Eileen Navarro MD, FACP Medical Officer, OCS, OTS, CDER, FDA
|
|
|
- Dylan Arnold Fleming
- 6 years ago
- Views:
Transcription
1 Eileen Navarro MD, FACP Medical Officer, OCS, OTS, CDER, FDA 1
2 WHAT MEDICAL REVIEWERS CAN DO WITH STANDARDIZED DATA AND METADATA RECEIVED IN MODULE 5 Eileen Navarro, MD, FACP OCS/OTS/CDER/FDA PharmaSUG
3 Disclaimer: This presentation reflects the views of the author and do not necessarily represent FDA s views or policies. 3

4 Clinical Review Goals Purpose of medical review is not to replicate all analyses but independently assess that -the clinical protocol was implemented as planned -the data needed was collected and documented -the analyses were appropriate and -the results provide information on the drug s efficacy and safety 4

5 BENEFIT > RISK Substantial evidence purported under labeled conditions of use KH Amendments 1962 ALL tests reasonably applicable to show drug to be safe under proposed labeling FDCA 1938 LABEL adequate directions for use 5
6 Content and Format of an Application (21 CFR ), ectd Module 5 (1) human pharmacokinetics (2) microbiology (3) clinical data (4) statistical section (7) pediatric use (8) CRF and CRT 6
7 Clinical Section 1. STUDY REPORTS - description of every study & the statistical analys used to evaluate it 2. Non NDA information relevant to evaluation of safety and effectiveness - from any source evidence (other investigations, commercial marketing experience, scientific literature, unpublished papers) 3. Integrated summaries Efficacy - to assess substantial evidence and support dosage modifications for subgroups Summary and updates of safety - all available information (animal data, summaries, abuse potential, subgroups based on biology rena hepatic, disease severity, 4 month updates) 4. Benefit/Risk assessment 5. Documentation of Human subject protection 6. Trial Audit reports or monitored studies and a list of such studies 21 CFR , ICH E3 7
8 Putting data in perspective 8
9 Clinical Filing Checklist (Day 45) : Are datasets available for all pivotal trials? Are they reliable, transparent, traceable to the CRF? Do the datasets reflect the Sponsor s report of dosage, treatment arms, adequate exposure of doses and duration? Are the datasets in a format to allow review of patient data? Are endpoints, adverse events evaluable? Is the raw data available to derive the composite endpoints? Do the data allow replication of findings? 9
10 Source Data Validation assess consistency of the data provided (e.g., compare information in CRF, CRT, and narrative summaries) for important AEs, assess narrative description, and may ask to see CRF, hospital records and laboratory, radiology, or pathology results 10

11 11 Standards Facilitate the Review Process 11


12 M5 12
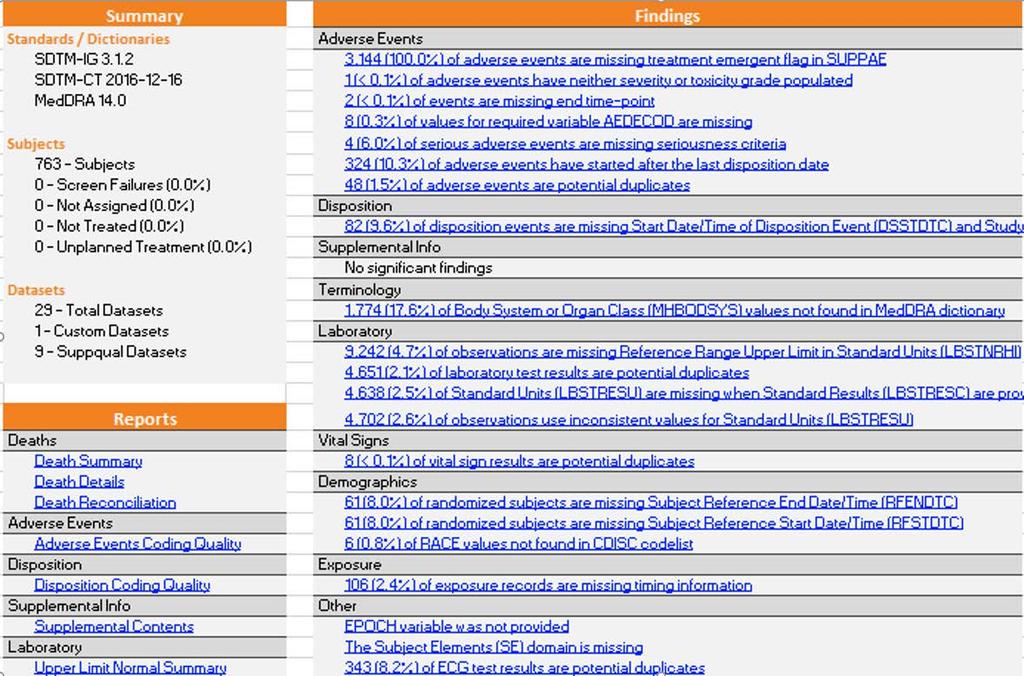
13 13

14 Standardized data and metadata facilitates subject reconciliation 14
15 Standard data help characterize the study population - who was excluded, who was enrolled, randomized, treated, analysed? 15

16 Standard data enables data trace across various domains 16


17 Standard data enables visualization of patient withdrawals or timing of adverse events 17
 Aes of special interest, grouped by system, special tests Individual detailed assessment of individual events Assess causality, drug interaction Suspected adverse reaction (temporality,")
18 Event Data Population quantitative and qualitative comparison by treatment arm, across subgroups death serious AE (SAE) AE leading to discontinuation (AEDC) discontinued patients lost to follow-up (LTFU) Aes of special interest, grouped by system, special tests Individual detailed assessment of individual events Assess causality, drug interaction Suspected adverse reaction (temporality, re-occurrence) Determine whether reported terms refer to the same event (do different codes really refer to the same event) Assess in context of other clinical procedures or events (blood transfusion, surgical procedure, etc) 18

19 Test Data Laboratory: distribution of baseline and change on treatment, central tendency and dispersal, outliers, cumulative rates, time to event, resolution, preclinical/class effects Special assessments and other analyses: hepatic, QT, immunogenicity, carcinogenicity, reprotox, effect on growth, population differences, drug-drug interaction, drug-disease interaction 19

20 Standard data leads to standard AE definition integration of clinical AND laboratory data 20

21 Evaluation for Hepatotoxicity - Hy s Law New Drug New Drug New Drug New Drug New Drug 21
22 Standard data enables insight into drug exposures 22
23 ? 23

24 Standard data helps reviewers develop new safety visualizations 24
25

26




27 Standard data enables shared tools for review collaboration 27


28 28 Standardized data enables subgroup analyses The risk of increased liver-related blood tests were higher in women, Asians and in patients that were older than
29 End to End Standardization CRF SDTM AdAM Page 29

30 Eileen Navarro OTS/OCS/CDER/FDA Silver Spring, MD
ectd: A Clinical Reviewer s Experience Sarah M. Connelly, MD Medical Officer Division of Antiviral Products FDA
 ectd: A Clinical Reviewer s Experience Sarah M. Connelly, MD Medical Officer Division of Antiviral Products FDA Disclaimer The views and opinions expressed in the following PowerPoint slides are those
ectd: A Clinical Reviewer s Experience Sarah M. Connelly, MD Medical Officer Division of Antiviral Products FDA Disclaimer The views and opinions expressed in the following PowerPoint slides are those
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval
 Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
Question and Answer Guide Regarding Notification on Practical Operations of Electronic Study Data Submissions
 Administrative Notice April 27, 2015 To: Prefectrual Health Department (Bureau) Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare Question
Administrative Notice April 27, 2015 To: Prefectrual Health Department (Bureau) Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare Question
Evolving Regulatory Guidance on Submission of Standardized Data. James R. Johnson, PhD RTP CDISC User Network
 Evolving Regulatory Guidance on Submission of Standardized Data James R. Johnson, PhD RTP CDISC User Network 2014-06-11 Originally Presented at PhUSE Conference 2013 Brussels, Belgium (Paper Number: RG03)
Evolving Regulatory Guidance on Submission of Standardized Data James R. Johnson, PhD RTP CDISC User Network 2014-06-11 Originally Presented at PhUSE Conference 2013 Brussels, Belgium (Paper Number: RG03)
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
Objectives Discuss the importance of proper data collection. Identify the types of data collected for clinical trials. List potential source documents
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance
 Vol. 9, No. 2, February 2013 Happy Trials to You What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance Reprinted from the Guide to Good Clinical Practice with permission of Thompson Publishing
Vol. 9, No. 2, February 2013 Happy Trials to You What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance Reprinted from the Guide to Good Clinical Practice with permission of Thompson Publishing
MEDICINES CONTROL COUNCIL
 MEDICINES CONTROL COUNCIL SAFETY REPORTING DURING CLINICAL TRIALS IN SOUTH AFRICA This document has been prepared to serve as a guideline to those reporting adverse events occurring during the use of registered
MEDICINES CONTROL COUNCIL SAFETY REPORTING DURING CLINICAL TRIALS IN SOUTH AFRICA This document has been prepared to serve as a guideline to those reporting adverse events occurring during the use of registered
Sonja Brajovic Medical Officer, Office of Surveillance and Epidemiology Center for Drug Evaluation and Research Food and Drug Administration
 FDA Perspective on MedDRA Coding Quality in Post marketing Safety Reports European Informal MedDRA User Group Webinar Sonja Brajovic Medical Officer, Office of Surveillance and Epidemiology Center for
FDA Perspective on MedDRA Coding Quality in Post marketing Safety Reports European Informal MedDRA User Group Webinar Sonja Brajovic Medical Officer, Office of Surveillance and Epidemiology Center for
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd.
 Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Extrapolation in Pediatric Product Development: Practical Application of the Principle of Scientific Necessity
 Extrapolation in Pediatric Product Development: Practical Application of the Principle of Scientific Necessity Robert Skip Nelson, MD PhD Deputy Director and Senior Pediatric Ethicist Office of Pediatric
Extrapolation in Pediatric Product Development: Practical Application of the Principle of Scientific Necessity Robert Skip Nelson, MD PhD Deputy Director and Senior Pediatric Ethicist Office of Pediatric
1.4 Applicable Regulatory Requirement(s) Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.
 Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.") 1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
Regulatory Pathways. Devices vs. Drugs Are there roles for registries? John Laschinger, MD CDRH/ODE/DCD/SHDB
 Regulatory Pathways Devices vs. Drugs Are there roles for registries? John Laschinger, MD CDRH/ODE/DCD/SHDB johnlaschinger@fda.hhs.gov 1 Disclosures and Disclaimer John C. Laschinger, M.D. I am a full
Regulatory Pathways Devices vs. Drugs Are there roles for registries? John Laschinger, MD CDRH/ODE/DCD/SHDB johnlaschinger@fda.hhs.gov 1 Disclosures and Disclaimer John C. Laschinger, M.D. I am a full
Optimizing Your Study Data Submissions to FDA Updates from CDER and CBER
 This single PDF file contains the slides for all three presentations in the webinar: Optimizing Your Study Data Submissions to FDA Updates from CDER and CBER Please page down to find the slides for all
This single PDF file contains the slides for all three presentations in the webinar: Optimizing Your Study Data Submissions to FDA Updates from CDER and CBER Please page down to find the slides for all
Guidance for Industry Postmarketing Studies and Clinical Trials Implementation of Section 505(o) of the Federal Food, Drug, and Cosmetic Act
 of the Federal Food, Drug, and Cosmetic Act") Guidance for Industry Postmarketing Studies and Clinical Trials Implementation of Section 505(o) of the Federal Food, Drug, and Cosmetic Act DRAFT GUIDANCE This guidance document is being distributed for
Guidance for Industry Postmarketing Studies and Clinical Trials Implementation of Section 505(o) of the Federal Food, Drug, and Cosmetic Act DRAFT GUIDANCE This guidance document is being distributed for
Good Clinical Practice
 Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Simonetta Viviani, MD BIO-VIPE Consulting Limited, Hong Kong
 Simonetta Viviani, MD BIO-VIPE Consulting Limited, Hong Kong DCVMN Clinical Development & Pharmacovigilance Training 17-21 July 2016, Bali, Indonesia Pratical tips on how to write a protocol Write the
Simonetta Viviani, MD BIO-VIPE Consulting Limited, Hong Kong DCVMN Clinical Development & Pharmacovigilance Training 17-21 July 2016, Bali, Indonesia Pratical tips on how to write a protocol Write the
8. Clinical Trial Assessment Phase II
 8. Clinical Trial Assessment Phase II Junko Sato, PhD Office of New Drug I, PMDA Disclaimer: The information within this presentation is based on the presenter s expertise and experience, and represents
8. Clinical Trial Assessment Phase II Junko Sato, PhD Office of New Drug I, PMDA Disclaimer: The information within this presentation is based on the presenter s expertise and experience, and represents
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
JMP JMP Pro JMP Genomics JMP Clinical JMP Add-Ins
 JMP FAMILY OF PRODUCTS: SOFTWARE FOR LIFE SCIENTISTS JMP JMP Pro JMP Genomics JMP Clinical JMP Add-Ins CLINICAL TRIAL DATA REVIEWS Randomized clinical trial remains the gold standard for evaluating efficacy
JMP FAMILY OF PRODUCTS: SOFTWARE FOR LIFE SCIENTISTS JMP JMP Pro JMP Genomics JMP Clinical JMP Add-Ins CLINICAL TRIAL DATA REVIEWS Randomized clinical trial remains the gold standard for evaluating efficacy
Investigator-Initiated INDs
 Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Exploration des données d essais cliniques en utilisant le standard CDISC
 Exploration des données d essais cliniques en utilisant le standard CDISC Florence Kussener French System Engineer Florence.Kussener@jmp.com SAS Institute AGENDA Outcomes of using CDISC standards Case
Exploration des données d essais cliniques en utilisant le standard CDISC Florence Kussener French System Engineer Florence.Kussener@jmp.com SAS Institute AGENDA Outcomes of using CDISC standards Case
Availability of Masked and De-identified Non-Summary Safety and Efficacy Data; Request for
 This document is scheduled to be published in the Federal Register on 06/04/2013 and available online at http://federalregister.gov/a/2013-13083, and on FDsys.gov 4160-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 06/04/2013 and available online at http://federalregister.gov/a/2013-13083, and on FDsys.gov 4160-01-P DEPARTMENT OF HEALTH AND HUMAN
Safety Assessment of Pharmaceutical Products. Christy Chuang-Stein, Pfizer Inc. BASS XIX
 Safety Assessment of Pharmaceutical Products Christy Chuang-Stein, Pfizer Inc. BASS XIX 1 Acknowledgement Some materials came from a joint presentation with Amy Xia (Amgen) at the conference Risk Assessment
Safety Assessment of Pharmaceutical Products Christy Chuang-Stein, Pfizer Inc. BASS XIX 1 Acknowledgement Some materials came from a joint presentation with Amy Xia (Amgen) at the conference Risk Assessment
Introduction to Clinical Research
 Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
GUIDELINE REGARDING COLLECTION, VERIFICATION, AND SUBMISSION OF THE REPORTS OF ADVERSE EVENTS / REACTIONS OCCURRING IN CLINICAL DRUG TRIALS
 1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE November 2004 Revised June 2009 SEX-RELATED CONSIDERATIONS IN THE CONDUCT OF CLINICAL
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE November 2004 Revised June 2009 SEX-RELATED CONSIDERATIONS IN THE CONDUCT OF CLINICAL
Putting CDISC Standards to Work How Converting to CDISC Standards Early in the Clinical Trial Process Will Make Your CDISC Investment Pay
 How Converting to CDISC Standards Early in the Clinical Trial Process Will Make Your CDISC Investment Pay Jon Roth, Vice President, Data Sciences & Biometrics Authorized Trainer for ADaM, CDISC Mark Vieder,
How Converting to CDISC Standards Early in the Clinical Trial Process Will Make Your CDISC Investment Pay Jon Roth, Vice President, Data Sciences & Biometrics Authorized Trainer for ADaM, CDISC Mark Vieder,
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
1 The Clinical Research Coordinator (CRC)... 1
... 1") TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
CDISC Standards: Current and Future
 CDISC 2010 CDISC Standards: Current and Future CDISC Japan Interchange Tokyo, Japan 20 July 2010 Rebecca D. Kush, PhD President and CEO, CDISC Clinical Research Standards (Content) (Protocol-driven Research;
CDISC 2010 CDISC Standards: Current and Future CDISC Japan Interchange Tokyo, Japan 20 July 2010 Rebecca D. Kush, PhD President and CEO, CDISC Clinical Research Standards (Content) (Protocol-driven Research;
Document Reuse: Theory and Practice
 Document Reuse: Theory and Practice Peggy Boe, RN Sr. Director, Medical Writing Image Solutions, Inc (ISI) Company logo here Best Practice: Single Sourcing Creating reusable text and information Requires
Document Reuse: Theory and Practice Peggy Boe, RN Sr. Director, Medical Writing Image Solutions, Inc (ISI) Company logo here Best Practice: Single Sourcing Creating reusable text and information Requires
How did it evolve? o Public disasters, serious fraud and abuse of human rights. o Trials of War criminals-nuremberg code 1949
 ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order
 Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
The Drug Development Process and Design of Clinical Trials
 The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 Clinical Trial Design Guidance Clinical Trial:
The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 Clinical Trial Design Guidance Clinical Trial:
5/23/2016. The Drug Development Process and Design of Clinical Trials. Clinical Trial Design Guidance. Regulatory Definitions
 The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services May 25, 2016 Clinical Trial Design Guidance Clinical Trial: a prospective
The Drug Development Process and Design of Clinical Trials Darlene Kitterman, MBA Director, Investigator Support & Integration Services May 25, 2016 Clinical Trial Design Guidance Clinical Trial: a prospective
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited.
. All rights reserved. Further dissemination without express written consent strictly prohibited.") Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
FDA > CDRH > CFR Title 21 Database Search
 Seite 1 von 7 FDA Home Page CDRH Home Page Search A-Z Index 510 (k) Registration Listing Adverse Events PMA Classification CLIA CFR Title 21 Advisory Committees Assembler Recalls Guidance Standards New
Seite 1 von 7 FDA Home Page CDRH Home Page Search A-Z Index 510 (k) Registration Listing Adverse Events PMA Classification CLIA CFR Title 21 Advisory Committees Assembler Recalls Guidance Standards New
Conducted Under an IND to Support a
 Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Defining Clinical Benefit in Clinical Trials: FDA Perspective
 Defining Clinical Benefit in Clinical Trials: FDA Perspective Jessica J. Lee, MD, MMSc Medical Team Leader Division of Gastroenterology and Inborn Errors Products Center for Drug Evaluation and Research
Defining Clinical Benefit in Clinical Trials: FDA Perspective Jessica J. Lee, MD, MMSc Medical Team Leader Division of Gastroenterology and Inborn Errors Products Center for Drug Evaluation and Research
Industrialized Clinical Data Standards Management Speed of automation, Power of accuracy and Transforming clinical data into business intelligence
 Industrialized Clinical Standards Management Speed of automation, Power of accuracy and Transforming clinical data into business intelligence Executive Summary The majority of commercially available legacy
Industrialized Clinical Standards Management Speed of automation, Power of accuracy and Transforming clinical data into business intelligence Executive Summary The majority of commercially available legacy
SAE Reporting Timelines, Causality Assessment And Compensation. Pawandeep Kaur Associate Medical Director CDSA
 SAE Reporting Timelines, Causality Assessment And Compensation Pawandeep Kaur Associate Medical Director CDSA Objective Background Important definitions SAE Reporting Timelines Causality Assessment Compensation
SAE Reporting Timelines, Causality Assessment And Compensation Pawandeep Kaur Associate Medical Director CDSA Objective Background Important definitions SAE Reporting Timelines Causality Assessment Compensation
Clinical Trials Management for Molecular Diagnostics. April 2016
 Clinical Trials Management for Molecular Diagnostics April 2016 Clinical Operations Responsibilities Accrue samples that have proper informed consent for use Retrospective cohorts Remnant samples Prospective
Clinical Trials Management for Molecular Diagnostics April 2016 Clinical Operations Responsibilities Accrue samples that have proper informed consent for use Retrospective cohorts Remnant samples Prospective
Definitions and Acronyms
 Definitions and Acronyms 510(k) A particular FDA Class For Medical Devices AE Adverse Event CFR Code of Federal Regulations CMS United States Center/College for Medicare and Medicaid Services CRA Clinical
Definitions and Acronyms 510(k) A particular FDA Class For Medical Devices AE Adverse Event CFR Code of Federal Regulations CMS United States Center/College for Medicare and Medicaid Services CRA Clinical
SERIOUS ADVERSE EVENTS
 EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
Food and Drug Administration (FDA) 101
 101") Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
Food and Drug Administration (FDA) 101 What is the Food and Drug Administration (FDA)? The FDA is an agency within the U.S. Department of Health and Human Services that is responsible for protecting the
Chapter 1. Pharmaceutical Industry Overview
 Chapter 1 Pharmaceutical Industry Overview 1.1 Introduction 2 1.2 Regulations 2 1.2.1 Health Insurance Portability and Accountability Act 2 1.2.2 The Code of Federal Regulations 3 1.2.3 Guidance for Industry
Chapter 1 Pharmaceutical Industry Overview 1.1 Introduction 2 1.2 Regulations 2 1.2.1 Health Insurance Portability and Accountability Act 2 1.2.2 The Code of Federal Regulations 3 1.2.3 Guidance for Industry
January (San Francisco, CA) January 8, 2018
 January 8, 2018") January 2017 J.P. Morgan 36 th Annual Management Healthcare Presentation Conference (San Francisco, CA) January 8, 2018 DISCLAIMER Certain information contained in this presentation relates to or is based
January 2017 J.P. Morgan 36 th Annual Management Healthcare Presentation Conference (San Francisco, CA) January 8, 2018 DISCLAIMER Certain information contained in this presentation relates to or is based
FDA Guidance, Clinical Pharmacology, and Regulatory Science
 Principles of Clinical Pharmacology NIH, April 25, 2013 FDA Guidance, Clinical Pharmacology, and Regulatory Science Carl Peck, MD UCSF Center for Drug Development Science Washington DC and San Francisco
Principles of Clinical Pharmacology NIH, April 25, 2013 FDA Guidance, Clinical Pharmacology, and Regulatory Science Carl Peck, MD UCSF Center for Drug Development Science Washington DC and San Francisco
December 18, 2014 Approval Letter - INTERCEPT Blood System for Platelets
 Page 1 of 5 December 18, 2014 Approval Letter - INTERCEPT Blood System for Platelets December 18, 2014 Cerus Corporation Attn: Ms. Carol M. Moore 2550 Stanwell Drive Concord, CA 94520 APPROVAL ORDER Re:
Page 1 of 5 December 18, 2014 Approval Letter - INTERCEPT Blood System for Platelets December 18, 2014 Cerus Corporation Attn: Ms. Carol M. Moore 2550 Stanwell Drive Concord, CA 94520 APPROVAL ORDER Re:
CDISC Data Standards Validation
 CDISC Data Standards Validation How can it be done? Peter Van Reusel Business Unit Director Business & Decision Life Sciences Tel +32 2 774 11 00 Fax +32 2 774 11 99 Mobile +32 476 54 59 17 peter.vanreusel@businessdecision.com
CDISC Data Standards Validation How can it be done? Peter Van Reusel Business Unit Director Business & Decision Life Sciences Tel +32 2 774 11 00 Fax +32 2 774 11 99 Mobile +32 476 54 59 17 peter.vanreusel@businessdecision.com
Expedited Reporting. Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014
 Expedited Reporting Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014 Journal of Clinical Research Best Practices Vol. 7, No. 1, January 2011 Can You
Expedited Reporting Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014 Journal of Clinical Research Best Practices Vol. 7, No. 1, January 2011 Can You
Good Clinical Practice
 Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
Data and Safety Monitoring Boards
 Data and Safety Monitoring Boards Purposes, Roles and Challenges IACCT2017 Nov 29, 2017, Tel Aviv, Israel Arthur Weinstein, MD, FACP, FRCP, MACR Attending Rheumatologist Emeritus, MedStar Washington Hospital
Data and Safety Monitoring Boards Purposes, Roles and Challenges IACCT2017 Nov 29, 2017, Tel Aviv, Israel Arthur Weinstein, MD, FACP, FRCP, MACR Attending Rheumatologist Emeritus, MedStar Washington Hospital
Pharmacovigilance and the Generic Industry. Mark Vieder RPh MBA Director, Safety & Medical Writing Biorasi
 and the Generic Industry Mark Vieder RPh MBA Director, Safety & Medical Writing Biorasi Disclaimer: This presentation has been prepared by Biorasi, LLC solely for the purpose of sharing information and
and the Generic Industry Mark Vieder RPh MBA Director, Safety & Medical Writing Biorasi Disclaimer: This presentation has been prepared by Biorasi, LLC solely for the purpose of sharing information and
FDA Drug Approval Process Vicki Seyfert-Margolis, Ph.D.
 Speaker Comparing The Effectiveness Of New Drugs: Should The FDA Be Asking 'Does It Work' Or 'Does It Work Better'? Vicki L. Seyfert-Margolis, PhD Senior Advisor, Science Innovation and Policy U.S. Food
Speaker Comparing The Effectiveness Of New Drugs: Should The FDA Be Asking 'Does It Work' Or 'Does It Work Better'? Vicki L. Seyfert-Margolis, PhD Senior Advisor, Science Innovation and Policy U.S. Food
EVENT TYPE TIMING REQUIRED FORM
 Reportable Events EVENT TYPE TIMING REQUIRED FORM Unanticipated Problem or Adverse Event 10 days Event Reporting Form Death (related) 10 days Event Reporting Form Death (not related) Renewal Summary with
Reportable Events EVENT TYPE TIMING REQUIRED FORM Unanticipated Problem or Adverse Event 10 days Event Reporting Form Death (related) 10 days Event Reporting Form Death (not related) Renewal Summary with
Human Research Protection Program Good Clinical Practice Guidance for Investigators Regulatory File Essential Documents
 Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Under this license, you are approved to manufacture aflibercept drug substance intermediate, drug substance, and formulated bulk at
 DEPARTMENT OF HEALTH AND HUMAN SERVICES Silver Spring MD 20993 Our STN: BL 125387/0 BLA APPROVAL November 18, 2011 Regeneron Pharmaceuticals, Inc. Attention: Laura Pologe, Ph.D. Associate Director, Regulatory
DEPARTMENT OF HEALTH AND HUMAN SERVICES Silver Spring MD 20993 Our STN: BL 125387/0 BLA APPROVAL November 18, 2011 Regeneron Pharmaceuticals, Inc. Attention: Laura Pologe, Ph.D. Associate Director, Regulatory
Guidance for Industry - Computerized Systems Used in Clinical Trials
 Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
Pharmacovigilance. Training session for patients and consumers involved in EMA activities, 25 November Presented by: Priya Bahri
 Pharmacovigilance Training session for patients and consumers involved in EMA activities, 25 November 2014 Presented by: Priya Bahri An agency of the European Union 2 Pharmacovigilance - the science concerned
Pharmacovigilance Training session for patients and consumers involved in EMA activities, 25 November 2014 Presented by: Priya Bahri An agency of the European Union 2 Pharmacovigilance - the science concerned
End-to-End Management of Clinical Trials Data
 End-to-End Management of Clinical Trials Data A Revolutionary Step Toward Supporting Clinical Trials Analysis Over the Next Decades of Clinical Research WHITE PAPER SAS White Paper Table of Contents Introduction....
End-to-End Management of Clinical Trials Data A Revolutionary Step Toward Supporting Clinical Trials Analysis Over the Next Decades of Clinical Research WHITE PAPER SAS White Paper Table of Contents Introduction....
Expanded Access to Investigational Imaging Drugs
 Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
PharmaSUG 2017 Paper DS14
 PharmaSUG 2017 Paper DS14 Considerations in Submitting Standardized Electronic Data Under the Animal Rule: The Use of SDTMIG and SENDIG Domains, and the Need for New Domains and Concepts Fred Wood, Accenture
PharmaSUG 2017 Paper DS14 Considerations in Submitting Standardized Electronic Data Under the Animal Rule: The Use of SDTMIG and SENDIG Domains, and the Need for New Domains and Concepts Fred Wood, Accenture
Long-Term Follow-Up in Gene Transfer Clinical Research
 Long-Term Follow-Up in Gene Transfer Clinical Research Jan P. Vleck, MD CIP Institutional Biosafety Committee Services A Division of WIRB www.ibcservicepoint.com ibcs@wirb.com What is LTFU? the collection
Long-Term Follow-Up in Gene Transfer Clinical Research Jan P. Vleck, MD CIP Institutional Biosafety Committee Services A Division of WIRB www.ibcservicepoint.com ibcs@wirb.com What is LTFU? the collection
Pharmacovigilance Playbook (Part 1 of 2)
") PHARMACOVIGILANCE AND RISK MANAGEMENT Pharmacovigilance Playbook (Part 1 of 2) Compiled By: Dr. Mufti Suhail Sayeed James Lind Institute www.jliedu.com www.jli.edu.in James Lind Institute www.jliedu.com
PHARMACOVIGILANCE AND RISK MANAGEMENT Pharmacovigilance Playbook (Part 1 of 2) Compiled By: Dr. Mufti Suhail Sayeed James Lind Institute www.jliedu.com www.jli.edu.in James Lind Institute www.jliedu.com
The Clinical Investigator Site Audit Process-Driven Good Clinical Practice
 The Clinical Investigator Site Audit Process-Driven Good Clinical Practice Terry Winchell* GCP Innovative Dynamics, LLC, 7400 Kansas Ave., Kansas City, Kansas 66111-2639, USA Summary This article describes
The Clinical Investigator Site Audit Process-Driven Good Clinical Practice Terry Winchell* GCP Innovative Dynamics, LLC, 7400 Kansas Ave., Kansas City, Kansas 66111-2639, USA Summary This article describes
Clinical Trials and the Code of Federal Regulations. Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014
 Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
What is New on the Regulatory Front?
 What is New on the Regulatory Front? Veronica Miller, PhD Forum for Collaborative Research UC Berkeley SPH Outline Innovation in regulatory science Regulatory challenges in NASH Opportunities for innovation
What is New on the Regulatory Front? Veronica Miller, PhD Forum for Collaborative Research UC Berkeley SPH Outline Innovation in regulatory science Regulatory challenges in NASH Opportunities for innovation
Guideline on good pharmacovigilance practices (GVP)
") 22 June 2012 EMA/816292/2011 (superseded version) Guideline on good pharmacovigilance practices (GVP) Module VII Periodic safety update report Draft finalised by the Agency in collaboration with Member
22 June 2012 EMA/816292/2011 (superseded version) Guideline on good pharmacovigilance practices (GVP) Module VII Periodic safety update report Draft finalised by the Agency in collaboration with Member
FDA Perspectives On Viral & Vector Shedding Studies. Daniel Takefman, Ph.D. Chief, Gene Therapy Branch FDA/CBER ICH Workshop: October 31, 2007
 FDA Perspectives On Viral & Vector Shedding Studies Daniel Takefman, Ph.D. Chief, Gene Therapy Branch FDA/CBER ICH Workshop: October 31, 2007 Overview Define shedding studies Outline basis for need of
FDA Perspectives On Viral & Vector Shedding Studies Daniel Takefman, Ph.D. Chief, Gene Therapy Branch FDA/CBER ICH Workshop: October 31, 2007 Overview Define shedding studies Outline basis for need of
Chin Koerner Executive Director US Regulatory and Development Policy
 Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
FDA Sponsor and Investigator Responsibility Checklist
 FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
Re: Docket No. FDA-2008-D-0386: International Conference on Harmonisation; Draft Guidance on E2F Development Safety Update Report; Availability.
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org November 03, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org November 03, 2008 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
INVESTIGATIONAL DEVICE EXEMPTION APPLICATION. IDE Title (if title being used)
") INVESTIGATIONAL DEVICE EXEMPTION APPLICATION IDE Title (if title being used) Name of Sponsor Investigator, MD X Professor, Department Icahn School of Medicine at Mount Sinai Date of Submission Form version
INVESTIGATIONAL DEVICE EXEMPTION APPLICATION IDE Title (if title being used) Name of Sponsor Investigator, MD X Professor, Department Icahn School of Medicine at Mount Sinai Date of Submission Form version
My Experiences as an FDA Statistician
 My Experiences as an FDA Statistician Yeh-Fong Chen, Ph.D. FDA/CDER/OB/DB3 CBA 2016-2017 Workshop series-3 Dec. 18, 2016 Disclaimer This presentation reflects the views of the author and should not be
My Experiences as an FDA Statistician Yeh-Fong Chen, Ph.D. FDA/CDER/OB/DB3 CBA 2016-2017 Workshop series-3 Dec. 18, 2016 Disclaimer This presentation reflects the views of the author and should not be
EIGHT BASIC ELEMENTS OF INFORMED CONSENT
 1/6 This guidance addresses: o Eight Basic elements of informed consent o Additional elements of informed consent o Other information required by the IRB EIGHT BASIC ELEMENTS OF INFORMED CONSENT Required
1/6 This guidance addresses: o Eight Basic elements of informed consent o Additional elements of informed consent o Other information required by the IRB EIGHT BASIC ELEMENTS OF INFORMED CONSENT Required
New Pharmacovigilance legislation. Post-authorisation safety studies. ENCePP Plenary meeting. 3 May 2012
 New Pharmacovigilance legislation Post-authorisation safety studies ENCePP Plenary meeting 3 May 2012 Presented by: Annalisa Rubino, PhV and Risk Management, EMA An agency of the European Union Why? Public
New Pharmacovigilance legislation Post-authorisation safety studies ENCePP Plenary meeting 3 May 2012 Presented by: Annalisa Rubino, PhV and Risk Management, EMA An agency of the European Union Why? Public
@ALSETF #EAP2015. Jess B. Rabourn CBI Expanded Access Conference July 22, 2015
 How to Win Physician Collaboration Models for Advancing Access to Lifesaving Therapies @ALSETF #EAP2015 Jess B. Rabourn CBI Expanded Access Conference July 22, 2015 How to Win Now that we want Expanded
How to Win Physician Collaboration Models for Advancing Access to Lifesaving Therapies @ALSETF #EAP2015 Jess B. Rabourn CBI Expanded Access Conference July 22, 2015 How to Win Now that we want Expanded
Jose Giron 5/19/16. Protecting and Promoting Your Health. Public Health Service Food and Drug Administration Silver Spring, MD WARNING LETTER
 U.S. Food and Drug Administration Protecting and Promoting Your Health Jose Giron 5/19/16 Department of Health and Human Services Public Health Service Food and Drug Administration Silver Spring, MD 20993
U.S. Food and Drug Administration Protecting and Promoting Your Health Jose Giron 5/19/16 Department of Health and Human Services Public Health Service Food and Drug Administration Silver Spring, MD 20993
Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory Challenges
 The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
SPECIAL EDITION. the backbone of clinical development
 SPECIAL EDITION Medical writing the backbone of clinical development 2017 Medical Writing: The Backbone of Clinical Development Medical Writing for Submission to Asia-Pacific Regulatory Authorities The
SPECIAL EDITION Medical writing the backbone of clinical development 2017 Medical Writing: The Backbone of Clinical Development Medical Writing for Submission to Asia-Pacific Regulatory Authorities The
1201 Maryland Avenue SW, Suite 900, Washington, DC ,
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org December 28, 2010 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org December 28, 2010 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
Volunteering for Clinical Trials
 Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Adverse Event Reporting
 Adverse Event Reporting AE Case Receipt When we receive a case, we induct it through a well-oiled process that reduces the number of subsequent queries, classifies events appropriately, and increases the
Adverse Event Reporting AE Case Receipt When we receive a case, we induct it through a well-oiled process that reduces the number of subsequent queries, classifies events appropriately, and increases the
Nice SUPPQUAL Variables to Have Beilei Xu, Merck & Co., Inc., Rahway, NJ Changhong Shi, Merck & Co., Inc., Rahway, NJ
 Paper RS05 Nice SUPPQUAL Variables to Have Beilei Xu, Merck & Co., Inc., Rahway, NJ Changhong Shi, Merck & Co., Inc., Rahway, NJ ABSTRACT A Supplemental Qualifier (SUPPQUAL) data set is a special Study
Paper RS05 Nice SUPPQUAL Variables to Have Beilei Xu, Merck & Co., Inc., Rahway, NJ Changhong Shi, Merck & Co., Inc., Rahway, NJ ABSTRACT A Supplemental Qualifier (SUPPQUAL) data set is a special Study
April 13, Background
 Pfizer Inc 235 East 42nd Street New York, NY 10017-5755 Tel 212 733 4210 Fax 646 383 9249 Email: marc.wilenzick@pfizer.com April 13, 2009 http://www.regulations.gov Christine Ireland Committee management
Pfizer Inc 235 East 42nd Street New York, NY 10017-5755 Tel 212 733 4210 Fax 646 383 9249 Email: marc.wilenzick@pfizer.com April 13, 2009 http://www.regulations.gov Christine Ireland Committee management
Objectives. The Regulatory Binder = Investigator Site File= Trial Center File 8/16/2010. Essential Documents: Maintaining the Site's Regulatory Binder
 Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Janus Clinical Trials Repository: Modernizing the Review Process through Innovation
 Janus Clinical Trials Repository: Modernizing the Review Process through Innovation Lilliam Rosario, Ph.D. Director Office of Computational Science Food and Drug Administration Janus Clinical Trials Repository
Janus Clinical Trials Repository: Modernizing the Review Process through Innovation Lilliam Rosario, Ph.D. Director Office of Computational Science Food and Drug Administration Janus Clinical Trials Repository
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
GCP Convergence Improves Transportability of Medical Device Clinical Data
 GCP Convergence Improves Transportability of Medical Device Clinical Data By Harmonization-by-Doing Working Group 4 The safety, performance and effectiveness of medical devices are often evaluated by well-controlled
GCP Convergence Improves Transportability of Medical Device Clinical Data By Harmonization-by-Doing Working Group 4 The safety, performance and effectiveness of medical devices are often evaluated by well-controlled
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155
 MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
10/17/2016. Learning Objectives. What is Drug Accountability. Patricia Novo, MPA, MPH Beth Jeffries, BS, CCRP
 PREPARATION FOR DRUG MANAGEMENT AND ACCOUNTABILITY IN A CTN CLINICAL TRIAL Presented by Patricia Novo, MPA, MPH Beth Jeffries, BS, CCRP CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
PREPARATION FOR DRUG MANAGEMENT AND ACCOUNTABILITY IN A CTN CLINICAL TRIAL Presented by Patricia Novo, MPA, MPH Beth Jeffries, BS, CCRP CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
Case studies in the design, analysis and interpretation of non-inferiority trials
 Case studies in the design, analysis and interpretation of non-inferiority trials Krishan Singh, Ph.D. GlaxoSmithKline EFSPI Verona, Nov '08 1 Outline Introduction & Background Case Studies Altabax a topical
Case studies in the design, analysis and interpretation of non-inferiority trials Krishan Singh, Ph.D. GlaxoSmithKline EFSPI Verona, Nov '08 1 Outline Introduction & Background Case Studies Altabax a topical
Overview and Application of the HCV Vertical Resistance Analysis Template
 PharmaSUG 2017 - Paper SS07 Overview and Application of the HCV Vertical Resistance Analysis Template Yan Xie, Abbvie Inc. ABSTRACT FDA proposed an updated hepatitis C virus (HCV) vertical resistance draft
PharmaSUG 2017 - Paper SS07 Overview and Application of the HCV Vertical Resistance Analysis Template Yan Xie, Abbvie Inc. ABSTRACT FDA proposed an updated hepatitis C virus (HCV) vertical resistance draft
Good Clinical Practice (GCP) & Clinical Trial Registries
 & Clinical Trial Registries") Good Clinical Practice (GCP) & Clinical Trial Registries The Fifth Annual Pharmaceutical Regulatory and Compliance Congress and Best Practice Forum November 14-17, 2004 Kate Maloney, RN, MS, CPHQ Manager,
Good Clinical Practice (GCP) & Clinical Trial Registries The Fifth Annual Pharmaceutical Regulatory and Compliance Congress and Best Practice Forum November 14-17, 2004 Kate Maloney, RN, MS, CPHQ Manager,
From Standards that Cost to Standards that Save: Cost Effective Standards Implementation
 From Standards that Cost to Standards that Save: Cost Effective Standards Implementation Jeffrey M. Abolafia, Rho, Chapel Hill, NC Frank DiIorio, CodeCrafters Inc., Philadelphia, PA Warning This presentation
From Standards that Cost to Standards that Save: Cost Effective Standards Implementation Jeffrey M. Abolafia, Rho, Chapel Hill, NC Frank DiIorio, CodeCrafters Inc., Philadelphia, PA Warning This presentation
Second Quarter 2017 Financial Results. August 8, 2017
 Second Quarter 2017 Financial Results August 8, 2017 Agios Conference Call Participants Prepared Remarks Introduction RENEE LECK, Sr. Manager, Investor Relations Business Highlights & 2017 Key Milestones
Second Quarter 2017 Financial Results August 8, 2017 Agios Conference Call Participants Prepared Remarks Introduction RENEE LECK, Sr. Manager, Investor Relations Business Highlights & 2017 Key Milestones
Quality Assurance QA STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits
 Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR
European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR