processing Read assembly: de novo, reference-guided, and hybrid Assembly programs chosen for testing Assembly improvement Polishing/Gap filling Contig integration Automated SNP calling for reference-guided assemblies Assembly quality assessment 9
10 Raw Reads TrimGalore! (FastQC and Cutadapt) Quality Reads de novo Assembly Spades ABySS Velvet EDENA DISCOVAR EPGA2 Pear Reference Assembly SMALT Pilon Bowtie QUAST Hybrid Assembly AlignGraph Polish/Gap Filling CISA QUAST 10
11 Quality Control and Pre-processing 11
12 Quality Control and Pre-processing Remove barcodes Remove poor quality reads Filter duplicates (Typically for speed considerations) 12
13 Quality Control and Pre-processing Sickle Sliding window trimming based on quality and length Quake Kmer based trimming and miscall correction Useful to preprocess files when using assemblers without baked in read correction Prinseq Sliding window trimming and cleaning based on statistical modeling FastQC Trim Galore! Our pick Uses cutadapt (adapter/barcode cleanup) plus sliding window trimming Summary statistics provided by FastQC 13
14 14
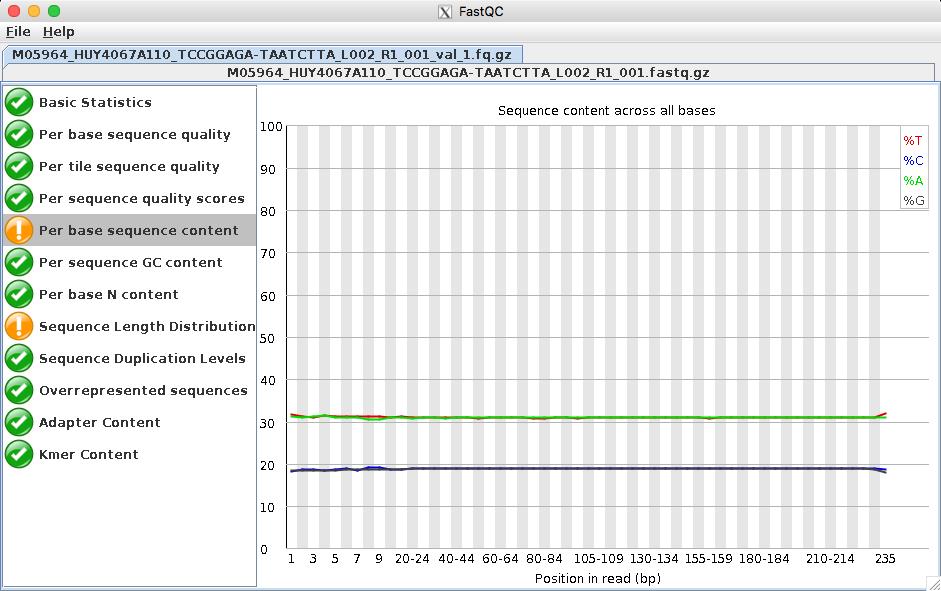
15 15
16 Read Assembly 16
17 Read Assembly De novo: from new SPAdes, ABySS, Velvet, EDENA, DISCOVAR, etc. Use only sequence information to order reads Reference-guided bwa/bowtie, SMALT PILON, etc. Reference must be closely related "fast" SNP detection Lots of "left over" reads 17
18 De novo Assembly 18
19 De novo Assembly Attempts to order reads using their sequence Two methods: 1. Overlap Layout Consensus 2. de Bruijn graphs 19

20 Overlap Layout Consensus "Old," computationally expensive Overlap: Build overlap graph Layout: Bundle stretches of the overlap graph into contigs Consensus: Pick most likely nucleotide sequences for each contig 20
21 de Bruijn Graphs Fast, memory and space efficient Resolution of ambiguous read placement Assembly method: Break reads into mers of size k (smaller than read length) Find overlaps of kmers (computationally easier than OLC) Resolve ambiguities using estimated genomic distances 21
22 SPAdes Short read assembler, takes single and paired ends High level view of SPAdes assembly: i. Assembly graph construction with multi-sized de Bruijn graphs (dbg) and bulge resolution ii. Integration of Pair-end data with dbg-derived paths to determine genomic distance iii. Paired assembly graph iv. Contigreconstruction 22
23 SPAdes improvements over other assemblers 1. SPAdes's assembly model uses multi-sized Paired de Bruijn graphs (PDBGs) 2. Bulges, tips and other ends are resolved using the paired end metadata 3. PDBGs are more space/memory/computationally efficient for building and unwinding 4. Built in read correction (BayesHammer), and chimera/miscall correction 23
24 ABySS Assembly By Short Sequencing Amounts of data generated from large-scale sequencing projects 3.5 billion paired-end reads from the genome of an African male publicly released by Illumina
25 ABySS: Algorithmic approach 1. All possible substrings of length k (termed k-mers) are generated from the sequence reads. The k-mer data set is then processed to remove read errors and initial contigs are built 2. Mate-pair information is used to extend contigs by resolving ambiguities in contig overlaps
26 Evaluation of ABySS performance Simulated data Experimental sequence data Polymorphic and novel human sequence identified in the assembled experimental data Comparison of short read assemblers

27 Velvet (Daniel Zerbino and Ewan Birney) Designed to deal with short read sequencing alignments Has 4 stages Hashing reads into k-mers Construction of the graph Error Removal Tips due to errors at edges of reads Bulges due to internal read errors removed by TourBus algorithm Erroneous connections due to cloning errors Simplification of repeats by separating paths sharing local overlaps
28 Velvet Pros Easy to install, stable Easy to run Fast (multithreading) Can take in long and short reads Can use a reference genome to anchor reads which normally map to repetitive regions (Columbus module) Cons Might need large amounts of RAM for large genomes, potentially > 512 GB for a human genome if at all possible.
