Comparative Study of Regulatory Requirements for Biologics Filing in United States and European Union
|
|
|
- Blaise Winfred Montgomery
- 6 years ago
- Views:
Transcription



1 Comparative Study of Regulatory Requirements for Biologics Filing in United States and European Union Mr. Shashi Kumar Yadav Assistant Professor Sri Indu Institute of Pharmacy Hyderabad
 in US Regulatory requirements for the development of Biologics in EU Marketing authorization process Preclinical Studies for Biologics Clinical Studies for Biologics The Marketing Authorization")
2 Outline Introduction Regulatory requirements for biologics in the US What are biologics Preclinical Studies for Biologics in US Clinical Studies for Biologics in US The Biologics License Application (BLA) in US Regulatory requirements for the development of Biologics in EU Marketing authorization process Preclinical Studies for Biologics Clinical Studies for Biologics The Marketing Authorization Application(MAA) Conclusion
3 Introduction Regulatory Affairs in the Pharmaceutical industry may be defined as "The interface between the pharmaceutical company and the regulatory agencies across the world. Each and every country has its own regulatory body.
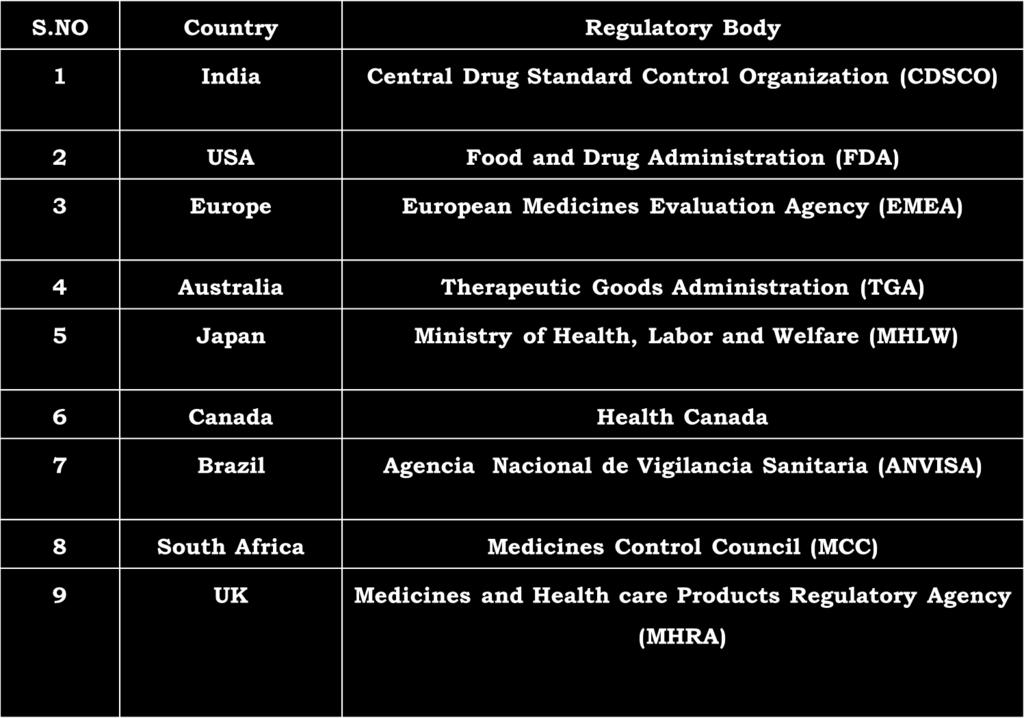
4 Introduction
5 Introduction US FDA: The FDA regulates biopharmaceuticals as drugs under the Federal Food, Drug, and Cosmetic Act. FDA is a part of the Department of Health and Human Services. Currently the Public Health Service Act authorizes the FDA to ensure the safety, purity, and potency of biologics. The FDA approves biologics for marketing under section 351 of the Act.
6 Introduction What Does the FDA Regulate? Food (with Agriculture Department) Drugs Biologics Medical Devices Cosmetics Anything That Produces Dangerous Radiation
7 Introduction FDA is comprised of several Offices and Centers Office of the Commissioner (OC) Office of Regulatory Affairs (ORA) Center for Devices and Radiological Health (CDRH) Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) Center for Food Safety and Applied Nutrition (CFSAN) Center for Veterinary Medicine (CVM) National Center for Toxicological Research (NCTR)
8 Introduction Three FDA Centers deal with medical products: Center for Drug Evaluation and Research (CDER) Center for Devices and Radiological Health (CDRH) Center for Biologics Evaluation and Research (CBER) Compounds characterized as biologics are reviewed by CBER
9 Regulatory requirements for the development of Biologics in the United States What are BIOLOGICS Biological Products or biologics were defined as Any virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product or analogous product applicable to the prevention, treatment, or cure of a disease or injuries in man.

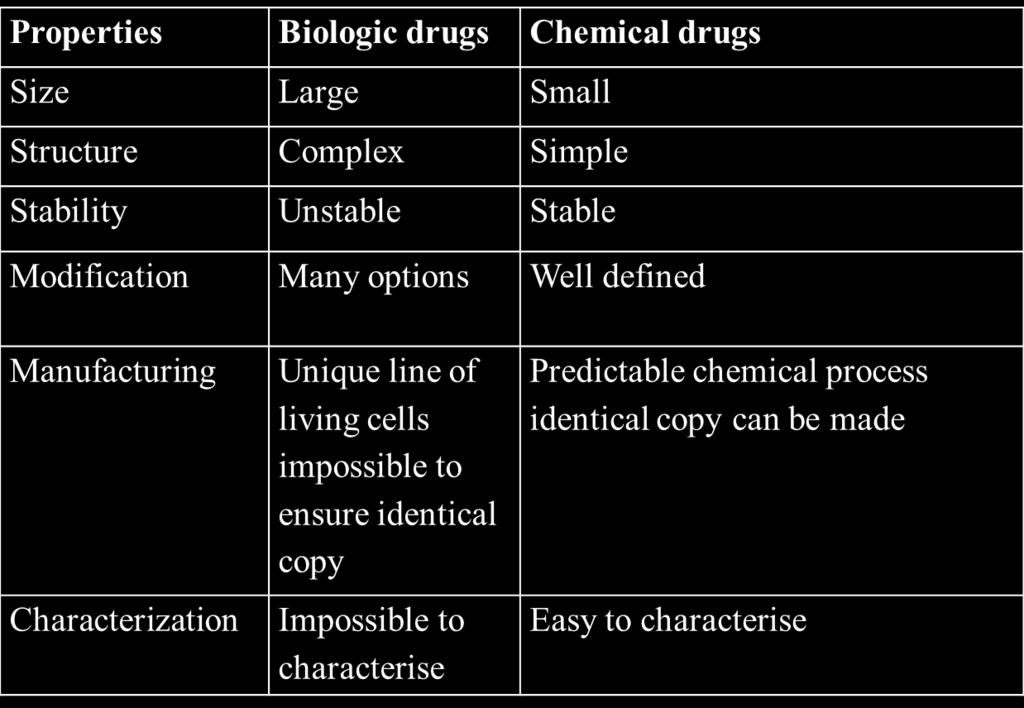
10 Differentiation
11 Preclinical Studies for Biologics in US For biologics, the FDA has adopted the ICH S6 guidance and FDAs GLP regulations typically apply. In May 2012, the FDA adopted the addendum to that ICH guidance.

12 Preclinical Studies for Biologics in US Species selection: Many biologics cannot be tested in commonly used animal species, such as rats and dogs, because of their biological activity and species or tissuespecific activity. In vitro binding assays and Functional tests, to identify a relevant species, In some cases, the chimpanzee is the only relevant species
13 Preclinical Studies for Biologics in US Immunogenicity: Many biologics elicit immune responses, which can affect preclinical study results. neutralizing or prolonging the biologic s activity, forming immune complexes, or cross-reacting with endogenous substances. Sponsors should obtain necessary samples for antibody testing during repeat-dose toxicity studies.
14 Preclinical Studies for Biologics in US Study design: 1) Primary pharmacodynamics studies In vitro binding assay In vivo studies 2) Secondary pharmacodynamics studies 3) Safety pharmacodynamics studies
15 FDA Review and Decision-Making FDA inaction in 30 days triggers the study under the IND to proceed or FDA issuance of clinical hold
16 Clinical Hold (21 C.F.R ) A clinical hold is an order issued by FDA to the sponsor of an IND to delay or to suspend a clinical investigation Partial or complete clinical hold Partial A delay or suspension of only part of the clinical work requested under the IND Complete A delay or suspension of all clinical work requested under an IND
17 Clinical Studies for Biologics in US The Investigational New Drug Application: The sponsor will submit the INDA to the FDA to perform clinical testing of a biologic in the United States An IND generally goes into effect 30 days after the FDA receives it. The IND must contain information from preclinical studies. the product s pharmacologic effects and mechanism of action and information on its ADME. Chemistry, Manufacturing, and Control (CMC) information.
18 Clinical Studies for Biologics in US Study Design Considerations: As with new drugs, clinical development of biologics typically involves three phases, Phase I, Phase II and Phase III. This programs must include an assessment of immunogenicity. With respect to immunogenicity, these studies should assess subjects antibody development, both directly after administration and at least 28 days thereafter.
19 Clinical Studies for Biologics in US Study Design Considerations: Phase I studies the initial introduction of the biologic to f humans to assess the product s metabolism, pharmacology, and safety at escalating doses. Determine Maximum Tolerated Dose (MTD) Unlike Phase I trials for drugs, Phase I studies of biologics frequently involve administration to patients rather than healthy volunteers.
20 Clinical Studies for Biologics in US Phase II trials Begin if Phase 1 studies do not reveal unacceptable toxicity Phase II trials are controlled studies that evaluate safety and short-term adverse events Biologics sponsors often combine phase II studies with phase I or phase III studies.
21 Clinical Studies for Biologics in US Phase III studies Begin if preliminary evidence of effectiveness is shown during phase II. Phase III studies are randomized, controlled, and performed at multiple study centers. Gather more information about safety and effectiveness in a defined population.
22 Clinical Studies for Biologics in US Meetings with the FDA Before and During the Clinical Trial Period: Sponsors can obtain several types of pre-approval meetings with the FDA. 21 C.F.R describes two types of such meetings. First, the sponsor can seek a pre-ind meeting to reach agreement with the FDA on the design of preclinical studies. Second, the sponsor may meet with the FDA to reach agreement on phase 2 study design.
23 The Biologics License Application (BLA) in US A BLA is used rather than a NDA though the official FDA form is designated 356h and is identical. Under 21 C.F.R , the BLA must contain, nonclinical and clinical data, a full description of manufacturing methods, stability data, proposed labeling, enclosures, and containers;
24
25 The Biologics License Application (BLA) in US Contents of the Biologics License Application: A BLA is used rather than a NDA though the official FDA form is designated 356h and is identical. Under 21 C.F.R , the BLA must contain, nonclinical and clinical data, a full description of manufacturing methods, stability data, proposed labeling, enclosures, and containers;
26 The Biologics License Application (BLA) in US Biologics License Application review process: After a sponsor submits a BLA, the FDA assembles a review team and then decides, within the first 60 days after submission, whether it can file the application or a refuse-to-file decision After the agency completes its review of the BLA, it will issue an approval letter, or a complete response letter (CRL), which states that the agency cannot approve the BLA in its current form. An applicant may file a resubmission to address the deficiencies. The review timeline for a resubmission depends on its content but is either 2 or 6 months from receipt.
27 The Biologics License Application (BLA) in US Approval Standard : The FDA must approve a BLA if it shows that the proposed product is safe, pure, and potent and the facilities where the product made, processed, packed, or held comply with good manufacturing practice (GMP).
28 Regulatory requirements for the development of Biologics in EU EUROPEAN UNION European Medicines Agency, an EU regulatory agency for the evaluation of medicinal products. In EU Directive 2001/83/EC of the European Parliament Regulates marketing authorization of biologics with the essential aim of governing the production, distribution, and use of biological products for protection of public health. The Committee for Medicinal Products for Human Use (CHMP) for assessment of all medicinal products for human use including biological products.
29 MARKETING AUTHORIZATION PROCESS Marketing authorisations Procedure in European Union divided in to Four types. National procedure Centralised procedure Mutual recognition procedure Decentralised procedure
30 Preclinical Studies for Biologics in EU The CHMP has adopted ICH S6 as a guideline governing preclinical testing of biologics. In July 2011, the CHMP adopted the addendum to this guideline, and the addendum came into effect in Europe in December 2011.
31 Preclinical Studies for Biologics in EU The addendum covers the following five topics: Species selection, Study design, Immunogenicity, Reproductive and developmental toxicity, and Carcinogenicity.

32 Preclinical Studies for Biologics in EU Species selection: According to the addendum, the sponsor use in vitro assays making qualitative and quantitative cross-species comparisons of relative target binding affinities, receptor ligand occupancy, and kinetics. Sponsors also should assess functional activity. Immunogenicity: The addendum provides more detail than ICH S6 regarding situations when the sponsor should measure antidrug antibodies (ADAs), namely when (1) there is evidence of altered PD activity, (2) there is evidence of immune-mediated reactions.
33 Preclinical Studies for Biologics in EU Reproductive and Developmental Toxicity: The addendum provides general advice on reproductive and developmental testing and then discusses more specific recommendations for fertility studies, embryo fetal development (EFD) studies and pre- and postnatal development (PPND) studies, and the timing of studies in nonhuman primates (NHPs). Typical carcinogenicity bioassays are generally inappropriate for biologics
34 Clinical Studies for Biologics in EU Clinical trials of biologics must comply with GCP, as described in Directive 2005 /28 /EC and the ICH E6 guideline, which the CHMP has adopted. CHMP has issued a guideline on quality requirements during the clinical trial period for investigational medicinal products (IMPs) containing biological or biotechnology-derived substances. The sponsor will submit the sponsor s Investigational Medicinal Product Dossier (IMPD) to the competent authority to perform clinical trials.
