Computational Genomics [2017] Faction 2: Genome Assembly Results, Protocol & Demo
|
|
|
- Justin Booker
- 6 years ago
- Views:
Transcription
![Computational Genomics [2017] Faction 2: Genome](/docs-images/78/78361000/images/1-0.jpg "Assembly Results, Protocol & Demo Christian Colon,")

1 Computational Genomics [2017] Faction 2: Genome Assembly Results, Protocol & Demo Christian Colon, Erisa Sula, Juichang Lu, Tian Jin, Lijiang Long, Rohini Mopuri, Bowen Yang, Saminda Wijeratne, Harrison Kim
2 Outline Objective Initial Workflow Pre-Assembly Tools Assembler Tools Post-Assembly Tools Final Workflow Result discussion
3 Objective Determine the best method to assemble the Salmonella genomes Evaluate and compare the available tools Assemble reads and combine results into super-assembly Compare results from different tools and find the best assemblies
4 Initial Workflow De Novo MaSuRCA Raw Reads Trim Reads Trimmomatic Prinseq Trim Galore De Novo Velvet SPAdes Abyss SOAPdenovo2 Mergers CISA Metassembler Scaffolding /Extensions SSPACE SOAPdenovo SOPRA Improvement Pilon GapFiller FGAP Reference Bwa mem Final Assembly
5 Trim Reads
6 Trim Galore! Adapter trimming (13bp Illumina default) (--illumina) Clip options for bp removal prior to actual trimming (bias removal) Length option to discard reads shorter than a set INT amount FastQC for read quality assessment Usage: $ trim_galore --illumina --clip_r clip_r three_prime_clip_r1 5 --three_prime_r2 5 --length paired read1.fq.gz read2.fq.gz -o output.dir
7 Assemblers
8 SPAdes Short read de Bruijn graph assembler, takes single and paired ends High level view of SPAdes assembly: Assembly graph construction with multi-sized de Bruijn graphs and bulge resolution Integration of paired-end data to determine genomic distance Contig reconstruction Error correction by BayesHammer Usage: $ spades.py-1 --pe1-1 <read_one> --pe1-2 <read_two> -t 4 -k <kmer list> -o <output directory>
9 MaSuRCA Example Configuration File Algorithm combines benefits of debruijn graphs with overlap layout consensus Generates Super Reads Input reads: raw reads generated from Illumina, no preprocessing Usage: $ masurca configure_file.txt Generates assemble.sh file in current directory $./assemble.sh Creates actual results
10 Velvet Manipulates de Bruijn graphs for de novo genome assembly Assembly steps: Read hashing and graph construction Error removal (tips; bubbles; and erroneous connections) Resolve repeats Velvet Optimiser: VelvetOptimiser is a multi-threaded Perl script for automatically optimising the three primary parameter options (K, -exp_cov, -cov_cutoff) for the Velvet de novo sequence assembler Usage:./Velvetoptimiser.pl -d out.dir -s start_kmer -e end_kmer -x step_size -f file_type -shortpaired -separate read1.file_type read2.file_type -t # of threads --optfunckmer n50
11 SOAPdenovo2 Short read, de novo assembler capable of working up to the size of the human genome Employs de Bruijn graphing algorithm SOAPdenovo2 is improved to accommodate reduced memory consumption in the graphing step, resolves repeats in contig assembly, and increased coverage in scaffolding Usage: SOAPdenovo-63mer all -s ~/data/config1 -K 63 -R -o graph_prefix Example Configuration File