29 EDENA EDENA is high-throughput novel software for de novo assembly of accurate contigs. It is suitable to characterize a bacterial genome which containing data sets with very short reads of the same length. This application is based on the classical assembly approach where all overlaps are computed and structured in a graph. EDENA has two features, exact matching and detection of spurious reads, to improve the assembly of very short sequences: There are four steps in this algorithm: 1. Remove redundant information 2. Overlapping phase 3. Cleaning and removing transitive and spurious edges 4. All contigs of a minimum size that are unambiguously represented in the graph are provided as output 29
30 DISCOVAR de novo At the time to Discovar s paper (Weisenfeld et al., 2014), the methods available to investigate GV did a good job in 90% of most cases. Calling variants was a challenging in the 10% remaining of the genome, specifically those occurring in low-complexity sequence, segmental duplications and high GC content regions. Discovar was specifically designed to address challenging variant types using a modified ALLPATHS assembly algorithm that identifies variants as it assemble the genome Compared with GATK, Discovar provides a better coverage of challenging variants and excellent coverage of ordinary variants The standalone de novo assembly algorithm(discovar de novo) was released later by popular demand.
31 DISCOVAR de novo Currently it takes as input Illumina reads of length 250 or longer produced on MiSeq or HiSeq 2500 (exclusively) and from a single PCR-free library. Yet, with adjustments, reads as short as 150bp may work with Discovar de novo, depending on fragment size and other factors.
32 DISCOVAR de novo Phase 1: Error correction in initial graph: Reads are error corrected Pairs are closed Pair closures are merged into unipaths Formation of graph is similar to ALLPATHS Compute approx graph by walking each path Select ideal paths for seeding Assemble neighbors around seed Find allpaths Glue together Unipath: maximal unbranched sequence
33 DISCOVAR de novo Phase 2: Optimization of graph: Graph is simplified and improved for accuracy Pull apart simple branches Pull apart complex branches Bubble popping (cleaning up variants) Graph reconstruction Orient assembly to reference
34 DISCOVAR de novo Formats assembly result into FASTA Graph can be viewed at end of assembly Variant paths (GVs) can be highlighted Comprehensive variation discovery in single human genomes. Weisenfeld NI, Yin S, Sharpe T, Lau B, Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, Nusbaum C, Lander ES, MacCallum I, Jaffe DB. Nat Genet Dec;46(12): doi: /ng Epub 2014 Oct 19.
35 EPGA2: Overview EPGA2 is an updated version from EPGA that does not require vast amounts of memory to handle large genome sequences as most assemblers do. It is able to be memory efficient and still display improved and higher coverage for contigs and scaffolds. As a first step, read errors are removed to help refine the precision of De Bruijn Graph and provide less work when assembling the contigs. Secondly, unlike EPGA which uses a hash table to store K-mers and its counts, EPGA2 has the user define the k-mer size(<32) and keeps k-mers that have a frequency greater than one. Lastly, to perfect EPGA2 it parallels the contigs so that if a portion of a contig has overlapping regions those sections will merge together. 35
36 EPGA2: Algorithm 1. Error Correction of Read Endings Using BLoom-filter-based Error correction Solution for high-throughput Sequencing reads (BLESS) 2. Uses Disk Streaming of K-mers (DSK) to Count K-mers and Partition Reads from the corrected sequences given from BLESS 3. Uses BCALM to Construct De Bruijn Graph -Stores k-mers in memory as clusters 4. Contig Assembly: starts with long nodes from the De Bruijn Graph to find the nodes before and after it 5. Parallel Contigs Merging 6. Scaffolding: order is determined by paired end reads 7. Gap Filling 36
37 PEAR (Paired-End read merger) A memory-efficient and accurate pair-end read merger that can generate reads from both ends of the target DNA fragments. It can assemble 95% of the reads with 35 bp mean overlap, with a false positive rate of This assembler is accurate on datasets with: short overlaps, and DNA target fragment sizes that are smaller than single-end read lengths. Does not require the target fragment size as input. Does not require preprocessing of the raw data or specifying the fragment size.
38 PEAR scores all possible overlaps for each pair of corresponding paired-end reads to determine the overlap with the highest Assembly Score (AS). Where: A scoring matrix that penalizes mismatches with a negative value β and rewards matches with a positive value α. Empirical tests using simulated data showed that setting and α =1.0 and β = -1.0 yield the best results. For the merged reads, PEAR computes the overlap that maximizes the AS. We denote the overlap that maximizes the AS by C*.
39 PEAR generates four FASTQ output files: Successfully merged reads Forward and reverse unmerged reads Discarded reads. The basic command to run PEAR is:./pear -f forward_read.fastq -r reverse_read.fastq -o output_prefix Requirements: GNU Autotools and GNU Libtool for compiling the source code. Repository:
40 Reference-guided Assembly 40
41 Reference-guided Assembly Reconstruct genome of interest using: i. Genome of a closely related organism ii. Raw reads of interest mapped for the above genome Genome is reconstructed by taking the consensus call for a give base Much faster than de novo, especially for strains of the same species Variant calling using same mapped data 41
42 SMALT SMALT is a pairwise sequence alignment program designed for the efficient and accurate mapping of DNA sequencing reads onto genomic referencesequences. The software employs a perfect hash index of short words, less than 15 nucleotides long, sampled at equidistant steps along the genomic reference sequences. For each read, potentially matching segments in the reference are identified from seed matches in the index and subsequently aligned with the read using a banded Smith-Waterman algorithm.
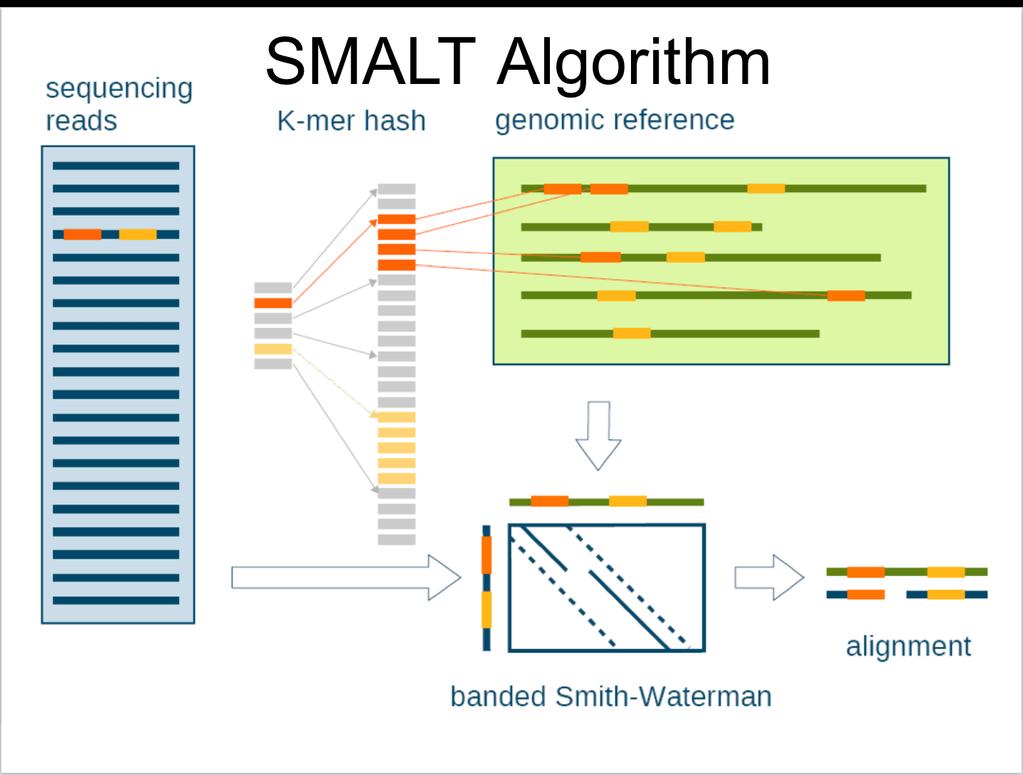
43
44 SMALT Paired-end bp reads produced by Illumina sequencing platforms for the human genome can be mapped at a rate of pairs per hour. Simulations suggest error rates below 0.03% when 96.7% of the reads are mapped. The user can adjust the tradeoff between sensitivity and speed by tuning the length and spacing of the hashed words.
45 Pilon Ostensibly not an assembler but designed as a tool to combine and automate the tasks of correcting of draft assemblies and variant calling. Pilon requires as input a FASTA file of the genome - either an existing draft assembly or a reference assembly from another strain - along with one or more BAM files of reads aligned to the input FASTA file. It then attempts to make improvements to the input reference using evidence from the reads. Pilon then outputs a FASTA file containing an improved representation of the genome from the read data and an optional VCF file detailing variation seen between the read data and the input genome. There is also an option to produce tracks that can be displayed in genome viewers such as IGV and GenomeView.
46 Pilon Correcting single base differences Resolving repeats using Jump Pairs Identifying and aligning small indels Fixing larger indel or block substitution events Gap filling Identification of local misassemblies, including optional opening of new gaps Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One Nov 19;9(11):e doi: /journal.pone ecollection Walker BJ 1, Abeel T 2, Shea T 1, Priest M 1, Abouelliel A 1, SakthikumarS 1, Cuomo CA 1, Zeng Q 1, Wortman J 1, Young SK 1, Earl AM 1.
47 Hybrid Assembly 47
48 AlignGraph AlignGraph is a "reference-assisted assembly approach" (1). Guided by a reference genome of a closely related organism, AlignGraph algorithm extends and joins de novo-assembled contigs or scaffolds This approach will likely become the default option for future projects Inputs: 1. Paired-end DNA reads in FASTA format 2. De novo contigs or scaffolds assembled by: Velvet, ABySS, ALLPATHS-LG, SOAPdenovo, etc. 3. Reference genome from a closely related organism 48
49 49
50 AlignGraph: Advantages Performance tests show AlignGraph is able to considerably improve the contigs and scaffolds from several assemblers (Velvet, ABySS, etc.) For example: AlignGraph allowed % of the contigs of A. thaliana and human to be extended => improvements of assembly metrics 50
51 Assembly Improvement 51
52 Assembly Improvement Polishing - Fixing short indels and miscalls Gap filling - Remove N's and extending contig ends Contig integration - Merging two assemblies Automated SNP calling for reference guided assemblies 52
53 Polishing and Gap Filling Polishing - PILON, IMAGE2 Map raw reads to check for consistency SPAdes does this automatically using bayeshammer during assembly Gap Filling - Sealer (ABySS), PILON, GMcloser, IMAGE2 Map raw reads to assembly and use low-coverage information Does not always work, some regions are just not sequenced 53
54 Contig Integration Attempt to build better assemblies by combining multiple assemblies Typically from different assemblers Some assembly algorithms are strict and produce shorter, low error contigs Some are greedy and produce longer contigs with a higher error rate Contig integration requires that input assemblies have high homology 54
55 Automated SNP Calling during Refinement Part of PILON's internal pipeline Uses the SNP calling pipeline from samtools on alignment generated during polishing and gap closing Applies some statistical models to suppress low quality SNP calls 55
56 Assembly Quality Assessment 56
57 Assembly Quality Assessment Quast Gold standard Produces summary statistics and graphics SuRankCo Machine-learning based approach Can be trained using assemblies of interest qatools Uses SAM/BAM mapping files Computes many of the same stats as Quast 57
58 Broad Typing using MLST SRST2 tool Short Read Sequence Typing Used to sort reads into Hi and Hhae 58
59 References ABySS: AlignGraph: BaoE, et al. (2014) AlignGraph: algorithm for secondary de novo genome assembly guided by closely related references, Bioinformatics, 30, i319-i328. ( DISCOVAR: WeisenfeldNI et al. Comprehensive variation discovery in single human genomes. Nat Genet Dec; 46(12): EDENA: EPGA2: PEAR: 59
60 References Pilon: Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One Nov 19; 9(11):e SPAdes: SMALT: Trim Galore!: Velvet: 1. Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs.genome Research. 2008;18(5): doi: /gr Images:
61 Thanks! Questions? 61
Genome Assembly. J Fass UCD Genome Center Bioinformatics Core Friday September, 2015
 Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
Assembly of Ariolimax dolichophallus using SOAPdenovo2
 Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
Workflow of de novo assembly
 Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Sequence assembly. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
A shotgun introduction to sequence assembly (with Velvet) MCB Brem, Eisen and Pachter
 MCB Brem, Eisen and Pachter") A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
De novo genome assembly with next generation sequencing data!! "
 De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
NOW GENERATION SEQUENCING. Monday, December 5, 11
 NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Computational assembly for prokaryotic sequencing projects
 Computational assembly for prokaryotic sequencing projects Lee Katz, Ph.D. Bioinformatician, Enteric Diseases Laboratory Branch January 21, 2015 Disclaimers The findings and conclusions in this presentation
Computational assembly for prokaryotic sequencing projects Lee Katz, Ph.D. Bioinformatician, Enteric Diseases Laboratory Branch January 21, 2015 Disclaimers The findings and conclusions in this presentation
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Lees J.A., Vehkala M. et al., 2016 In Review
 Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes Lees J.A., Vehkala M. et al., 2016 In Review Journal Club Triinu Kõressaar 16.03.2016 Introduction Bacterial
Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes Lees J.A., Vehkala M. et al., 2016 In Review Journal Club Triinu Kõressaar 16.03.2016 Introduction Bacterial
White paper on de novo assembly in CLC Assembly Cell 4.0
 White Paper White paper on de novo assembly in CLC Assembly Cell 4.0 June 7, 2016 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
White Paper White paper on de novo assembly in CLC Assembly Cell 4.0 June 7, 2016 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Introduction: Methods:
 Eason 1 Introduction: Next Generation Sequencing (NGS) is a term that applies to many new sequencing technologies. The drastic increase in speed and cost of these novel methods are changing the world of
Eason 1 Introduction: Next Generation Sequencing (NGS) is a term that applies to many new sequencing technologies. The drastic increase in speed and cost of these novel methods are changing the world of
Mate-pair library data improves genome assembly
 De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
BST227 Introduction to Statistical Genetics. Lecture 8: Variant calling from high-throughput sequencing data
 BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
BST227 Introduction to Statistical Genetics Lecture 8: Variant calling from high-throughput sequencing data 1 PC recap typical genome Differs from the reference genome at 4-5 million sites ~85% SNPs ~15%
Incorporating Molecular ID Technology. Accel-NGS 2S MID Indexing Kits
 Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Next Generation Sequencing Lecture Saarbrücken, 19. March Sequencing Platforms
 Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Illumina (Solexa) Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme
 Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme") Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Variation detection based on second generation sequencing data. Xin LIU Department of Science and Technology, BGI
 Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly
 Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
A Roadmap to the De-novo Assembly of the Banana Slug Genome
 A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
De novo Genome Assembly
 De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
RADSeq Data Analysis. Through STACKS on Galaxy. Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé
 RADSeq Data Analysis Through STACKS on Galaxy Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé RAD sequencing: next-generation tools for an old problem INTRODUCTION source: Karim Gharbi
RADSeq Data Analysis Through STACKS on Galaxy Yvan Le Bras Anthony Bretaudeau Cyril Monjeaud Gildas Le Corguillé RAD sequencing: next-generation tools for an old problem INTRODUCTION source: Karim Gharbi
CloG: a pipeline for closing gaps in a draft assembly using short reads
 CloG: a pipeline for closing gaps in a draft assembly using short reads Xing Yang, Daniel Medvin, Giri Narasimhan Bioinformatics Research Group (BioRG) School of Computing and Information Sciences Miami,
CloG: a pipeline for closing gaps in a draft assembly using short reads Xing Yang, Daniel Medvin, Giri Narasimhan Bioinformatics Research Group (BioRG) School of Computing and Information Sciences Miami,
High Throughput Sequencing Technologies. UCD Genome Center Bioinformatics Core Monday 15 June 2015
 High Throughput Sequencing Technologies UCD Genome Center Bioinformatics Core Monday 15 June 2015 Sequencing Explosion www.genome.gov/sequencingcosts http://t.co/ka5cvghdqo Sequencing Explosion 2011 PacBio
High Throughput Sequencing Technologies UCD Genome Center Bioinformatics Core Monday 15 June 2015 Sequencing Explosion www.genome.gov/sequencingcosts http://t.co/ka5cvghdqo Sequencing Explosion 2011 PacBio
L3: Short Read Alignment to a Reference Genome
 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
Genomics AGRY Michael Gribskov Hock 331
 Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
ABSTRACT COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION. David Kelley, Doctor of Philosophy, 2011
 ABSTRACT Title of dissertation: COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION David Kelley, Doctor of Philosophy, 2011 Dissertation directed by: Professor Steven Salzberg Department
ABSTRACT Title of dissertation: COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION David Kelley, Doctor of Philosophy, 2011 Dissertation directed by: Professor Steven Salzberg Department
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES. Sari S. Khaleel
 PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES by Sari S. Khaleel A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements
PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES by Sari S. Khaleel A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements
Genomic DNA ASSEMBLY BY REMAPPING. Course overview
 ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John DiCarlo Yexun Wang
 Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Introduction to NGS Analysis Tools
 National Center for Emerging and Zoonotic Infectious Diseases Introduction to NGS Analysis Tools Heather Carleton, PhD, MPH Team Lead, Enteric Diseases Bioinformatics, Enteric Diseases Laboratory Branch,
National Center for Emerging and Zoonotic Infectious Diseases Introduction to NGS Analysis Tools Heather Carleton, PhD, MPH Team Lead, Enteric Diseases Bioinformatics, Enteric Diseases Laboratory Branch,
Introductie en Toepassingen van Next-Generation Sequencing in de Klinische Virologie. Sander van Boheemen Medical Microbiology
 Introductie en Toepassingen van Next-Generation Sequencing in de Klinische Virologie Sander van Boheemen Medical Microbiology Next-generation sequencing Next-generation sequencing (NGS), also known as
Introductie en Toepassingen van Next-Generation Sequencing in de Klinische Virologie Sander van Boheemen Medical Microbiology Next-generation sequencing Next-generation sequencing (NGS), also known as
De novo genome assembly. Dr Torsten Seemann
 De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
HiSeqTM 2000 Sequencing System
 IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment HiSeqTM 2000 Sequencing System Performance
IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment HiSeqTM 2000 Sequencing System Performance
Genomics and Transcriptomics of Spirodela polyrhiza
 Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
BIOINFORMATICS 1 SEQUENCING TECHNOLOGY. DNA story. DNA story. Sequencing: infancy. Sequencing: beginnings 26/10/16. bioinformatic challenges
 BIOINFORMATICS 1 or why biologists need computers SEQUENCING TECHNOLOGY bioinformatic challenges http://www.bioinformatics.uni-muenster.de/teaching/courses-2012/bioinf1/index.hbi Prof. Dr. Wojciech Makałowski"
BIOINFORMATICS 1 or why biologists need computers SEQUENCING TECHNOLOGY bioinformatic challenges http://www.bioinformatics.uni-muenster.de/teaching/courses-2012/bioinf1/index.hbi Prof. Dr. Wojciech Makałowski"
Bioinformatics Support of Genome Sequencing Projects. Seminar in biology
 Bioinformatics Support of Genome Sequencing Projects Seminar in biology Introduction The Big Picture Biology reminder Enzyme for DNA manipulation DNA cloning DNA mapping Sequencing genomes Alignment of
Bioinformatics Support of Genome Sequencing Projects Seminar in biology Introduction The Big Picture Biology reminder Enzyme for DNA manipulation DNA cloning DNA mapping Sequencing genomes Alignment of
Axiom mydesign Custom Array design guide for human genotyping applications
 TECHNICAL NOTE Axiom mydesign Custom Genotyping Arrays Axiom mydesign Custom Array design guide for human genotyping applications Overview In the past, custom genotyping arrays were expensive, required
TECHNICAL NOTE Axiom mydesign Custom Genotyping Arrays Axiom mydesign Custom Array design guide for human genotyping applications Overview In the past, custom genotyping arrays were expensive, required
Read Quality Assessment & Improvement. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
Why can GBS be complicated? Tools for filtering, error correction and imputation.
 Why can GBS be complicated? Tools for filtering, error correction and imputation. Edward Buckler USDA-ARS Cornell University http://www.maizegenetics.net Many Organisms Are Diverse Humans are at the lower
Why can GBS be complicated? Tools for filtering, error correction and imputation. Edward Buckler USDA-ARS Cornell University http://www.maizegenetics.net Many Organisms Are Diverse Humans are at the lower
Welcome to the NGS webinar series
 Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
Introduction to Bioinformatics
 Introduction to Bioinformatics Alla L Lapidus, Ph.D. SPbSU St. Petersburg Term Bioinformatics Term Bioinformatics was invented by Paulien Hogeweg (Полина Хогевег) and Ben Hesper in 1970 as "the study of
Introduction to Bioinformatics Alla L Lapidus, Ph.D. SPbSU St. Petersburg Term Bioinformatics Term Bioinformatics was invented by Paulien Hogeweg (Полина Хогевег) and Ben Hesper in 1970 as "the study of
Gap Filling for a Human MHC Haplotype Sequence
 American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
Haploid Assembly of Diploid Genomes
 Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Alignment methods. Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics
 Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
BLAST. compared with database sequences Sequences with many matches to high- scoring words are used for final alignments
 BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
BLAST 100 times faster than dynamic programming. Good for database searches. Derive a list of words of length w from query (e.g., 3 for protein, 11 for DNA) High-scoring words are compared with database
Meraculous-2D: Haplotype-sensitive Assembly of Highly Heterozygous genomes.
 Meraculous-2D: Haplotype-sensitive Assembly of Highly Heterozygous genomes. Eugene Goltsman [1], Isaac Ho [1], Daniel Rokhsar [1,2,3] [1] DOE Joint Genome Institute, 2800 Mitchell Drive, Walnut Creek,
Meraculous-2D: Haplotype-sensitive Assembly of Highly Heterozygous genomes. Eugene Goltsman [1], Isaac Ho [1], Daniel Rokhsar [1,2,3] [1] DOE Joint Genome Institute, 2800 Mitchell Drive, Walnut Creek,
Introduction to Bioinformatics
 Introduction to Bioinformatics Richard Corbett Canada s Michael Smith Genome Sciences Centre Vancouver, British Columbia June 28, 2017 Our mandate is to advance knowledge about cancer and other diseases
Introduction to Bioinformatics Richard Corbett Canada s Michael Smith Genome Sciences Centre Vancouver, British Columbia June 28, 2017 Our mandate is to advance knowledge about cancer and other diseases
Genome Assembly Workshop Titles and Abstracts
 Genome Assembly Workshop Titles and Abstracts TUESDAY, MARCH 15, 2011 08:15 AM Richard Durbin, Wellcome Trust Sanger Institute A generic sequence graph exchange format for assembly and population variation
Genome Assembly Workshop Titles and Abstracts TUESDAY, MARCH 15, 2011 08:15 AM Richard Durbin, Wellcome Trust Sanger Institute A generic sequence graph exchange format for assembly and population variation
Next Generation Sequencing Technologies
 Next Generation Sequencing Technologies Julian Pierre, Jordan Taylor, Amit Upadhyay, Bhanu Rekepalli Abstract: The process of generating genome sequence data is constantly getting faster, cheaper, and
Next Generation Sequencing Technologies Julian Pierre, Jordan Taylor, Amit Upadhyay, Bhanu Rekepalli Abstract: The process of generating genome sequence data is constantly getting faster, cheaper, and
Human genome sequence
 NGS: the basics Human genome sequence June 26th 2000: official announcement of the completion of the draft of the human genome sequence (truly finished in 2004) Francis Collins Craig Venter HGP: 3 billion
NGS: the basics Human genome sequence June 26th 2000: official announcement of the completion of the draft of the human genome sequence (truly finished in 2004) Francis Collins Craig Venter HGP: 3 billion
CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management
 Sequence Data Analysis and Management") CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management Scott Sammons Technology Officer Office of Advanced Molecular Detection National Center for Emerging and Zoonotic Infectious
CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management Scott Sammons Technology Officer Office of Advanced Molecular Detection National Center for Emerging and Zoonotic Infectious
Outline. DNA Sequencing. Whole Genome Shotgun Sequencing. Sequencing Coverage. Whole Genome Shotgun Sequencing 3/28/15
 Outline Introduction Lectures 22, 23: Sequence Assembly Spring 2015 March 27, 30, 2015 Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
Outline Introduction Lectures 22, 23: Sequence Assembly Spring 2015 March 27, 30, 2015 Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter
 Method ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter Shaun D. Jackman, 1 Benjamin P. Vandervalk, 1 Hamid Mohamadi, Justin Chu, Sarah Yeo, S. Austin Hammond, Golnaz Jahesh,
Method ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter Shaun D. Jackman, 1 Benjamin P. Vandervalk, 1 Hamid Mohamadi, Justin Chu, Sarah Yeo, S. Austin Hammond, Golnaz Jahesh,
Molecular Biology: DNA sequencing
 Molecular Biology: DNA sequencing Author: Prof Marinda Oosthuizen Licensed under a Creative Commons Attribution license. SEQUENCING OF LARGE TEMPLATES As we have seen, we can obtain up to 800 nucleotides
Molecular Biology: DNA sequencing Author: Prof Marinda Oosthuizen Licensed under a Creative Commons Attribution license. SEQUENCING OF LARGE TEMPLATES As we have seen, we can obtain up to 800 nucleotides
Genome Assembly, part II. Tandy Warnow
 Genome Assembly, part II Tandy Warnow How to apply de Bruijn graphs to genome assembly Phillip E C Compeau, Pavel A Pevzner & Glenn Tesler A mathematical concept known as a de Bruijn graph turns the formidable
Genome Assembly, part II Tandy Warnow How to apply de Bruijn graphs to genome assembly Phillip E C Compeau, Pavel A Pevzner & Glenn Tesler A mathematical concept known as a de Bruijn graph turns the formidable
ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCE DATA. by Aakrosh Ratan
 The Pennsylvania State University The Graduate School College of Engineering ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCE DATA A Dissertation in Computer Science and Engineering by Aakrosh Ratan c
The Pennsylvania State University The Graduate School College of Engineering ASSEMBLY ALGORITHMS FOR NEXT-GENERATION SEQUENCE DATA A Dissertation in Computer Science and Engineering by Aakrosh Ratan c
Next Generation Sequencing. Jeroen Van Houdt - Leuven 13/10/2017
 Next Generation Sequencing Jeroen Van Houdt - Leuven 13/10/2017 Landmarks in DNA sequencing 1953 Discovery of DNA double helix structure 1977 A Maxam and W Gilbert "DNA seq by chemical degradation" F Sanger"DNA
Next Generation Sequencing Jeroen Van Houdt - Leuven 13/10/2017 Landmarks in DNA sequencing 1953 Discovery of DNA double helix structure 1977 A Maxam and W Gilbert "DNA seq by chemical degradation" F Sanger"DNA
Variant Detection in Next Generation Sequencing Data. John Osborne Sept 14, 2012
 + Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
+ Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
BENG 183 Trey Ideker. Genome Assembly and Physical Mapping
 BENG 183 Trey Ideker Genome Assembly and Physical Mapping Reasons for sequencing Complete genome sequencing!!! Resequencing (Confirmatory) E.g., short regions containing single nucleotide polymorphisms
BENG 183 Trey Ideker Genome Assembly and Physical Mapping Reasons for sequencing Complete genome sequencing!!! Resequencing (Confirmatory) E.g., short regions containing single nucleotide polymorphisms
Corset: enabling differential gene expression analysis for de novo assembled transcriptomes
 Davidson and Oshlack Genome Biology 2014, 15:410 METHOD Open Access : enabling differential gene expression analysis for de novo assembled transcriptomes Nadia M Davidson 1 and Alicia Oshlack 1,2* Abstract
Davidson and Oshlack Genome Biology 2014, 15:410 METHOD Open Access : enabling differential gene expression analysis for de novo assembled transcriptomes Nadia M Davidson 1 and Alicia Oshlack 1,2* Abstract
The Genome Analysis Centre. Building Excellence in Genomics and Computational Bioscience
 Building Excellence in Genomics and Computational Bioscience Wheat genome sequencing: an update from TGAC Sequencing Technology Development now Plant & Microbial Genomics Group Leader Matthew Clark matt.clark@tgac.ac.uk
Building Excellence in Genomics and Computational Bioscience Wheat genome sequencing: an update from TGAC Sequencing Technology Development now Plant & Microbial Genomics Group Leader Matthew Clark matt.clark@tgac.ac.uk
The first generation DNA Sequencing
 The first generation DNA Sequencing Slides 3 17 are modified from faperta.ugm.ac.id/newbie/download/pak_tar/.../instrument20072.ppt slides 18 43 are from Chengxiang Zhai at UIUC. The strand direction http://en.wikipedia.org/wiki/dna
The first generation DNA Sequencing Slides 3 17 are modified from faperta.ugm.ac.id/newbie/download/pak_tar/.../instrument20072.ppt slides 18 43 are from Chengxiang Zhai at UIUC. The strand direction http://en.wikipedia.org/wiki/dna
Package DNABarcodes. March 1, 2018
 Type Package Package DNABarcodes March 1, 2018 Title A tool for creating and analysing DNA barcodes used in Next Generation Sequencing multiplexing experiments Version 1.9.0 Date 2014-07-23 Author Tilo
Type Package Package DNABarcodes March 1, 2018 Title A tool for creating and analysing DNA barcodes used in Next Generation Sequencing multiplexing experiments Version 1.9.0 Date 2014-07-23 Author Tilo
Each cell of a living organism contains chromosomes
 COVER FEATURE Genome Sequence Assembly: Algorithms and Issues Algorithms that can assemble millions of small DNA fragments into gene sequences underlie the current revolution in biotechnology, helping
COVER FEATURE Genome Sequence Assembly: Algorithms and Issues Algorithms that can assemble millions of small DNA fragments into gene sequences underlie the current revolution in biotechnology, helping
Bioinformatics small variants Data Analysis. Guidelines. genomescan.nl
 Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Barcode Sequence Alignment and Statistical Analysis (Barcas) tool
 tool") Barcode Sequence Alignment and Statistical Analysis (Barcas) tool 2016.10.05 Mun, Jihyeob and Kim, Seon-Young Korea Research Institute of Bioscience and Biotechnology Barcode-Sequencing Ø Genome-wide screening
Barcode Sequence Alignment and Statistical Analysis (Barcas) tool 2016.10.05 Mun, Jihyeob and Kim, Seon-Young Korea Research Institute of Bioscience and Biotechnology Barcode-Sequencing Ø Genome-wide screening
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Data Intensive Biomedical Research: The EU RL VTEC efforts to take up the NGS challenge. EU RL for E. coli Annual Workshop 2015
 Data Intensive Biomedical Research: The EU RL VTEC efforts to take up the NGS challenge EU RL for E. coli Annual Workshop 2015 NGS adoption: Worldwide Source: Omicsmap.com November, 2015 Data Production
Data Intensive Biomedical Research: The EU RL VTEC efforts to take up the NGS challenge EU RL for E. coli Annual Workshop 2015 NGS adoption: Worldwide Source: Omicsmap.com November, 2015 Data Production
High Throughput Sequencing Technologies. J Fass UCD Genome Center Bioinformatics Core Monday June 16, 2014
 High Throughput Sequencing Technologies J Fass UCD Genome Center Bioinformatics Core Monday June 16, 2014 Sequencing Explosion www.genome.gov/sequencingcosts http://t.co/ka5cvghdqo Sequencing Explosion
High Throughput Sequencing Technologies J Fass UCD Genome Center Bioinformatics Core Monday June 16, 2014 Sequencing Explosion www.genome.gov/sequencingcosts http://t.co/ka5cvghdqo Sequencing Explosion
Title: High-quality genome assembly of channel catfish, Ictalurus punctatus
 Author s response to reviews Title: High-quality genome assembly of channel catfish, Ictalurus punctatus Authors: Qiong Shi (shiqiong@genomics.cn) Xiaohui Chen (xhchenffri@hotmail.com) Liqiang Zhong (lqzhongffri@hotmail.com)
Author s response to reviews Title: High-quality genome assembly of channel catfish, Ictalurus punctatus Authors: Qiong Shi (shiqiong@genomics.cn) Xiaohui Chen (xhchenffri@hotmail.com) Liqiang Zhong (lqzhongffri@hotmail.com)
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline Workflows
 Genes 2012, 3, 545-575; doi:10.3390/genes3030545 Article OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline
Genes 2012, 3, 545-575; doi:10.3390/genes3030545 Article OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline
Analysis of barcode sequencing
 Analysis of barcode sequencing Department of Functional Genomics, UST Jihyeob Mun 2016.12.07 Pooled library screen analysis experience knowledge gene A is a target? High-throughput Simplicity Fail Pooled
Analysis of barcode sequencing Department of Functional Genomics, UST Jihyeob Mun 2016.12.07 Pooled library screen analysis experience knowledge gene A is a target? High-throughput Simplicity Fail Pooled
Read Mapping and Variant Calling. Johannes Starlinger
 Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
HLA and Next Generation Sequencing it s all about the Data
 HLA and Next Generation Sequencing it s all about the Data John Ord, NHSBT Colindale and University of Cambridge BSHI Annual Conference Manchester September 2014 Introduction In 2003 the first full public
HLA and Next Generation Sequencing it s all about the Data John Ord, NHSBT Colindale and University of Cambridge BSHI Annual Conference Manchester September 2014 Introduction In 2003 the first full public
Variant calling in NGS experiments
 Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
RIPTIDE HIGH THROUGHPUT RAPID LIBRARY PREP (HT-RLP)
") Application Note: RIPTIDE HIGH THROUGHPUT RAPID LIBRARY PREP (HT-RLP) Introduction: Innovations in DNA sequencing during the 21st century have revolutionized our ability to obtain nucleotide information
Application Note: RIPTIDE HIGH THROUGHPUT RAPID LIBRARY PREP (HT-RLP) Introduction: Innovations in DNA sequencing during the 21st century have revolutionized our ability to obtain nucleotide information
Next Generation Sequencing: An Overview
 Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Illumina Read QC. UCD Genome Center Bioinformatics Core Monday 29 August 2016
 Illumina Read QC UCD Genome Center Bioinformatics Core Monday 29 August 2016 QC should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical processes involved
Illumina Read QC UCD Genome Center Bioinformatics Core Monday 29 August 2016 QC should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical processes involved
Introduction to taxonomic analysis of metagenomic amplicon and shotgun data with QIIME. Peter Sterk EBI Metagenomics Course 2014
 Introduction to taxonomic analysis of metagenomic amplicon and shotgun data with QIIME Peter Sterk EBI Metagenomics Course 2014 1 Taxonomic analysis using next-generation sequencing Objective we want to
Introduction to taxonomic analysis of metagenomic amplicon and shotgun data with QIIME Peter Sterk EBI Metagenomics Course 2014 1 Taxonomic analysis using next-generation sequencing Objective we want to
Alignment. J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
 Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
ALGORITHMS IN BIO INFORMATICS. Chapman & Hall/CRC Mathematical and Computational Biology Series A PRACTICAL INTRODUCTION. CRC Press WING-KIN SUNG
 Chapman & Hall/CRC Mathematical and Computational Biology Series ALGORITHMS IN BIO INFORMATICS A PRACTICAL INTRODUCTION WING-KIN SUNG CRC Press Taylor & Francis Group Boca Raton London New York CRC Press
Chapman & Hall/CRC Mathematical and Computational Biology Series ALGORITHMS IN BIO INFORMATICS A PRACTICAL INTRODUCTION WING-KIN SUNG CRC Press Taylor & Francis Group Boca Raton London New York CRC Press
Outline. Evolution. Adaptive convergence. Common similarity problems. Chapter 7: Similarity searches on sequence databases
 Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing
Chapter 7: Similarity searches on sequence databases All science is either physics or stamp collection. Ernest Rutherford Outline Why is similarity important BLAST Protein and DNA Interpreting BLAST Individualizing