35 Clinical Studies for Biologics in EU The sponsor then must apply for approval from both the ethics committee in the country competent authorities of the Member States. The opinion of the ethics committee should be issued within 60 days. The trial may begin only if (1) the ethics committee has issued a favorable opinion and (2) no competent authority has informed the applicant for non acceptance.
36 Clinical Studies for Biologics in EU Phase I studies typically investigate (1) initial safety and tolerability; (2) PK, (3) PD; and (4) drug activity. Phase I studies may be conducted in healthy volunteers or certain types of patients. If the medicine has significant potential toxicity (e.g., cytotoxic products), the trial will usually be conducted In patients. Phase II trials usually allowing for evaluation of the medicine s safety and efficacy. A major goal of this phase is to determine the dose(s) for phase III trials. Phase III typically involves therapeutic confirmatory studies and explore the dose response relationship
37 The Marketing Authorization Application(MAA): Contents and Approval Standard The approval standards for biotechnology products are the same as for drugs. Both types of products must be safe and effective and have appropriate quality. Many biologics fall under the scope of the centralized marketing authorization procedure, which is mandatory for medicines developed through biotechnological methods. Other special rules apply certain types of biological medicines. For example, for plasma-derived medicinal products, the applicant must provide an information dossier, the Plasma Master File. MAAs for vaccines other than for influenza need to contain a Vaccine Antigen Master File.
38 conclusion The study introduces the legal and regulatory aspects pertaining to biological products in the United States and in the European Union. The Drug approvals in the US, Europe are the most demanding in the world. The primary purpose of the rules governing medicinal products in US, Europe is to safeguard public health. It is the role of public regulatory authorities to ensure that pharmaceutical companies comply with regulations. There are legislations that require drugs to be developed, tested, trailed, and manufactured in accordance to the guidelines so that they are safe and patient s well - being is protected.
39 BIBLIOGRAPHY Richard Guarino - New Drug Approval Process, Fifth Edition; Informa health care;2009: Douglas J. Pisano, David S. Mantus- FDA Regulatory Affairs: A Guide For Prescription Drugs, Medical Devices, And Biologics; CRC Press; Second Edition; 2008: 28-29, on/guidances/ucm pdf ocedural_guideline/2012/12/wc pdf Steven Lee, Ph.D, ceo, A-Bio Pharma Pte Ltd-Process development and manufacturing (CMC) for biologics development-an overview-. November Gary Walsh- Pharmaceutical biotechnology concepts and application; Wiley Publications; 2007:1-11.


40
KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar
 KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar Kyung-Min Baek, Ph.D. Recombinant Protein Products Division Korea Food and Drug Administration(KFDA) Biopharmaceuticals A biopharmaceutical
KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar Kyung-Min Baek, Ph.D. Recombinant Protein Products Division Korea Food and Drug Administration(KFDA) Biopharmaceuticals A biopharmaceutical
Non-clinical Assessment Requirements
 Non-clinical Assessment Requirements Presented by: Maria Nieto-Gutierrez Safety and Efficacy of Medicines/Human Medicines Development and Evaluation An agency of the European Union Contents: Relevance
Non-clinical Assessment Requirements Presented by: Maria Nieto-Gutierrez Safety and Efficacy of Medicines/Human Medicines Development and Evaluation An agency of the European Union Contents: Relevance
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR
European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR
Pharmacology. Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University.
 Pharmacology Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University. 1 PHARMACODYNAMIC STUDIES A. Primary pharmacodynamics primary action in target
Pharmacology Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University. 1 PHARMACODYNAMIC STUDIES A. Primary pharmacodynamics primary action in target
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited.
. All rights reserved. Further dissemination without express written consent strictly prohibited.") Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
Guidance for Industry
 Reprinted from FDA s website by Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product DRAFT GUIDANCE This guidance document is being distributed for comment
Reprinted from FDA s website by Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product DRAFT GUIDANCE This guidance document is being distributed for comment
Structure and content of an IMPD. What is required for first into man trial?
 What is required for first into man? The EU IMPD Thomas Sudhop, MD Scope Structure and content of an IMPD What is required for first into man trial? Only for IMPs that do not have a marketing authorisation
What is required for first into man? The EU IMPD Thomas Sudhop, MD Scope Structure and content of an IMPD What is required for first into man trial? Only for IMPs that do not have a marketing authorisation
Regulatory Considerations and Trends Europe and the U.S.
 Regulatory Considerations and Trends Europe and the U.S. Professor Kjell Strandberg MD PhD Chairman NDA Advisory Board, NDA Regulatory Science Ltd UK Former CPMP Member and Director General Medical Products
Regulatory Considerations and Trends Europe and the U.S. Professor Kjell Strandberg MD PhD Chairman NDA Advisory Board, NDA Regulatory Science Ltd UK Former CPMP Member and Director General Medical Products
Baek, Kyung-min. Recombinant Protein Products Division. Ministry of Food and Drug Safety
 Baek, Kyung-min Recombinant Protein Products Division Ministry of Food and Drug Safety About Ministry of Food and Drug Safety Regulation for Biosimilar Principle of Biosimilar Approach Status of Biosimilar
Baek, Kyung-min Recombinant Protein Products Division Ministry of Food and Drug Safety About Ministry of Food and Drug Safety Regulation for Biosimilar Principle of Biosimilar Approach Status of Biosimilar
Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory Challenges
 The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
Food and Drug Administration (FDA) 101
 101") Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
US FDA: CMC Issues for INDs
 ISBTC Global Regulatory Summit October 29, 2008 US FDA: CMC Issues for INDs Keith Wonnacott, Ph.D. keith.wonnacott@fda.hhs.gov US Food and Drug Administration Center for Biologics Evaluation and Research
ISBTC Global Regulatory Summit October 29, 2008 US FDA: CMC Issues for INDs Keith Wonnacott, Ph.D. keith.wonnacott@fda.hhs.gov US Food and Drug Administration Center for Biologics Evaluation and Research
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/94528/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) ANNEX TO GUIDELINE
European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/94528/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) ANNEX TO GUIDELINE
The Role of Chemists in the FDA Drug Approval Process
 The Role of Chemists in the FDA Drug Approval Process 231 st ACS National Meeting Atlanta, GA M. Scott Furness, Ph.D. March 26, 2006 Introduction Presentation Outline FDA Organization CDER Organization
The Role of Chemists in the FDA Drug Approval Process 231 st ACS National Meeting Atlanta, GA M. Scott Furness, Ph.D. March 26, 2006 Introduction Presentation Outline FDA Organization CDER Organization
PRECLINICAL DRUG DISCOVERY AND DEVELOPMENT BIOBOOT CAMP 2016
 PRECLINICAL DRUG DISCOVERY AND DEVELOPMENT BIOBOOT CAMP 2016 1 FROM DISCOVERY TO COMMERCIALIZATION At least 10 years on average Average spend of $1.4B Less than 12% of drugs entering Phase I are approved
PRECLINICAL DRUG DISCOVERY AND DEVELOPMENT BIOBOOT CAMP 2016 1 FROM DISCOVERY TO COMMERCIALIZATION At least 10 years on average Average spend of $1.4B Less than 12% of drugs entering Phase I are approved
Regulation of Microbiota- Based Products
 Regulation of Microbiota- Based Products LCDR Matthew Steele, PhD Team Leader, Regulatory Review Branch 1 Division of Vaccines and Related Products Applications CBER/OVRR My presentation is an informal
Regulation of Microbiota- Based Products LCDR Matthew Steele, PhD Team Leader, Regulatory Review Branch 1 Division of Vaccines and Related Products Applications CBER/OVRR My presentation is an informal
Track III: International Clinical Trials: Global Compliance Norms and EU Focus
 Track III: International Clinical Trials: Global Compliance Norms and EU Focus EU Focus Emmanuelle Voisin, PhD Principal, Voisin Consulting May 2008 Rationale Clinical trials in EU important part of health
Track III: International Clinical Trials: Global Compliance Norms and EU Focus EU Focus Emmanuelle Voisin, PhD Principal, Voisin Consulting May 2008 Rationale Clinical trials in EU important part of health
COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP)
") European Medicines Agency Evaluation of Medicines for Human Use London, 16 June 2005 EMEA/CHMP/94526/2005 COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP) ANNEX GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL PRODUCTS
European Medicines Agency Evaluation of Medicines for Human Use London, 16 June 2005 EMEA/CHMP/94526/2005 COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP) ANNEX GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL PRODUCTS
S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers
 S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers Current version dated 8 June 2016 International Council for Harmonisation of
S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers Current version dated 8 June 2016 International Council for Harmonisation of
Preclinical Development Drugs. Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015
 Preclinical Development Drugs Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015 Product Development: Development process: File Approval Drug Discovery Preclinical Phase 1 Phase 2 Phase 3 Lifecycle
Preclinical Development Drugs Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015 Product Development: Development process: File Approval Drug Discovery Preclinical Phase 1 Phase 2 Phase 3 Lifecycle
Adoption by CHMP for release for 3-month public consultation 18 November End of consultation (deadline for comments) 28 February 2011
 28 February 2011") 18 October 2012 EMA/651649/2010 Committee for Medicinal Products for Human Use Reflection paper on considerations given to designation of a single stereo isomeric form (enantiomer), a complex, a derivative,
18 October 2012 EMA/651649/2010 Committee for Medicinal Products for Human Use Reflection paper on considerations given to designation of a single stereo isomeric form (enantiomer), a complex, a derivative,
SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007
 European Medicines Agency SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007 COMMENTS FROM The Biotechnology
European Medicines Agency SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007 COMMENTS FROM The Biotechnology
Explanatory note on general fees payable to the European Medicines Agency
 Explanatory note on general fees payable to the European Medicines Agency The fees, fee exemptions and definitions described in this explanatory note apply as of 1 June 2017 and are based on Council Regulation
Explanatory note on general fees payable to the European Medicines Agency The fees, fee exemptions and definitions described in this explanatory note apply as of 1 June 2017 and are based on Council Regulation
GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN MAKING AN ASSESSMENT OF CLINICAL INVESTIGATION NOTIFICATION
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
Non-clinical documentation Overview of Requirements
 3 rd EMEA-. Non-clinical Aspects Non-clinical documentation Overview of Requirements EMEA Pre-Authorisation Human Unit 3 rd EMEA-. Non-clinical Aspects Outline Overview of Legal and Regulatory requirements
3 rd EMEA-. Non-clinical Aspects Non-clinical documentation Overview of Requirements EMEA Pre-Authorisation Human Unit 3 rd EMEA-. Non-clinical Aspects Outline Overview of Legal and Regulatory requirements
Regulatory Approval of Modern Gene-Based Cancer Immunotherapies CAR T Cells A product perspective
 Regulatory Approval of Modern Gene-Based Cancer Immunotherapies CAR T Cells A product perspective ASQ509 Biomed/Biotech SIG 2/1/18 Xiaobin Victor Lu Division of Cellular and Gene Therapies Office of Tissues
Regulatory Approval of Modern Gene-Based Cancer Immunotherapies CAR T Cells A product perspective ASQ509 Biomed/Biotech SIG 2/1/18 Xiaobin Victor Lu Division of Cellular and Gene Therapies Office of Tissues
Transmission to CHMP July Adoption by CHMP for release for consultation 20 July Start of consultation 31 August 2017
 1 2 3 29 August 2017 EMA/CHMP/ICH/544278/1998 Committee for Human Medicinal Products 4 ICH S5 (R3) guideline on reproductive toxicology: 5 detection of toxicity to reproduction for human 6 pharmaceuticals
1 2 3 29 August 2017 EMA/CHMP/ICH/544278/1998 Committee for Human Medicinal Products 4 ICH S5 (R3) guideline on reproductive toxicology: 5 detection of toxicity to reproduction for human 6 pharmaceuticals
Optimisation de votre programme de développement
 Optimisation de votre programme de développement Cedric Lamy, PhD. Cedric.lamy@crl.com Charles River Overview 65-year history: Founded in 1946, publicly traded (NYSE:CRL) Investment in skilled staff: ~7,500
Optimisation de votre programme de développement Cedric Lamy, PhD. Cedric.lamy@crl.com Charles River Overview 65-year history: Founded in 1946, publicly traded (NYSE:CRL) Investment in skilled staff: ~7,500
BIOPHARMA SOLUTIONS TM Expedite Your Drug Development Program
 BIOPHARMA SOLUTIONS TM Expedite Your Drug Program Maximize the Value of Your Asset The journey of drug development can be complex stressful. But it doesn t have to be that way. Join more than 600 biopharmaceutical
BIOPHARMA SOLUTIONS TM Expedite Your Drug Program Maximize the Value of Your Asset The journey of drug development can be complex stressful. But it doesn t have to be that way. Join more than 600 biopharmaceutical
CTA/NDA Regulatory Landscape in China. Jack Xie, PhD, DABT SOT 2016
 CTA/NDA Regulatory Landscape in China Jack Xie, PhD, DABT SOT 2016 Disclaimer The content of the following presentation represents solely author s view and may not reflect any position of Roche or China
CTA/NDA Regulatory Landscape in China Jack Xie, PhD, DABT SOT 2016 Disclaimer The content of the following presentation represents solely author s view and may not reflect any position of Roche or China
Guideline for the Quality, Safety, and Efficacy Assurance of Follow-on Biologics
 Provisional Translation (as of April 19, 2013) PFSB/ELD Notification No. 0304007 March 4, 2009 To: Prefectural Health Department (Bureau) From: Evaluation and Licensing Division, Pharmaceutical and Food
Provisional Translation (as of April 19, 2013) PFSB/ELD Notification No. 0304007 March 4, 2009 To: Prefectural Health Department (Bureau) From: Evaluation and Licensing Division, Pharmaceutical and Food
Glossary of Abbreviations
 Glossary of Abbreviations ANDA APhA Abbreviated New Drug Application American Pharmaceutical Association API Active Pharmaceutical Ingredient BA/BE Bioavailability/Bioequivalence BE Bioequivalence Bio
Glossary of Abbreviations ANDA APhA Abbreviated New Drug Application American Pharmaceutical Association API Active Pharmaceutical Ingredient BA/BE Bioavailability/Bioequivalence BE Bioequivalence Bio
Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for an orphan designation
 2 March 2010 EMA/COMP/15893/2009 Final Committee for Orphan Medicinal Products (COMP) Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for
2 March 2010 EMA/COMP/15893/2009 Final Committee for Orphan Medicinal Products (COMP) Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for
Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations. Audrey Chang, PhD, Senior Director Development Services
 Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations Audrey Chang, PhD, Senior Director Development Services Definition of Biologics: PHS Act, section 351 Virus, therapeutic
Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations Audrey Chang, PhD, Senior Director Development Services Definition of Biologics: PHS Act, section 351 Virus, therapeutic
Combination Products at US FDA
 Multimodal Therapies for Brain Disorders: Session II Regulatory and Reimbursement Considerations Combination Products at US FDA Patricia Y. Love, MD, MBA Deputy Director Office of Combination Products,
Multimodal Therapies for Brain Disorders: Session II Regulatory and Reimbursement Considerations Combination Products at US FDA Patricia Y. Love, MD, MBA Deputy Director Office of Combination Products,
Introduction to CMC Regulatory Affairs
 Introduction to CMC Regulatory Affairs Bharathi Mamidipudi Regulatory Affairs Consultant II Syner-G Pharma Consulting, LLC Northeastern University, Boston November 10, 2016 My Background Experience ~4
Introduction to CMC Regulatory Affairs Bharathi Mamidipudi Regulatory Affairs Consultant II Syner-G Pharma Consulting, LLC Northeastern University, Boston November 10, 2016 My Background Experience ~4
Documentation requirements for an initial consultation
 Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
THE COMMON TECHNICAL DOCUMENT: TAKING INDIAN NDA PROCESS TOWARDS GLOBALIZATION
 Review Article Bhalodiya H. A.*, Boda J. M., Shah J. S., Patel P. B., Vaghela J. P. Department of Quality Assurance, S. J. Thakkar Pharmacy College, Rajkot-360005, Gujarat, India. *Corresponding author
Review Article Bhalodiya H. A.*, Boda J. M., Shah J. S., Patel P. B., Vaghela J. P. Department of Quality Assurance, S. J. Thakkar Pharmacy College, Rajkot-360005, Gujarat, India. *Corresponding author
Early Phase Education WHITEPAPER. Three major items to consider when moving from preclinical to clinical development
 Early Phase Education WHITEPAPER Three major items to consider when moving from preclinical to clinical development Three major items to consider when moving from preclinical to clinical development Dr.
Early Phase Education WHITEPAPER Three major items to consider when moving from preclinical to clinical development Three major items to consider when moving from preclinical to clinical development Dr.
ICH Topic E16 Genomic Biomarkers Related to Drug Response: Context, Structure and Format of Qualification Submissions. Step 3
 European Medicines Agency June 2009 EMEA/CHMP/ICH/380636/2009 ICH Topic E16 Genomic Biomarkers Related to Drug Response: Context, Structure and Format of Qualification Submissions Step 3 NOTE FOR GUIDANCE
European Medicines Agency June 2009 EMEA/CHMP/ICH/380636/2009 ICH Topic E16 Genomic Biomarkers Related to Drug Response: Context, Structure and Format of Qualification Submissions Step 3 NOTE FOR GUIDANCE
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS OF MEDICINAL PRODUCTS
 GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS OF MEDICINAL PRODUCTS") European Medicines Agency London, 24 January 2008 Doc. Ref. EMEA/CHMP/SWP/258498/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS
European Medicines Agency London, 24 January 2008 Doc. Ref. EMEA/CHMP/SWP/258498/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS
FDA Public Hearing: Approval Pathway for Biosimilar. Products. November 2-3, 2010
 FDA Public Hearing: Approval Pathway for Biosimilar and Interchangeable Biological Products November 2-3, 2010 1 The Biotechnology Industry Organization Over 1,100 members, including biotechnology companies,
FDA Public Hearing: Approval Pathway for Biosimilar and Interchangeable Biological Products November 2-3, 2010 1 The Biotechnology Industry Organization Over 1,100 members, including biotechnology companies,
ICH Topic M 3 (R2) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals.
 Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals.") European Medicines Agency June 2009 CPMP/ICH/286/95 ICH Topic M 3 (R2) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals Step 4 NOTE FOR
European Medicines Agency June 2009 CPMP/ICH/286/95 ICH Topic M 3 (R2) Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals Step 4 NOTE FOR
"From Bedside to Bench and Back: regulatory requirements for collaborations between pharma industry and academia
 "From Bedside to Bench and Back: regulatory requirements for collaborations between pharma industry and academia Damir Hamamdžić D.V.M., Ph.D. Office of Research Regulatory Affairs Rutgers University RWJMS
"From Bedside to Bench and Back: regulatory requirements for collaborations between pharma industry and academia Damir Hamamdžić D.V.M., Ph.D. Office of Research Regulatory Affairs Rutgers University RWJMS
International Transfers of Personal Data at sanofi-aventis R & D
 International Transfers of Personal Data at sanofi-aventis R & D Pierre-Yves Lastic, PhD Senior Director, Standards Management & Data Privacy Sanofi-aventis R&D CONFERENCE ON INTERNATIONAL TRANSFERS OF
International Transfers of Personal Data at sanofi-aventis R & D Pierre-Yves Lastic, PhD Senior Director, Standards Management & Data Privacy Sanofi-aventis R&D CONFERENCE ON INTERNATIONAL TRANSFERS OF
Paradigm Shift in Comparability Assessment:
 Paradigm Shift in Comparability Assessment: How Quality by Design (QbD) and Process Analytical Technology (PAT) can improve Structure-Activity Relationship (SAR) evaluation and its relevance to comparability
Paradigm Shift in Comparability Assessment: How Quality by Design (QbD) and Process Analytical Technology (PAT) can improve Structure-Activity Relationship (SAR) evaluation and its relevance to comparability
Center for Drug Evaluation and Research. CDER Small Business and Industry Assistance. (CDER SBIA) and New Drug Review.
 and New Drug Review.") Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
Center for Drug Evaluation and Research Small Business and Industry Assistance (CDER SBIA) and New Drug Review LT Renu Lal, Pharm.D. CDER Small Business and Industry Assistance Division of Drug Information
COPYRIGHTED MATERIAL. IN THISchapter, we first give definitions of the assay and the bioassay. INTRODUCTION TO ASSAY DEVELOPMENT
 CHAPTER1 INTRODUCTION TO ASSAY DEVELOPMENT IN THISchapter, we first give definitions of the assay and the bioassay. Drug discovery and development processes are then reviewed to show the role bioassay
CHAPTER1 INTRODUCTION TO ASSAY DEVELOPMENT IN THISchapter, we first give definitions of the assay and the bioassay. Drug discovery and development processes are then reviewed to show the role bioassay
Guidance for Industry
 Guidance for Industry Comparability Protocols - Protein Drug Products and Biological Products - Chemistry, Manufacturing, and Controls Information DRAFT GUIDANCE This guidance document is being distributed
Guidance for Industry Comparability Protocols - Protein Drug Products and Biological Products - Chemistry, Manufacturing, and Controls Information DRAFT GUIDANCE This guidance document is being distributed
REQUEST FOR AUTHORISATION TO THE COMPETENT AUTHORITY: REQUEST FOR OPINION OF THE ETHICS COMMITTEE:
 Annex 1: Clinical trial Application Form REQUEST FOR AUTHORISATION OF A CLINICAL TRIAL ON A MEDICINAL PRODUCT FOR HUMAN USE TO THE COMPETENT AUTHORITIES AND FOR OPINION OF THE ETHICS COMMITTEES IN THE
Annex 1: Clinical trial Application Form REQUEST FOR AUTHORISATION OF A CLINICAL TRIAL ON A MEDICINAL PRODUCT FOR HUMAN USE TO THE COMPETENT AUTHORITIES AND FOR OPINION OF THE ETHICS COMMITTEES IN THE
Beth Hutchins, PhD PhRMA ICH Gene Therapy Discussion Group
 ICH Considerations on General Principles to Address the Risk of Inadvertent Germline Integration of Gene Therapy Vectors and Current Topics on Gene Therapy in USA Beth Hutchins, PhD PhRMA ICH Gene Therapy
ICH Considerations on General Principles to Address the Risk of Inadvertent Germline Integration of Gene Therapy Vectors and Current Topics on Gene Therapy in USA Beth Hutchins, PhD PhRMA ICH Gene Therapy
Mitochondrial Manipulation Technologies: Preclinical Considerations
 Mitochondrial Manipulation Technologies: Preclinical Considerations Wei Liang, Ph.D. FDA / CBER / OCTGT Wei.liang@fda.hhs.gov Ethical and Social Policy Considerations of Novel Techniques for Prevention
Mitochondrial Manipulation Technologies: Preclinical Considerations Wei Liang, Ph.D. FDA / CBER / OCTGT Wei.liang@fda.hhs.gov Ethical and Social Policy Considerations of Novel Techniques for Prevention
The Drug Development Process and Design of Clinical Trials
 The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 Clinical Trial Design Guidance Clinical Trial:
The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 Clinical Trial Design Guidance Clinical Trial:
FDA Oversight of Gene Therapy
 FDA Oversight of Gene Therapy Celia M. Witten, Ph.D., M.D. Deputy Director, FDA CBER Rare Disease and Orphan Products Breakthrough Summit NORD October 17, 2016 Hyatt Regency Crystal City Crystal City,
FDA Oversight of Gene Therapy Celia M. Witten, Ph.D., M.D. Deputy Director, FDA CBER Rare Disease and Orphan Products Breakthrough Summit NORD October 17, 2016 Hyatt Regency Crystal City Crystal City,
elearning Catalog DIAglobal.org/eLearning
 elearning Catalog DIAglobal.org/eLearning Continuing education DIA s Continuing education program has been approved by a wide range of leading accrediting bodies that oversee the awarding of continuing
elearning Catalog DIAglobal.org/eLearning Continuing education DIA s Continuing education program has been approved by a wide range of leading accrediting bodies that oversee the awarding of continuing
Regulatory considerations for manufacturing and testing of investigational chimeric antigen receptor (CAR) T-cell products
 T-cell products") Regulatory considerations for manufacturing and testing of investigational chimeric antigen receptor (CAR) T-cell products Xiaobin Victor Lu Product Reviewer Gene Therapies Branch DCGT/OCTGT/CBER/FDA MEASUREMENT
Regulatory considerations for manufacturing and testing of investigational chimeric antigen receptor (CAR) T-cell products Xiaobin Victor Lu Product Reviewer Gene Therapies Branch DCGT/OCTGT/CBER/FDA MEASUREMENT
Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE
 Inspection: VOICE") Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute
Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute
Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India
 Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India 1 2 Content Message Foreword 1. Introduction 2. Background & Objectives 3. Applicable Regulations and Guidelines
Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India 1 2 Content Message Foreword 1. Introduction 2. Background & Objectives 3. Applicable Regulations and Guidelines
CANADA (HEALTH CANADA)
") 1 GMP GAZETTE TM May 2016 HPFBI CANADA (HEALTH CANADA) No updates NNHPD NHPs Final Monograph for Antiseptic Skin Cleanser Who`s Affected? Companies seeking NPN or DIN for topical antiseptic hand cleansers
1 GMP GAZETTE TM May 2016 HPFBI CANADA (HEALTH CANADA) No updates NNHPD NHPs Final Monograph for Antiseptic Skin Cleanser Who`s Affected? Companies seeking NPN or DIN for topical antiseptic hand cleansers
EU scientific regulatory support mechanisms and initiatives for innovation in drug development: the EMA perspective
 EU scientific regulatory support mechanisms and initiatives for innovation in drug development: the EMA perspective FAMHP Workshop, Brussels, 2 nd May 2016 Presented by Zahra Hanaizi Product Development
EU scientific regulatory support mechanisms and initiatives for innovation in drug development: the EMA perspective FAMHP Workshop, Brussels, 2 nd May 2016 Presented by Zahra Hanaizi Product Development
Drug Development Services
 Drug Development Services Speaker Introduction Nik Burlew Vice President Regulatory and Strategic Development 25 Years in Pharma: Development, MFG and QA. Consulting for the last 13 years primarily to
Drug Development Services Speaker Introduction Nik Burlew Vice President Regulatory and Strategic Development 25 Years in Pharma: Development, MFG and QA. Consulting for the last 13 years primarily to
Preclinical Development Discovery to IND
 Preclinical Development Discovery to IND Biobootcamp CBSA / Fitzsimons / FBBp / Holland & Hart Lauren C. Costantini, Ph.D. www.lccconsulting.net This Discussion Research and Development Product development
Preclinical Development Discovery to IND Biobootcamp CBSA / Fitzsimons / FBBp / Holland & Hart Lauren C. Costantini, Ph.D. www.lccconsulting.net This Discussion Research and Development Product development
Drug Development: Why Does it Cost so Much? Lewis J. Smith, MD Professor of Medicine Director, Center for Clinical Research Associate VP for Research
 Drug Development: Why Does it Cost so Much? Lewis J. Smith, MD Professor of Medicine Director, Center for Clinical Research Associate VP for Research Drug Development Process by which new chemical entities
Drug Development: Why Does it Cost so Much? Lewis J. Smith, MD Professor of Medicine Director, Center for Clinical Research Associate VP for Research Drug Development Process by which new chemical entities
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
Envigo Corporate & Industry Overview. Rutgers University
 Envigo Corporate & Industry Overview Rutgers University Joseph Bedford, Ph.D. Corporate Vice President, Strategic Marketing November 15, 2016 envigo.com 1 Who we are Envigo is a global contract research
Envigo Corporate & Industry Overview Rutgers University Joseph Bedford, Ph.D. Corporate Vice President, Strategic Marketing November 15, 2016 envigo.com 1 Who we are Envigo is a global contract research
Injectable modified release products
 Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CPMP/EWP/280/96 Corr1) Injectable modified release products Dr Sotiris Michaleas, National Expert for the
Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CPMP/EWP/280/96 Corr1) Injectable modified release products Dr Sotiris Michaleas, National Expert for the
Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective
 Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective Patrick G. Swann, Ph.D. Deputy Director Division of Monoclonal Antibodies Office of Biotechnology Products Office of Pharmaceutical
Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective Patrick G. Swann, Ph.D. Deputy Director Division of Monoclonal Antibodies Office of Biotechnology Products Office of Pharmaceutical
BIOPHARMACEUTICAL PROCESS EVALUATED FOR VIRAL CLEARANCE
 The purpose of Viral Clearance evaluation is to assess the capability of a manufacturing production process to inactivate and/or remove potential viral contaminants. Experience and knowledge in selecting
The purpose of Viral Clearance evaluation is to assess the capability of a manufacturing production process to inactivate and/or remove potential viral contaminants. Experience and knowledge in selecting
A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999
 EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Pharmaceuticals and cosmetics Final Revision 0 NOTICE TO APPLICANTS A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999 This guideline
EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Pharmaceuticals and cosmetics Final Revision 0 NOTICE TO APPLICANTS A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999 This guideline
POLICY AND PROCEDURES. Office of Pharmaceutical Quality. Table of Contents
 POLICY AND PROCEDURES Office of Pharmaceutical Quality NDA Classification Codes Table of Contents PURPOSE...1 BACKGROUND...1 POLICY...1 NDA Classification Codes...2 RESPONSIBILITIES...7 REFERENCES...9
POLICY AND PROCEDURES Office of Pharmaceutical Quality NDA Classification Codes Table of Contents PURPOSE...1 BACKGROUND...1 POLICY...1 NDA Classification Codes...2 RESPONSIBILITIES...7 REFERENCES...9
The rules governing medicinal products in the European Union. Presentation and content of the dossier Edition
 The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
Samatasvir (IDX719), a Potent Pan-Genotypic NS5A Inhibitor, for the Treatment of Hepatitis C Virus (HCV) Infection
, a Potent Pan-Genotypic NS5A Inhibitor, for the Treatment of Hepatitis C Virus (HCV) Infection") Samatasvir (IDX719), a Potent Pan-Genotypic NS5A Inhibitor, for the Treatment of Hepatitis C Virus (HCV) Infection Douglas Mayers, MD December 11, 2013 1 Idenix: Advancing All-Oral, Pan-Genotypic Combination
Samatasvir (IDX719), a Potent Pan-Genotypic NS5A Inhibitor, for the Treatment of Hepatitis C Virus (HCV) Infection Douglas Mayers, MD December 11, 2013 1 Idenix: Advancing All-Oral, Pan-Genotypic Combination
Global Development Challenges: Classical and Advanced Therapy Medicinal products
 Global Development Challenges: Classical and Advanced Therapy Medicinal products Beatriz Silva Lima imed, Lisbon University and Infarmed,, Portugal CHMP, CAT, SAWP Member and SWP Chair NONCLINICAL STUDIES
Global Development Challenges: Classical and Advanced Therapy Medicinal products Beatriz Silva Lima imed, Lisbon University and Infarmed,, Portugal CHMP, CAT, SAWP Member and SWP Chair NONCLINICAL STUDIES
GMO Technology Conference
 GMO Technology Conference The regulation of Clinical Trials on humans involving therapies containing or consisting of genetically modified organisms The Printworks, Dublin Castle 10 th & 11 th October
GMO Technology Conference The regulation of Clinical Trials on humans involving therapies containing or consisting of genetically modified organisms The Printworks, Dublin Castle 10 th & 11 th October
Clinical Trials application process, legislation & guidelines
 Clinical Trials application process, legislation & guidelines IMB Clinical Trials Seminar 19 th June 2012 Elaine Breslin MB BCh (NUI), PhD, FRCPI Clinical Assessment Manager 19/06/2012 Slide 1 IMB Mission
Clinical Trials application process, legislation & guidelines IMB Clinical Trials Seminar 19 th June 2012 Elaine Breslin MB BCh (NUI), PhD, FRCPI Clinical Assessment Manager 19/06/2012 Slide 1 IMB Mission
What s the difference? Challenges in pre-clinical development of biologics
 Biologics vs Small MW NCEs What s the difference? Challenges in pre-clinical development of biologics Peter Lloyd Joint Conference of EU Human Pharmacological Societies and 20 th Anniversary of AGAH 31
Biologics vs Small MW NCEs What s the difference? Challenges in pre-clinical development of biologics Peter Lloyd Joint Conference of EU Human Pharmacological Societies and 20 th Anniversary of AGAH 31
FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010
 FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010 FDA Nanotechnology Task Force Carlos Peña, PhD Office of the Commissioner & Subhas Malghan, PhD Center for Devices and
FDA Nanotechnology Regulatory Science Program Science Board Presentation August 2010 FDA Nanotechnology Task Force Carlos Peña, PhD Office of the Commissioner & Subhas Malghan, PhD Center for Devices and
GCP Basics - refresher
 p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
Nonclinical Safety Evaluation of Biotechnology-Derived Therapeutics
 Toxicology for Industrial and Regulatory Scientists Nonclinical Safety Evaluation of Biotechnology-Derived Therapeutics Melanie Hartsough, Ph.D. Senior Consultant Biologics Consulting Group, Inc. April
Toxicology for Industrial and Regulatory Scientists Nonclinical Safety Evaluation of Biotechnology-Derived Therapeutics Melanie Hartsough, Ph.D. Senior Consultant Biologics Consulting Group, Inc. April
September 12, Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852
 Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852") September 12, 2013 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: : Draft Guidance for Industry on Pediatric Study Plans: Content
September 12, 2013 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Room 1061 Rockville, MD 20852 Re: : Draft Guidance for Industry on Pediatric Study Plans: Content
Post-approval Change Management Protocols - Current Status and Next Steps on the Way towards a Global Tool
 Post-approval Change Management Protocols - Current Status and Next Steps on the Way towards a Global Tool Dr. Markus Goese Lead EU CMC Regulatory Policy F. Hoffmann-La Roche Ltd, Basel - Switzerland Presentation
Post-approval Change Management Protocols - Current Status and Next Steps on the Way towards a Global Tool Dr. Markus Goese Lead EU CMC Regulatory Policy F. Hoffmann-La Roche Ltd, Basel - Switzerland Presentation
Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd
![Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd](/thumbs/77/76275664.jpg "Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd") Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations [mabs -monoclonal antibodies ] Peter Lloyd Head of Pharmacokinetics-Pharmacodynamics Novartis Biologics
Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations [mabs -monoclonal antibodies ] Peter Lloyd Head of Pharmacokinetics-Pharmacodynamics Novartis Biologics
Regulatory Perspective
 Regulatory Perspective Presented by Dr Maria Isaac MASc, MD, PhD, Pharmaceutical Medicine Physician, Psychiatrist Senior Scientific Officer An agency of the European Union Disclaimer The views expressed
Regulatory Perspective Presented by Dr Maria Isaac MASc, MD, PhD, Pharmaceutical Medicine Physician, Psychiatrist Senior Scientific Officer An agency of the European Union Disclaimer The views expressed
Guideline on similar biological medicinal products containing interferon beta
 1 2 3 15 December 2011 EMA/CHMP/BMWP/652000/2010 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Guideline on similar biological medicinal products containing interferon beta 7 Draft Draft
1 2 3 15 December 2011 EMA/CHMP/BMWP/652000/2010 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Guideline on similar biological medicinal products containing interferon beta 7 Draft Draft
-Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009
 No.1394/2007 -Regulation (EC) No. 668/2009") Introduction to Advanced Therapy Medicinal Products Regulation -Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009 -Directive 2009/120/EC Dr. Maura O Donovan F.R.C.O.G. MA MD M.R.C.P.I. CAT member
Introduction to Advanced Therapy Medicinal Products Regulation -Regulation (EC) No.1394/2007 -Regulation (EC) No. 668/2009 -Directive 2009/120/EC Dr. Maura O Donovan F.R.C.O.G. MA MD M.R.C.P.I. CAT member
Global Clinical Trials in Korea
 Global Clinical Trials in Korea In-Sook Park Department of Drug Evaluation Korea Food & Drug Administration Contents Regulatory changes relevant to Clinical Trials in Korea Current Status of Clinical Trials
Global Clinical Trials in Korea In-Sook Park Department of Drug Evaluation Korea Food & Drug Administration Contents Regulatory changes relevant to Clinical Trials in Korea Current Status of Clinical Trials
Guide for National Scientific and Regulatory Advice
 Guide for National Scientific and Regulatory Advice ADV-G0017-3 DATE 18 JULY 2017 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes only. CONTENTS
Guide for National Scientific and Regulatory Advice ADV-G0017-3 DATE 18 JULY 2017 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes only. CONTENTS
February 28, Churchill Place Canary Wharf London E14 5EU United Kingdom
 February 28, 2017 Submission of comments on 'Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products' (EMEA/CHMP/SWP/28367/07
February 28, 2017 Submission of comments on 'Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products' (EMEA/CHMP/SWP/28367/07
Chin Koerner Executive Director US Regulatory and Development Policy
 Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
The interface between Good Clinical Practice and Good Manufacturing Practice
 1 The interface between Good Clinical Practice and Good Manufacturing Practice your partner in compliance 1 The interface between GCP and GMP Generally, studies are designed and planned by physicians who
1 The interface between Good Clinical Practice and Good Manufacturing Practice your partner in compliance 1 The interface between GCP and GMP Generally, studies are designed and planned by physicians who
Nonproprietary Naming of Biological Products
 Nonproprietary Naming of Biological Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document
Nonproprietary Naming of Biological Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document
Combination Drugs: Regulatory Guidance and Expectations
 Combination Drugs: Regulatory Guidance and Expectations Philip Gatti, Ph.D. Pharmacologist/CDER Division of Cardiovascular and Renal Products CDER/FDA May 16, 2017 Disclaimer This presentation reflects
Combination Drugs: Regulatory Guidance and Expectations Philip Gatti, Ph.D. Pharmacologist/CDER Division of Cardiovascular and Renal Products CDER/FDA May 16, 2017 Disclaimer This presentation reflects
EU and FDA GMP Regulations: Overview and Comparison
 THE QUALITY ASSURANCE JOURNAL, VOL. 2, 55 60 (1997) EU and FDA GMP Regulations: Overview and Comparison The increasing emphasis on global supply of drug products, as well as starting materials and investigational
THE QUALITY ASSURANCE JOURNAL, VOL. 2, 55 60 (1997) EU and FDA GMP Regulations: Overview and Comparison The increasing emphasis on global supply of drug products, as well as starting materials and investigational
HCT/P Regulation vs 361 Products
 HCT/P Regulation - 351 vs 361 Products Presented by: Paul Gadiock February 15, 2017 Arent Fox LLP Washington, DC New York, NY Los Angeles, CA San Francisco, CA 1 Presentation Overview Introduction Public
HCT/P Regulation - 351 vs 361 Products Presented by: Paul Gadiock February 15, 2017 Arent Fox LLP Washington, DC New York, NY Los Angeles, CA San Francisco, CA 1 Presentation Overview Introduction Public
Public release of clinical information in drug submissions and medical device applications
 Public release of clinical information in drug submissions and medical device applications Health Products and Food Branch March 10, 2017 Health Canada is the federal department responsible for helping
Public release of clinical information in drug submissions and medical device applications Health Products and Food Branch March 10, 2017 Health Canada is the federal department responsible for helping
US FDA Drug Approval Strategies for Pharmaceutical Industry
 Int. J. Pharm. Sci. Rev. Res., 25(1), Mar Apr 2014; Article. 24, Pages: 137-146 Research Article US FDA Drug Approval Strategies for Pharmaceutical Industry Accepted on: 25-12-2013; Finalized on: 28-02-2014.
Int. J. Pharm. Sci. Rev. Res., 25(1), Mar Apr 2014; Article. 24, Pages: 137-146 Research Article US FDA Drug Approval Strategies for Pharmaceutical Industry Accepted on: 25-12-2013; Finalized on: 28-02-2014.
Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products
 Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Table of contents 1.Introduction...2 2.Scope...3 3.Discussion...4 3.1. Chemistry, manufacturing, and controls...4
Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Table of contents 1.Introduction...2 2.Scope...3 3.Discussion...4 3.1. Chemistry, manufacturing, and controls...4
New TB Drugs Approval in Thailand
 New TB Drugs Approval in Thailand Tharnkamol Chanprapaph,, Ph.D. Drug Control Division, Thai Food and Drug Administration Third Annual Open Forum on Key Issues in Tuberculosis Drug Development, New Delhi,
New TB Drugs Approval in Thailand Tharnkamol Chanprapaph,, Ph.D. Drug Control Division, Thai Food and Drug Administration Third Annual Open Forum on Key Issues in Tuberculosis Drug Development, New Delhi,
Regulatory Pathways for Rare Diseases
 Regulatory Pathways for Rare Diseases Celia M. Witten, Ph.D., M.D. Deputy Director, FDA Center for Biologics Evaluation and Research Emerging Technologies for Rare Diseases: Clinical and Regulatory Case
Regulatory Pathways for Rare Diseases Celia M. Witten, Ph.D., M.D. Deputy Director, FDA Center for Biologics Evaluation and Research Emerging Technologies for Rare Diseases: Clinical and Regulatory Case
GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs)
") GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs) DRAFT GUIDANCE This guidance document is for feedback purposes only Comments and suggestions regarding this draft document should be submitted within
GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs) DRAFT GUIDANCE This guidance document is for feedback purposes only Comments and suggestions regarding this draft document should be submitted within