12 ABySS Usage: abyss-pe name= <name> k=<kmer size> in= reads1.fa reads2.fa
13 Merger


14 CISA Integrate the assemblies into a hybrid set of contigs. CISA runs in four phases Phase 1: Identification of the representative contigs and possible extensions Phase 2: The uncertain regions located in the end of contigs are clipped Phase 3: blastn is performed to merge the contigs iteratively and identify repetitive regions. Phase 4: blastn with overlap larger than the maximum size of the repetitive regions. Usage: Merging Reads: $ python Merge.py <config> Running CISA : $ python CISA.py <config>

15 Metassembler Merging and optimizing de novo genome assemblies. Ranking assemblies by N50 size descending usually gives the best superassembly. Usage: $ metassemble --conf <conf-file> --outd <output-dir>
16 Scaffolding
17 SSPACE w/o extension Uses pre-assembled contigs from a de novo assembler to generate scaffolds Estimates the gap size between contigs to construct scaffolds based on their spatial relationship Can also be ran with extension to improve contigs prior to scaffolding Uses BWA to map the reads to the contigs The position and orientation of the reads are stored to determine the spatial relationship of the contigs Usage: $./SSPACE_Standard_v3.0.pl -l library_1.txt -s CISA1.fa -k 5 -a n 15 -z 0 -b SSPACE_Output1 -p 1

18 SSPACE w/ extension Uses BWA to map our trimmed reads to the contigs to determine what reads were unmapped in the assembly of the contigs Uses these unmapped reads to extend the contigs prior to scaffolding If enough of unmapped reads contain the same nucleotide, it will be added to the sequence Usage: $./SSPACE_Standard_v3.0.pl -l library_1.txt -s CISA1.fa -x 1 -m 50 -o 20 -r 0.9 -k 5 -a n 15 -z 0 -p 1 -b SSPACE_Output1
19 SOAPdenovo2 SOAPdenovo is a novel short-read assembly method that can build a de novo draft assembly for the human-sized genomes. Couldn t figure out how to isolate scaffolding tool within SOAPdenovo so that it could be used with other assemblies (specific for SOAPdenovo assemblies) Ran it only on the SOAPdenovo contigs
20 Improvement
21 GapFiller GapFiller is a stand-alone program for closing gaps within pre-assembled scaffolds. The input data is given by pre-assembled scaffold sequences (FASTA) and NGS paired-read data (FASTA or FASTQ). The final gap-filled scaffolds are provided in FASTA format. Gaps are iteratively filled from the left and right edge by incorporating one overhang nucleotide at a time, provided the position is sufficiently covered. Usage: $ perl GapFiller.pl -l <library.txt> -s <genome.fasta> (<library.txt>: <libname> <forward_fq> <reverse_fq> <insert_size> <standard_dev> FR )
22 FGAP Via alternative assemblies or incorporating alternative data, this tool focuses on deriving sequences best suited for closing gaps. The tool depends upon the functionalities of matlab and blast tools for working out potential sequences. We used the trimmed reads from the preassembly step as alternative data for the tool. Usage: $ run_fgap.sh <Matlab-libs> -d <genome.fasta> -a <fasta-dataset> -b <blast-libs> (<fasta-dataset>: <dataset1.fasta>,<dataset2.fasta>,...,<datasetn.fasta> )

23 PILON Pilon is a software tool which can be used to: Automatically improve draft assemblies Find variation among strains, including large event detection Requirement Input a FASTA file of the genome along with one or more BAM files of reads aligned to the input FASTA file. Pilon uses read alignment analysis to identify inconsistencies between the input genome and the evidence in the reads. Usage: $java Xmx15G jar pilon-1.16.jar --genome <genome.fasta> --frags <mapping.bam> --variant
24 All-in-one Tool


25 Unicycler Integrate SPAdes, samtools, Bowtie2, Samtools, and Blast+, pilon. Takes paired end reads and long reads (optional) to perform hybrid assembly. Uses graph to do scaffolding. Usage: $ unicycler -1 short_reads_1.fastq.gz -2 short_reads_2.fastq.gz -l long_reads_optional.fq.gz -o out.dir
26 Reference based assembly
27 Pipeline for reference base assembly #!/bin/bash # reference_base_assembly_pipeline sample_prefix=sp0001 read1='$sample_prefix'_r1_val_1.fq.gz' read2='$sample_prefix'_r2_val_2.fq.gz' fasta_file= fasta #bwa mapping bwa mem $fasta_file $read1 $read2 > $sample_prefix'.sam' #samtools sort samtools sort -O bam -T temp1 $sample_prefix'.sam' > $sample_prefix'.bam' #samtools index samtools index $sample_prefix'.bam' #samtools mpileup samtools mpileup -f fasta -gu $sample_prefix'.bam' bcftools call -c -O b -o $sample_prefix'.raw.bcf' #convert file to fastq format bcftools view -O v $sample_prefix'.raw.bcf' vcfutils.pl vcf2fq > $sample_prefix'.fastq' #convert fastq to fasta python3 convert_fastq_to_fasta.py -q $sample_prefix'.fastq' -a $sample_prefix'.fasta'
28 Mapping coverage map using BRIG shows a small region with no coverage in some samples
29
30
31

32 A detailed look at the region with no reads

33 The region with no read mapping is a deletion of the reference Region: 375, Around 39k
34 De novo assembly supports a transposon-like structure De novo assembly Reference Backbone Large insertion(~39kb) Repetitive region 46bp
35 Caveats in reference based assembly Genome_2 Inversion Genome_2 1. Genome_1 Genome_1 Genome_2 Insertion Genome_2 2. Genome_1 Genome_1






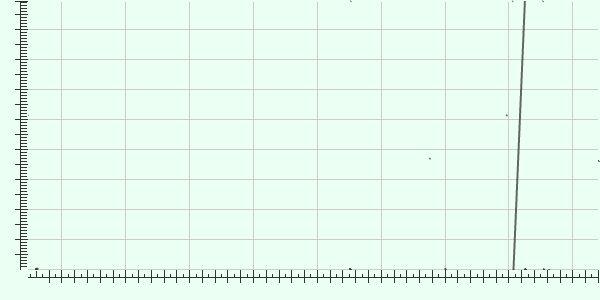
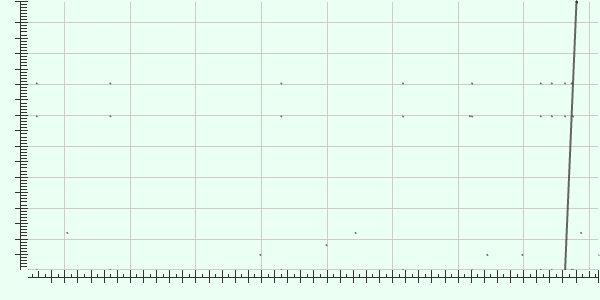
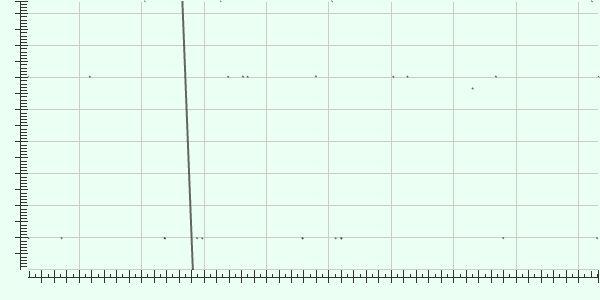
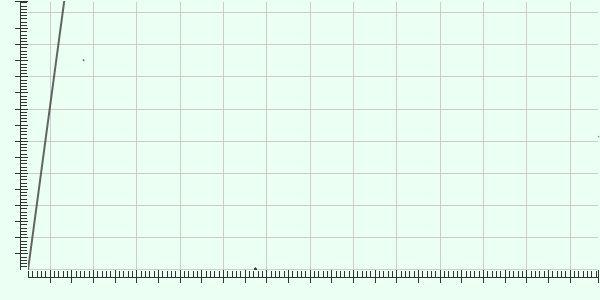
36 Alignment of de novo assembly with reference shows no inversion or insertion
37 Pre Assembly Results
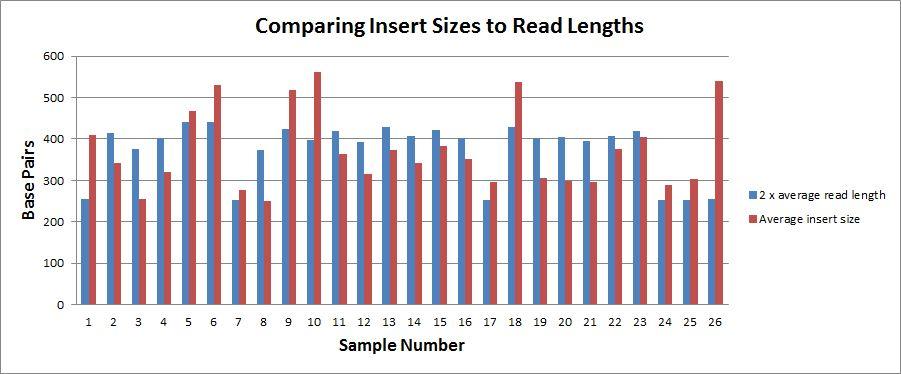
38
39
40 De novo Assembly Results
41 Use of Quast Reference Genome: -R <fasta file> Genome Annotation File: -G <gff, gtf, bed> Scaffold splitting: -s


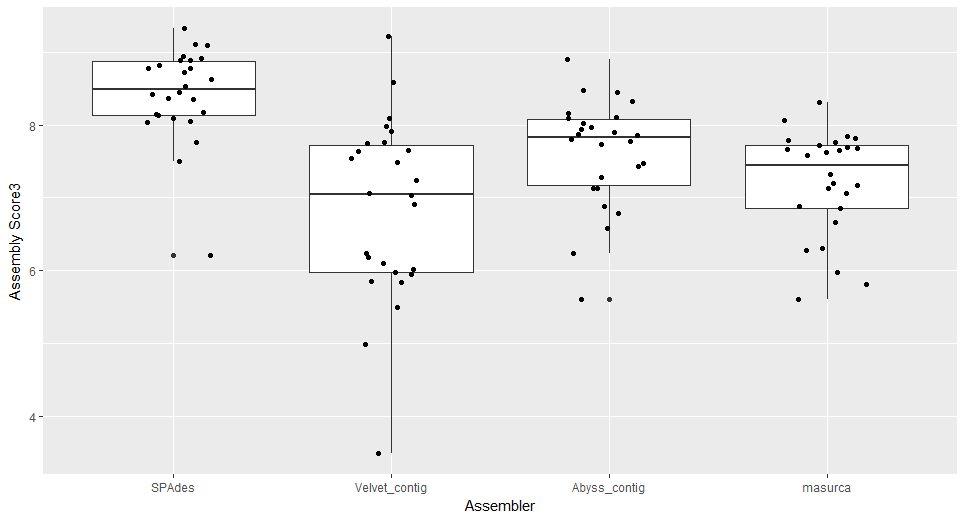
42 Selection of Assembly Score

43 Performance of Different Assemblers

44 Performance of Post-Assembly Tools
45 Performance of Post-Assembly Tools

46 Performance of Unicycler
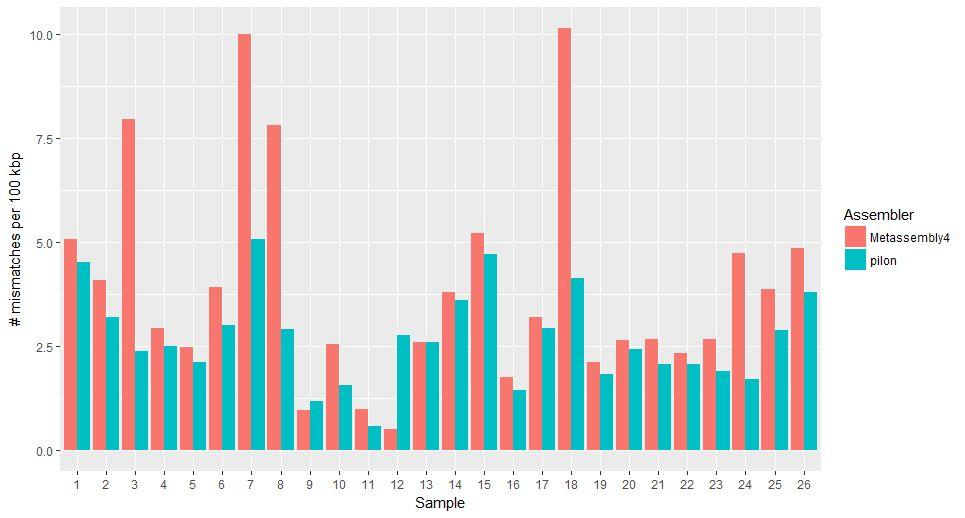
47 Performance of Pilon

48 Large Deletion or Insertion? Possible.
49 Final Workflow De Novo MaSuRCA Raw Reads Trim Reads Trim Galore De Novo Velvet SPAdes Abyss Mergers Metassembler Improvement Pilon De Novo Unicycler Reference BWA mem Final Assembly
50 References Vicedomini R, Vezzi F, Scalabrin S, Arvestad L, Policriti A GAM-NGS: genomic assemblies merger for next generation sequencing. BMC Bioinformatics 14(Suppl 7):S / S7-S6. Wences, A. H. & Schatz, M. C. Metassembler: merging and optimizing de novo genome assemblies. Genome Biology 16, 207 (2015). Zimin AV, Smith DR, Sutton G, Yorke Ja: Assembly reconciliation. Bioinformatics (Oxford, England). 2008, 24: /bioinformatics/btm542. Lin S-H, Liao Y-C. CISA: Contig Integrator for Sequence Assembly of Bacterial Genomes. Watson M, ed. PLoS ONE. 2013;8(3):e Aleksey V. Zimin, Guillaume Marçais, Daniela Puiu, Michael Roberts, Steven L. Salzberg, James A. Yorke; The MaSuRCA genome assembler. Bioinformatics 2013; 29 (21): doi: /bioinformatics/btt476 Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18. Tanja Magoc, Stephan Pabinger, Stefan Canzar, Xinyue Liu, Qi Su, Daniela Puiu, Luke J. Tallon, Steven L. Salzberg; GAGE-B: an evaluation of genome assemblers for bacterial organisms. Bioinformatics 2013; 29 (14): doi: /bioinformatics/btt273
Workflow of de novo assembly
 Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Introduction: Methods:
 Eason 1 Introduction: Next Generation Sequencing (NGS) is a term that applies to many new sequencing technologies. The drastic increase in speed and cost of these novel methods are changing the world of
Eason 1 Introduction: Next Generation Sequencing (NGS) is a term that applies to many new sequencing technologies. The drastic increase in speed and cost of these novel methods are changing the world of
Assembly of Ariolimax dolichophallus using SOAPdenovo2
 Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
Assembly of Ariolimax dolichophallus using SOAPdenovo2 Charles Markello, Thomas Matthew, and Nedda Saremi Image taken from Banana Slug Genome Project, S. Weber SOAPdenovo Assembly Tool Short Oligonucleotide
A Roadmap to the De-novo Assembly of the Banana Slug Genome
 A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
A Roadmap to the De-novo Assembly of the Banana Slug Genome Stefan Prost 1 1 Department of Integrative Biology, University of California, Berkeley, United States of America April 6th-10th, 2015 Outline
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De novo genome assembly with next generation sequencing data!! "
 De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
De novo genome assembly with next generation sequencing data!! " Jianbin Wang" HMGP 7620 (CPBS 7620, and BMGN 7620)" Genomics lectures" 2/7/12" Outline" The need for de novo genome assembly! The nature
Genome Assembly. J Fass UCD Genome Center Bioinformatics Core Friday September, 2015
 Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
Genome Assembly J Fass UCD Genome Center Bioinformatics Core Friday September, 2015 From reads to molecules What s the Problem? How to get the best assemblies for the smallest expense (sequencing) and
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
De novo whole genome assembly Lecture 1 Qi Sun Bioinformatics Facility Cornell University Data generation Sequencing Platforms Short reads: Illumina Long reads: PacBio; Oxford Nanopore Contiging/Scaffolding
Gap Filling for a Human MHC Haplotype Sequence
 American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
Computational assembly for prokaryotic sequencing projects
 Computational assembly for prokaryotic sequencing projects Lee Katz, Ph.D. Bioinformatician, Enteric Diseases Laboratory Branch January 21, 2015 Disclaimers The findings and conclusions in this presentation
Computational assembly for prokaryotic sequencing projects Lee Katz, Ph.D. Bioinformatician, Enteric Diseases Laboratory Branch January 21, 2015 Disclaimers The findings and conclusions in this presentation
De novo whole genome assembly
 De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
De novo whole genome assembly Lecture 1 Qi Sun Minghui Wang Bioinformatics Facility Cornell University DNA Sequencing Platforms Illumina sequencing (100 to 300 bp reads) Overlapping reads ~180bp fragment
Variation detection based on second generation sequencing data. Xin LIU Department of Science and Technology, BGI
 Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Genome Assembly, part II. Tandy Warnow
 Genome Assembly, part II Tandy Warnow How to apply de Bruijn graphs to genome assembly Phillip E C Compeau, Pavel A Pevzner & Glenn Tesler A mathematical concept known as a de Bruijn graph turns the formidable
Genome Assembly, part II Tandy Warnow How to apply de Bruijn graphs to genome assembly Phillip E C Compeau, Pavel A Pevzner & Glenn Tesler A mathematical concept known as a de Bruijn graph turns the formidable
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly
 Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Bioinformatics Advance Access published October 8, 2012 COPE: An accurate k-mer based pair-end reads connection tool to facilitate genome assembly Binghang Liu 1,2,, Jianying Yuan 2,, Siu-Ming Yiu 1,3,
Genomics and Transcriptomics of Spirodela polyrhiza
 Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Mate-pair library data improves genome assembly
 De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
De Novo Sequencing on the Ion Torrent PGM APPLICATION NOTE Mate-pair library data improves genome assembly Highly accurate PGM data allows for de Novo Sequencing and Assembly For a draft assembly, generate
Sequence assembly. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
Sequence assembly Jose Blanca COMAV institute bioinf.comav.upv.es Sequencing project Unknown sequence { experimental evidence result read 1 read 4 read 2 read 5 read 3 read 6 read 7 Computational requirements
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
Next Generation Sequencing Technologies
 Next Generation Sequencing Technologies Julian Pierre, Jordan Taylor, Amit Upadhyay, Bhanu Rekepalli Abstract: The process of generating genome sequence data is constantly getting faster, cheaper, and
Next Generation Sequencing Technologies Julian Pierre, Jordan Taylor, Amit Upadhyay, Bhanu Rekepalli Abstract: The process of generating genome sequence data is constantly getting faster, cheaper, and
RNA-Seq Workshop AChemS Sunil K Sukumaran Monell Chemical Senses Center Philadelphia
 RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
A shotgun introduction to sequence assembly (with Velvet) MCB Brem, Eisen and Pachter
 MCB Brem, Eisen and Pachter") A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
A shotgun introduction to sequence assembly (with Velvet) MCB 247 - Brem, Eisen and Pachter Hot off the press January 27, 2009 06:00 AM Eastern Time llumina Launches Suite of Next-Generation Sequencing
L3: Short Read Alignment to a Reference Genome
 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
CloG: a pipeline for closing gaps in a draft assembly using short reads
 CloG: a pipeline for closing gaps in a draft assembly using short reads Xing Yang, Daniel Medvin, Giri Narasimhan Bioinformatics Research Group (BioRG) School of Computing and Information Sciences Miami,
CloG: a pipeline for closing gaps in a draft assembly using short reads Xing Yang, Daniel Medvin, Giri Narasimhan Bioinformatics Research Group (BioRG) School of Computing and Information Sciences Miami,
Quality assessment and control of sequence data
 Quality assessment and control of sequence data Naiara Rodríguez-Ezpeleta Workshop on Genomics 2015 Cesky Krumlov fastq format fasta Most basic file format to represent nucleotide or amino-acid sequences
Quality assessment and control of sequence data Naiara Rodríguez-Ezpeleta Workshop on Genomics 2015 Cesky Krumlov fastq format fasta Most basic file format to represent nucleotide or amino-acid sequences
Introduction to NGS Analysis Tools
 National Center for Emerging and Zoonotic Infectious Diseases Introduction to NGS Analysis Tools Heather Carleton, PhD, MPH Team Lead, Enteric Diseases Bioinformatics, Enteric Diseases Laboratory Branch,
National Center for Emerging and Zoonotic Infectious Diseases Introduction to NGS Analysis Tools Heather Carleton, PhD, MPH Team Lead, Enteric Diseases Bioinformatics, Enteric Diseases Laboratory Branch,
De novo genome assembly. Dr Torsten Seemann
 De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
De novo genome assembly Dr Torsten Seemann IMB Winter School - Brisbane Mon 1 July 2013 Introduction Ideal world I would not need to give this talk! Human DNA Non-existent USB3 device AGTCTAGGATTCGCTA
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
RNAseq Differential Gene Expression Analysis Report
 RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John DiCarlo Yexun Wang
 Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
Supplementary Materials for: Detecting very low allele fraction variants using targeted DNA sequencing and a novel molecular barcode-aware variant caller Chang Xu Mohammad R Nezami Ranjbar Zhong Wu John
RNA-Seq with the Tuxedo Suite
 RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
White paper on de novo assembly in CLC Assembly Cell 4.0
 White Paper White paper on de novo assembly in CLC Assembly Cell 4.0 June 7, 2016 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
White Paper White paper on de novo assembly in CLC Assembly Cell 4.0 June 7, 2016 Sample to Insight QIAGEN Aarhus Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.qiagenbioinformatics.com
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
NOW GENERATION SEQUENCING. Monday, December 5, 11
 NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Bioinformatics pipeline development to support Helicobacter pylori genome analysis Master s thesis in Computer Science
 Bioinformatics pipeline development to support Helicobacter pylori genome analysis Master s thesis in Computer Science SEYEDEH SHAGHAYEGH HOSSEINI Department of Computer Science and Engineering CHALMERS
Bioinformatics pipeline development to support Helicobacter pylori genome analysis Master s thesis in Computer Science SEYEDEH SHAGHAYEGH HOSSEINI Department of Computer Science and Engineering CHALMERS
Title: High-quality genome assembly of channel catfish, Ictalurus punctatus
 Author s response to reviews Title: High-quality genome assembly of channel catfish, Ictalurus punctatus Authors: Qiong Shi (shiqiong@genomics.cn) Xiaohui Chen (xhchenffri@hotmail.com) Liqiang Zhong (lqzhongffri@hotmail.com)
Author s response to reviews Title: High-quality genome assembly of channel catfish, Ictalurus punctatus Authors: Qiong Shi (shiqiong@genomics.cn) Xiaohui Chen (xhchenffri@hotmail.com) Liqiang Zhong (lqzhongffri@hotmail.com)
Barcode Sequence Alignment and Statistical Analysis (Barcas) tool
 tool") Barcode Sequence Alignment and Statistical Analysis (Barcas) tool 2016.10.05 Mun, Jihyeob and Kim, Seon-Young Korea Research Institute of Bioscience and Biotechnology Barcode-Sequencing Ø Genome-wide screening
Barcode Sequence Alignment and Statistical Analysis (Barcas) tool 2016.10.05 Mun, Jihyeob and Kim, Seon-Young Korea Research Institute of Bioscience and Biotechnology Barcode-Sequencing Ø Genome-wide screening
Analysis of Structural Variants using 3 rd generation Sequencing
 Analysis of Structural Variants using 3 rd generation Sequencing Michael Schatz January 12, 2016 Bioinformatics / PAG XXIV @mike_schatz / #PAGXXIV Analysis of Structural Variants using 3 rd generation
Analysis of Structural Variants using 3 rd generation Sequencing Michael Schatz January 12, 2016 Bioinformatics / PAG XXIV @mike_schatz / #PAGXXIV Analysis of Structural Variants using 3 rd generation
arxiv: v1 [q-bio.gn] 25 Nov 2015
![arxiv: v1 [q-bio.gn] 25 Nov 2015](/thumbs/76/73490664.jpg "arxiv: v1 [q-bio.gn] 25 Nov 2015") MetaScope - Fast and accurate identification of microbes in metagenomic sequencing data Benjamin Buchfink 1, Daniel H. Huson 1,2 & Chao Xie 2,3 arxiv:1511.08753v1 [q-bio.gn] 25 Nov 2015 1 Department of
MetaScope - Fast and accurate identification of microbes in metagenomic sequencing data Benjamin Buchfink 1, Daniel H. Huson 1,2 & Chao Xie 2,3 arxiv:1511.08753v1 [q-bio.gn] 25 Nov 2015 1 Department of
Lees J.A., Vehkala M. et al., 2016 In Review
 Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes Lees J.A., Vehkala M. et al., 2016 In Review Journal Club Triinu Kõressaar 16.03.2016 Introduction Bacterial
Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes Lees J.A., Vehkala M. et al., 2016 In Review Journal Club Triinu Kõressaar 16.03.2016 Introduction Bacterial
Introduction to Bioinformatics
 Introduction to Bioinformatics Alla L Lapidus, Ph.D. SPbSU St. Petersburg Term Bioinformatics Term Bioinformatics was invented by Paulien Hogeweg (Полина Хогевег) and Ben Hesper in 1970 as "the study of
Introduction to Bioinformatics Alla L Lapidus, Ph.D. SPbSU St. Petersburg Term Bioinformatics Term Bioinformatics was invented by Paulien Hogeweg (Полина Хогевег) and Ben Hesper in 1970 as "the study of
Illumina (Solexa) Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme
 Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme") Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads
 Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads Authors Rei Kajitani 1, Kouta Toshimoto 1,2, Hideki Noguchi 3, Atsushi Toyoda 3,4, Yoshitoshi Ogura 5, Miki
Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads Authors Rei Kajitani 1, Kouta Toshimoto 1,2, Hideki Noguchi 3, Atsushi Toyoda 3,4, Yoshitoshi Ogura 5, Miki
Genomic DNA ASSEMBLY BY REMAPPING. Course overview
 ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
Read Mapping and Variant Calling. Johannes Starlinger
 Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
ABSTRACT COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION. David Kelley, Doctor of Philosophy, 2011
 ABSTRACT Title of dissertation: COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION David Kelley, Doctor of Philosophy, 2011 Dissertation directed by: Professor Steven Salzberg Department
ABSTRACT Title of dissertation: COMPUTATIONAL METHODS TO IMPROVE GENOME ASSEMBLY AND GENE PREDICTION David Kelley, Doctor of Philosophy, 2011 Dissertation directed by: Professor Steven Salzberg Department
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Genomics AGRY Michael Gribskov Hock 331
 Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Variant Detection in Next Generation Sequencing Data. John Osborne Sept 14, 2012
 + Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
+ Variant Detection in Next Generation Sequencing Data John Osborne Sept 14, 2012 + Overview My Bias Talk slanted towards analyzing whole genomes using Illumina paired end reads with open source tools
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
SCIENCE CHINA Life Sciences
 SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 143 155 RESEARCH PAPER doi: 10.1007/s11427-013-4442-z Comparative study of de novo assembly and genome-guided assembly strategies for
SCIENCE CHINA Life Sciences SPECIAL TOPIC February 2013 Vol.56 No.2: 143 155 RESEARCH PAPER doi: 10.1007/s11427-013-4442-z Comparative study of de novo assembly and genome-guided assembly strategies for
Bioinformatics small variants Data Analysis. Guidelines. genomescan.nl
 Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES. Sari S. Khaleel
 PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES by Sari S. Khaleel A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements
PRE- AND POST-PROCESSING TOOLS FOR NEXT-GENERATION SEQUENCING DE NOVO ASSEMBLIES by Sari S. Khaleel A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements
Supplementary Materials and Methods
 Supplementary Materials and Methods Scripts to run VirGA can be downloaded from https://bitbucket.org/szparalab, and documentation for their use is found at http://virga.readthedocs.org/. VirGA outputs
Supplementary Materials and Methods Scripts to run VirGA can be downloaded from https://bitbucket.org/szparalab, and documentation for their use is found at http://virga.readthedocs.org/. VirGA outputs
Outline. DNA Sequencing. Whole Genome Shotgun Sequencing. Sequencing Coverage. Whole Genome Shotgun Sequencing 3/28/15
 Outline Introduction Lectures 22, 23: Sequence Assembly Spring 2015 March 27, 30, 2015 Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
Outline Introduction Lectures 22, 23: Sequence Assembly Spring 2015 March 27, 30, 2015 Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management
 Sequence Data Analysis and Management") CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management Scott Sammons Technology Officer Office of Advanced Molecular Detection National Center for Emerging and Zoonotic Infectious
CDC s Advanced Molecular Detection (AMD) Sequence Data Analysis and Management Scott Sammons Technology Officer Office of Advanced Molecular Detection National Center for Emerging and Zoonotic Infectious
Hybrid Error Correction and De Novo Assembly with Oxford Nanopore
 Hybrid Error Correction and De Novo Assembly with Oxford Nanopore Michael Schatz Jan 13, 2015 PAG Bioinformatics @mike_schatz / #PAGXXIII Oxford Nanopore MinION Thumb drive sized sequencer powered over
Hybrid Error Correction and De Novo Assembly with Oxford Nanopore Michael Schatz Jan 13, 2015 PAG Bioinformatics @mike_schatz / #PAGXXIII Oxford Nanopore MinION Thumb drive sized sequencer powered over
Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline Workflows
 Genes 2012, 3, 545-575; doi:10.3390/genes3030545 Article OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline
Genes 2012, 3, 545-575; doi:10.3390/genes3030545 Article OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Next Generation Sequence Analysis and Computational Genomics Using Graphical Pipeline
N ext-generation sequencing (NGS) technologies have become common practice in life science1. Benefited
 technologies have become common practice in life science1. Benefited") OPEN SUBJECT AREAS: DATA PROCESSING HIGH-THROUGHPUT SCREENING BIOINFORMATICS Received 31 July 2014 Accepted 20 October 2014 Published 7 November 2014 Correspondence and requests for materials should be
OPEN SUBJECT AREAS: DATA PROCESSING HIGH-THROUGHPUT SCREENING BIOINFORMATICS Received 31 July 2014 Accepted 20 October 2014 Published 7 November 2014 Correspondence and requests for materials should be
Bionano Access 1.1 Software User Guide
 Bionano Access 1.1 Software User Guide Document Number: 30142 Document Revision: B For Research Use Only. Not for use in diagnostic procedures. Copyright 2017 Bionano Genomics, Inc. All Rights Reserved.
Bionano Access 1.1 Software User Guide Document Number: 30142 Document Revision: B For Research Use Only. Not for use in diagnostic procedures. Copyright 2017 Bionano Genomics, Inc. All Rights Reserved.
Analysis of barcode sequencing
 Analysis of barcode sequencing Department of Functional Genomics, UST Jihyeob Mun 2016.12.07 Pooled library screen analysis experience knowledge gene A is a target? High-throughput Simplicity Fail Pooled
Analysis of barcode sequencing Department of Functional Genomics, UST Jihyeob Mun 2016.12.07 Pooled library screen analysis experience knowledge gene A is a target? High-throughput Simplicity Fail Pooled
Approaches for in silico finishing of microbial genome sequences
 Review Article Genetics and Molecular Biology, 40, 3, 553-576 (2017) Copyright 2017, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-gmb-2016-0230 Approaches
Review Article Genetics and Molecular Biology, 40, 3, 553-576 (2017) Copyright 2017, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-gmb-2016-0230 Approaches
arxiv: v1 [q-bio.gn] 20 Apr 2013
![arxiv: v1 [q-bio.gn] 20 Apr 2013](/thumbs/72/66557559.jpg "arxiv: v1 [q-bio.gn] 20 Apr 2013") BIOINFORMATICS Vol. 00 no. 00 2013 Pages 1 7 Informed and Automated k-mer Size Selection for Genome Assembly Rayan Chikhi 1 and Paul Medvedev 1,2 1 Department of Computer Science and Engineering, The Pennsylvania
BIOINFORMATICS Vol. 00 no. 00 2013 Pages 1 7 Informed and Automated k-mer Size Selection for Genome Assembly Rayan Chikhi 1 and Paul Medvedev 1,2 1 Department of Computer Science and Engineering, The Pennsylvania
OHSU Digital Commons. Oregon Health & Science University. Benjamin Cordier. Scholar Archive
 Oregon Health & Science University OHSU Digital Commons Scholar Archive 5-19-2017 Evaluation Of Background Prediction For Variant Detection In A Clinical Context: Towards Improved Ngs Monitoring Of Minimal
Oregon Health & Science University OHSU Digital Commons Scholar Archive 5-19-2017 Evaluation Of Background Prediction For Variant Detection In A Clinical Context: Towards Improved Ngs Monitoring Of Minimal
Why can GBS be complicated? Tools for filtering, error correction and imputation.
 Why can GBS be complicated? Tools for filtering, error correction and imputation. Edward Buckler USDA-ARS Cornell University http://www.maizegenetics.net Many Organisms Are Diverse Humans are at the lower
Why can GBS be complicated? Tools for filtering, error correction and imputation. Edward Buckler USDA-ARS Cornell University http://www.maizegenetics.net Many Organisms Are Diverse Humans are at the lower
Haploid Assembly of Diploid Genomes
 Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Incorporating Molecular ID Technology. Accel-NGS 2S MID Indexing Kits
 Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Incorporating Molecular ID Technology Accel-NGS 2S MID Indexing Kits Molecular Identifiers (MIDs) MIDs are indices used to label unique library molecules MIDs can assess duplicate molecules in sequencing
Next Generation Sequencing Lecture Saarbrücken, 19. March Sequencing Platforms
 Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Long and short/small RNA-seq data analysis
 Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
NGS sequence preprocessing. José Carbonell Caballero
 NGS sequence preprocessing José Carbonell Caballero jcarbonell@cipf.es Contents Data Format Quality Control Sequence capture Fasta and fastq formats Sequence quality encoding Evaluation of sequence quality
NGS sequence preprocessing José Carbonell Caballero jcarbonell@cipf.es Contents Data Format Quality Control Sequence capture Fasta and fastq formats Sequence quality encoding Evaluation of sequence quality
RNASEQ WITHOUT A REFERENCE
 RNASEQ WITHOUT A REFERENCE Experimental Design Assembly in Non-Model Organisms And other (hopefully useful) Stuff Meg Staton mstaton1@utk.edu University of Tennessee Knoxville, TN I. Project Design Things
RNASEQ WITHOUT A REFERENCE Experimental Design Assembly in Non-Model Organisms And other (hopefully useful) Stuff Meg Staton mstaton1@utk.edu University of Tennessee Knoxville, TN I. Project Design Things
Metagenomics is the study of all micro-organisms coexistent in an environmental area, including
 JOURNAL OF COMPUTATIONAL BIOLOGY Volume 22, Number 2, 2015 # Mary Ann Liebert, Inc. Pp. 159 177 DOI: 10.1089/cmb.2014.0251 DIME: A Novel Framework for De Novo Metagenomic Sequence Assembly XUAN GUO, 1
JOURNAL OF COMPUTATIONAL BIOLOGY Volume 22, Number 2, 2015 # Mary Ann Liebert, Inc. Pp. 159 177 DOI: 10.1089/cmb.2014.0251 DIME: A Novel Framework for De Novo Metagenomic Sequence Assembly XUAN GUO, 1
Single Nucleotide Polymorphisms Caused by Assembly Errors
 Genomics Insights O r i g i n a l R e s e a r c h Open Access Full open access to this and thousands of other papers at http://www.la-press.com. Single Nucleotide Polymorphisms Caused by Assembly Errors
Genomics Insights O r i g i n a l R e s e a r c h Open Access Full open access to this and thousands of other papers at http://www.la-press.com. Single Nucleotide Polymorphisms Caused by Assembly Errors
Ensembl Tools. EBI is an Outstation of the European Molecular Biology Laboratory.
 Ensembl Tools EBI is an Outstation of the European Molecular Biology Laboratory. Questions? We ve muted all the mics Ask questions in the Chat box in the webinar interface I will check the Chat box periodically
Ensembl Tools EBI is an Outstation of the European Molecular Biology Laboratory. Questions? We ve muted all the mics Ask questions in the Chat box in the webinar interface I will check the Chat box periodically
Read Quality Assessment & Improvement. UCD Genome Center Bioinformatics Core Tuesday 14 June 2016
 Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
Read Quality Assessment & Improvement UCD Genome Center Bioinformatics Core Tuesday 14 June 2016 QA&I should be interactive Error modes Each technology has unique error modes, depending on the physico-chemical
De novo metatranscriptome assembly and coral gene expression profile of Montipora capitata with growth anomaly
 Additional File 1 De novo metatranscriptome assembly and coral gene expression profile of Montipora capitata with growth anomaly Monika Frazier, Martin Helmkampf, M. Renee Bellinger, Scott Geib, Misaki
Additional File 1 De novo metatranscriptome assembly and coral gene expression profile of Montipora capitata with growth anomaly Monika Frazier, Martin Helmkampf, M. Renee Bellinger, Scott Geib, Misaki
TSSpredator User Guide v 1.00
 TSSpredator User Guide v 1.00 Alexander Herbig alexander.herbig@uni-tuebingen.de Kay Nieselt kay.nieselt@uni-tuebingen.de June 3, 2013 1 Getting Started TSSpredator is a tool for the comparative detection
TSSpredator User Guide v 1.00 Alexander Herbig alexander.herbig@uni-tuebingen.de Kay Nieselt kay.nieselt@uni-tuebingen.de June 3, 2013 1 Getting Started TSSpredator is a tool for the comparative detection
De novo Genome Assembly
 De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
De novo Genome Assembly A/Prof Torsten Seemann Winter School in Mathematical & Computational Biology - Brisbane, AU - 3 July 2017 Introduction The human genome has 47 pieces MT (or XY) The shortest piece
Next-Generation Sequencing in practice
 Next-Generation Sequencing in practice Bioinformatics analysis techniques and some medical applications Salvatore Alaimo, MSc. Email: alaimos@dmi.unict.it Overview Next Generation Sequencing: how it works
Next-Generation Sequencing in practice Bioinformatics analysis techniques and some medical applications Salvatore Alaimo, MSc. Email: alaimos@dmi.unict.it Overview Next Generation Sequencing: how it works
ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter
 Method ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter Shaun D. Jackman, 1 Benjamin P. Vandervalk, 1 Hamid Mohamadi, Justin Chu, Sarah Yeo, S. Austin Hammond, Golnaz Jahesh,
Method ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter Shaun D. Jackman, 1 Benjamin P. Vandervalk, 1 Hamid Mohamadi, Justin Chu, Sarah Yeo, S. Austin Hammond, Golnaz Jahesh,
Quality assessment and control of sequence data. Naiara Rodríguez-Ezpeleta
 Quality assessment and control of sequence data Naiara Rodríguez-Ezpeleta Workshop on Genomics 2014 Quality control is important Some of the artefacts/problems that can be detected with QC Sequencing Sequence
Quality assessment and control of sequence data Naiara Rodríguez-Ezpeleta Workshop on Genomics 2014 Quality control is important Some of the artefacts/problems that can be detected with QC Sequencing Sequence
Lectures 18, 19: Sequence Assembly. Spring 2017 April 13, 18, 2017
 Lectures 18, 19: Sequence Assembly Spring 2017 April 13, 18, 2017 1 Outline Introduction Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
Lectures 18, 19: Sequence Assembly Spring 2017 April 13, 18, 2017 1 Outline Introduction Sequence Assembly Problem Different Solutions: Overlap-Layout-Consensus Assembly Algorithms De Bruijn Graph Based
Reference genomes and common file formats
 Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Analysing 454 amplicon resequencing experiments using the modular and database oriented Variant Identification Pipeline
 SOFTWARE Software Analysing 454 amplicon resequencing experiments using the modular and database oriented Variant Identification Pipeline Open Access Joachim M De Schrijver* 1, Kim De Leeneer 2, Steve
SOFTWARE Software Analysing 454 amplicon resequencing experiments using the modular and database oriented Variant Identification Pipeline Open Access Joachim M De Schrijver* 1, Kim De Leeneer 2, Steve
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow
 From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
HLA and Next Generation Sequencing it s all about the Data
 HLA and Next Generation Sequencing it s all about the Data John Ord, NHSBT Colindale and University of Cambridge BSHI Annual Conference Manchester September 2014 Introduction In 2003 the first full public
HLA and Next Generation Sequencing it s all about the Data John Ord, NHSBT Colindale and University of Cambridge BSHI Annual Conference Manchester September 2014 Introduction In 2003 the first full public
Corset: enabling differential gene expression analysis for de novo assembled transcriptomes
 Davidson and Oshlack Genome Biology 2014, 15:410 METHOD Open Access : enabling differential gene expression analysis for de novo assembled transcriptomes Nadia M Davidson 1 and Alicia Oshlack 1,2* Abstract
Davidson and Oshlack Genome Biology 2014, 15:410 METHOD Open Access : enabling differential gene expression analysis for de novo assembled transcriptomes Nadia M Davidson 1 and Alicia Oshlack 1,2* Abstract
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro
 Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro Philip Morris International R&D, Philip Morris Products S.A., Neuchatel, Switzerland Introduction Nicotiana sylvestris
Sequencing the genomes of Nicotiana sylvestris and Nicotiana tomentosiformis Nicolas Sierro Philip Morris International R&D, Philip Morris Products S.A., Neuchatel, Switzerland Introduction Nicotiana sylvestris
Utilization of defined microbial communities enables effective evaluation of meta-genomic assemblies
 Greenwald et al. BMC Genomics (2017) 18:296 DOI 10.1186/s12864-017-3679-5 RESEARCH ARTICLE Open Access Utilization of defined microbial communities enables effective evaluation of meta-genomic assemblies
Greenwald et al. BMC Genomics (2017) 18:296 DOI 10.1186/s12864-017-3679-5 RESEARCH ARTICLE Open Access Utilization of defined microbial communities enables effective evaluation of meta-genomic assemblies
Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome
 CORRECTION NOTICE Nat. Biotechnol. 34, 64 69 (2016) Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome Volodymyr Kuleshov, Chao Jiang, Wenyu Zhou, Fereshteh Jahanbani,
CORRECTION NOTICE Nat. Biotechnol. 34, 64 69 (2016) Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome Volodymyr Kuleshov, Chao Jiang, Wenyu Zhou, Fereshteh Jahanbani,
Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition
 RESEARCH ARTICLE Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition Moses M. Muraya 1,2, Thomas Schmutzer 1 *, Chris Ulpinnis
RESEARCH ARTICLE Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition Moses M. Muraya 1,2, Thomas Schmutzer 1 *, Chris Ulpinnis
RNA Seq: Methods and Applica6ons. Prat Thiru
 RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression