Bioinformatics in next generation sequencing projects
|
|
|
- Barry Hudson
- 6 years ago
- Views:
Transcription

1 Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet May 2013
2 Standard sequence library generation

3 Illumina Sequencing Technology
")


4 Illumina (Solexa) Sequencing
5 Illumina paired-end and index-read sequencing

6 Once sequenced the problem becomes computational Computational analyses is the bottleneck Rapid improvement in sequencing Still need for customized analysis for most projects
7 Overview of computational analyses genome sequence assembled contig RNA-Seq expression levels ChIP-Seq peak calling Primary Analyses: Image analysis Base calling Mapping (Assembly) Data type specific analyses (e.g. peak calling, calculate expression) Custom project specific analyses
 Text File (GB) Platform-specific analysis using the vendors")
8 Preliminary Analyses Sequences and Real Time Analysis Quality scores Raw Image (TB) Text File (GB) Platform-specific analysis using the vendors programs
9 Sequenced reads Fasta file: >EAS54_6_R1_2_1_413_324 CCCTTCTTGTCTTCAGCGTTTCTCC Read identifier Fastq file: - EAS269:1:120:1786:18#0/1 GAACTCTGCCTTTTTCAGTGATGAGGAAAGGAGTTCTCTCTGGTCCCCAG +HWI - EAS269:1:120:1786:18#0/1 aaab^_u_aa [ U [ _Z ] a `WU_^X `GT^_ \ TM^ ^ \ \ Z \ YQVVXUBBBB Quality scores csfasta file >1_39_146_F3 T >1_39_194_F3 T SOLiD, QV file >1_39_146_F >1_39_194_F
10 Phred Quality Score, Q Each base call has an estimate of the probability of being wrong (error probability, p) Q = -10 * log 10 (p) Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in % 20 1 in % 30 1 in % 40 1 in % 50 1 in %

11 FastQ encodings

12 Fastq quality control (FastQC) Video tutorial:

13 Quality scores for each sequence position

14 Quality scores for each sequence position: A good run

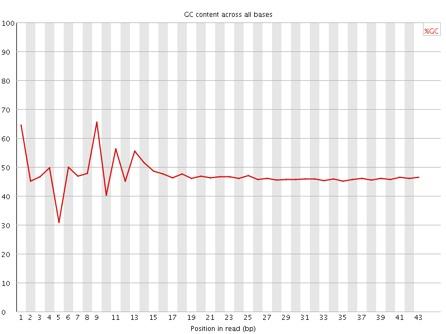
15 GC for reads

16 Percent A,C,G,T at each position

17 Relative enrichment of kmers
18 Overview of computational analyses genome sequence assembled contig RNA-Seq expression levels ChIP-Seq peak calling Primary Analyses: Image analysis Base calling Mapping Assembly Data type specific analyses (e.g. peak calling, calculate expression) Custom project specific analyses

19 Short Read Assembly Velvet and SOAPdenovo de novo genomic assembler specially designed for short read sequencing technologies Nature 2009
20 Two principal approaches for transcriptome reconstruction
21 Genome-independent transcriptome reconstruction Default k = 25 Garbherr et al. Nature Biotechnology, July 2011

22 Finding novel non-annotated genes or transcript variants
23 Mapping of millions of short reads Task: Map millions of short sequences ( nt) onto a genome (3 000 Mbp ) or transcriptome Mismatches (sequencing errors and SNPs) Unique / Repetitive matches Indels (Normal variation, CNVs) Large rearrangements (translocations) BLAST, BLAT tools not designed for these tasks
24 Mapping of RNA-Seq reads STAR Garber et al Nat Methods
25 Mapping of splice junctions Exon n GTAAGT AG Exon n+1 1. compile sets of junctions 2. map reads towards genome + junction compilation + Genome Chromosome Fasta Files Known and putative splice junctions Fasta File
26 Tophat first Method A B C identify candidate exons via genomic mapping A B A C B C Generate possible pairings of exons A B A C B C Align unmappable reads to possible junctions
27 Longer reads By segmenting the long reads, and mapping the segments independently, we can look harder for junctions we might have missed with shorter reads >HWI-EAS229_75_30DY0AAXX:7:1:0:949 GATGTTCTCAGTGTCC GATGTAATCAGTGTCC AACCCTCTCAGTGTCC Running time independent of intron size Very long (100Kb+) intron
28 Mapping to transcriptome Gene: 5 UTR Exons Introns 3 UTR W C DNA (genome) Transcription pre-mrna AAAAA RNA processing (splicing, polyadenylation) mrna AAAAA
29 Microexons and junction coverage 2 or more splice junctions within the same read in-house mapping tophat mapping
30 Microexons and junction coverage 2 or more splice junctions within the same read in-house mapping tophat mapping Different read length will have different problems!
31 Example of STAR aligned single-cell RNA-Seq data Mapping'speed 308'M'reads'/'hour %'uniquely'mapping 60 %'multimapping 25 %'unmapped splice junctions with GT/AG with GC/AG 215 with AT/AC
32 Storing mapped Alignments Formats for storing alignments should include: genomic coordinates mismatches, insertion, deletions etc. quality information
33 Samtools Sequence Alignment Map (SAM) Generic Alignment format Supports long and short reads Human readable, flexible and compact Emerging standard Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. BioinformaScs, 25, [PMID: ] h"p://samtools.sourceforge.net/
34 SAM Example Bit field, where 16 means reverse strand Alignment structure. Here: 22 aligned bases, then 731 bases intron, then 28 aligned bases Start position HWI - EAS269:1:114:1242:1582#0 16 chr Y M731N28M * 0 0 ATTTCGACCATGATCATCGAACCTTCCCCTGGATCCACTTCCACGATCAC #9 ; -7 +2@4 : 2=20-14= : ><?< ; : BB? : 4<BB?ABBBBABCBBBBC=BB NM: i : 0 XS: A:-
35 CIGAR Format M, match/ mismatch I, insertion D, deletion S, softclip... Ref: GCATTCAGATGCAGTACGC Read: cctcag--gcagtagtg Pos: 5 CIGAR: 2S4M3D6M3S 50M
36 Samtools for SAM/BAM files Library and software package (C, Java) Creating, sorting, indexing SAM & BAM Visualizing alignments in command SNP calling Short indel detection BAM (Binary representation of SAM) ~25% file size reduction
37 Read mapping statistics e.g. using RSeQC (package) Density of Reads Nucleotide Frequency A T G C GC content (%) Position of Read
38 Read mapping statistics: Read mapping across genes read number percentile of gene body (5' >3')
39 Read mapping statistics splicing junctions complete_novel 9% partial_novel 2% known 89%
40 Read mapping statistics: duplicate and unique reads Frequency Number of Reads (log10) Sequence base Mapping base Reads %
41 Read mapping statistics: q values on mapped reads Phred Quality Score Position of Read
42 Overview of computational analyses genome sequence assembled contig RNA-Seq expression levels ChIP-Seq peak calling Primary Analyses: Image analysis Base calling Mapping Assembly Data type specific analyses (e.g. peak calling, calculate expression) Custom project specific analyses
43 Visualization Integrated Genome Viewer (Broad Inst.) Custom tracks at UCSC Genome Browser
44 Peak characteristics differ with signal

45 Peak characteristics differ with signal H3K4me3: Sharp promoter peaks H3K36me3: Broad transcription elongation signal
46 Important file formats Sequences: FastQ Aligned reads: SAM/BAM Genome annotations: Bed, Gff Coverage: Wig, (Tdf)
47 BED format chrom - The name of the chromosome (e.g. chr3, chry, chr2_random) or scaffold (e.g. scaffold10671). chromstart - The starsng posison of the feature in the chromosome or scaffold. The first base in a chromosome is numbered 0. chromend - The ending posison of the feature in the chromosome or scaffold. The chromend base is not included in the display of the feature. For example, the first 100 bases of a chromosome are defined as chromstart=0, chromend=100, and span the bases numbered track name=pairedreads description="clone Paired Reads" usescore=1 chr
48 BED continued track name=pairedreads description="clone Paired Reads" usescore=1 chr cloneb ,399, 0,3601 strand - Defines the strand - either '+' or '-'. thickstart - The starting position at which the feature is drawn thickly (for example, the start codon in gene displays). thickend - The ending position at which the feature is drawn thickly (for example, the stop codon in gene displays). itemrgb - An RGB value of the form R,G,B (e.g. 255,0,0). If the track line itemrgb attribute is set to "On", this RBG value will determine the display color of the data contained in this BED line. NOTE: It is recommended that a simple color scheme (eight colors or less) be used with this attribute to avoid overwhelming the color resources of the Genome Browser and your Internet browser. blockcount - The number of blocks (exons) in the BED line. blocksizes - A comma-separated list of the block sizes. The number of items in this list should correspond to blockcount. blockstarts - A comma-separated list of block starts. All of the blockstart positions should be calculated relative to chromstart. The number of items in this list should correspond to blockcount.
49 WIG format (coverage format) Wiggle format (WIG) allows the display of continuous-valued data in a track format Variable step variablestep chrom=chr is equivalent to: variablestep chrom=chr2 span= Fixed step fixedstep chrom=chr3 start= step=
50 Data Repositories Short Read Archive (fastq) [discontinued!] European Nucleotide Archive Gene Expression Omnibus (bed, wig, fastq)
51 SEQAnswers, an active forum for discussions on next-generation sequencing methods and bioinformatics

52

53 Genome-independent transcriptome reconstruction: accuracy and coverage Garbherr et al. Nature Biotechnology, July 2011
54 Genome-independent transcriptome reconstruction: accuracy and coverage Garbherr et al. Nature Biotechnology, July 2011
Sanger vs Next-Gen Sequencing
 Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Tools and Algorithms in Bioinformatics GCBA815/MCGB815/BMI815, Fall 2017 Week-8: Next-Gen Sequencing RNA-seq Data Analysis Babu Guda, Ph.D. Professor, Genetics, Cell Biology & Anatomy Director, Bioinformatics
Reference genomes and common file formats
 Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Reference genomes and common file formats Overview Reference genomes and GRC Fasta and FastQ (unaligned sequences) SAM/BAM (aligned sequences) Summarized genomic features BED (genomic intervals) GFF/GTF
Alignment. J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014
 Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Alignment J Fass UCD Genome Center Bioinformatics Core Wednesday December 17, 2014 From reads to molecules Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG
Introduction to transcriptome analysis using High Throughput Sequencing technologies. D. Puthier 2012
 Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Introduction to transcriptome analysis using High Throughput Sequencing technologies D. Puthier 2012 A typical RNA-Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
RNA-seq Data Analysis
 Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Lecture 3. Clustering; Function/Pathway Enrichment analysis RNA-seq Data Analysis Qi Sun Bioinformatics Facility Biotechnology Resource Center Cornell University Lecture 1. Map RNA-seq read to genome Lecture
Genomic DNA ASSEMBLY BY REMAPPING. Course overview
 ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
ASSEMBLY BY REMAPPING Laurent Falquet, The Bioinformatics Unravelling Group, UNIFR & SIB MA/MER @ UniFr Group Leader @ SIB Course overview Genomic DNA PacBio Illumina methylation de novo remapping Annotation
Ecole de Bioinforma(que AVIESAN Roscoff 2014 GALAXY INITIATION. A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech
 GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
GALAXY INITIATION A. Lermine U900 Ins(tut Curie, INSERM, Mines ParisTech How does Next- Gen sequencing work? DNA fragmentation Size selection and clonal amplification Massive parallel sequencing ACCGTTTGCCG
Bioinformatics small variants Data Analysis. Guidelines. genomescan.nl
 Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
Next Generation Sequencing Bioinformatics small variants Data Analysis Guidelines genomescan.nl GenomeScan s Guidelines for Small Variant Analysis on NGS Data Using our own proprietary data analysis pipelines
RNA-Seq Software, Tools, and Workflows
 RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Software, Tools, and Workflows Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 1, 2016 Some mrna-seq Applications Differential gene expression analysis Transcriptional profiling Assumption:
RNA-Seq Workshop AChemS Sunil K Sukumaran Monell Chemical Senses Center Philadelphia
 RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
RNA-Seq Workshop AChemS 2017 Sunil K Sukumaran Monell Chemical Senses Center Philadelphia Benefits & downsides of RNA-Seq Benefits: High resolution, sensitivity and large dynamic range Independent of prior
Genomics and Transcriptomics of Spirodela polyrhiza
 Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Genomics and Transcriptomics of Spirodela polyrhiza Doug Bryant Bioinformatics Core Facility & Todd Mockler Group, Donald Danforth Plant Science Center Desired Outcomes High-quality genomic reference sequence
Introduction to RNA sequencing
 Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Introduction to RNA sequencing Bioinformatics perspective Olga Dethlefsen NBIS, National Bioinformatics Infrastructure Sweden November 2017 Olga (NBIS) RNA-seq November 2017 1 / 49 Outline Why sequence
Ensembl Tools. EBI is an Outstation of the European Molecular Biology Laboratory.
 Ensembl Tools EBI is an Outstation of the European Molecular Biology Laboratory. Questions? We ve muted all the mics Ask questions in the Chat box in the webinar interface I will check the Chat box periodically
Ensembl Tools EBI is an Outstation of the European Molecular Biology Laboratory. Questions? We ve muted all the mics Ask questions in the Chat box in the webinar interface I will check the Chat box periodically
About Strand NGS. Strand Genomics, Inc All rights reserved.
 About Strand NGS Strand NGS-formerly known as Avadis NGS, is an integrated platform that provides analysis, management and visualization tools for next-generation sequencing data. It supports extensive
About Strand NGS Strand NGS-formerly known as Avadis NGS, is an integrated platform that provides analysis, management and visualization tools for next-generation sequencing data. It supports extensive
De Novo Assembly of High-throughput Short Read Sequences
 De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
De Novo Assembly of High-throughput Short Read Sequences Chuming Chen Center for Bioinformatics and Computational Biology (CBCB) University of Delaware NECC Third Skate Genome Annotation Workshop May 23,
DATA FORMATS AND QUALITY CONTROL
 HTS Summer School 12-16th September 2016 DATA FORMATS AND QUALITY CONTROL Romina Petersen, University of Cambridge (rp520@medschl.cam.ac.uk) Luigi Grassi, University of Cambridge (lg490@medschl.cam.ac.uk)
HTS Summer School 12-16th September 2016 DATA FORMATS AND QUALITY CONTROL Romina Petersen, University of Cambridge (rp520@medschl.cam.ac.uk) Luigi Grassi, University of Cambridge (lg490@medschl.cam.ac.uk)
Next-Generation Sequencing. Technologies
 Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
Next-Generation Next-Generation Sequencing Technologies Sequencing Technologies Nicholas E. Navin, Ph.D. MD Anderson Cancer Center Dept. Genetics Dept. Bioinformatics Introduction to Bioinformatics GS011062
RNA-Sequencing analysis
 RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
RNA-Sequencing analysis Markus Kreuz 25. 04. 2012 Institut für Medizinische Informatik, Statistik und Epidemiologie Content: Biological background Overview transcriptomics RNA-Seq RNA-Seq technology Challenges
Mapping strategies for sequence reads
 Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Mapping strategies for sequence reads Ernest Turro University of Cambridge 21 Oct 2013 Quantification A basic aim in genomics is working out the contents of a biological sample. 1. What distinct elements
Next Gen Sequencing. Expansion of sequencing technology. Contents
 Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Next Gen Sequencing Contents 1 Expansion of sequencing technology 2 The Next Generation of Sequencing: High-Throughput Technologies 3 High Throughput Sequencing Applied to Genome Sequencing (TEDed CC BY-NC-ND
Whole Transcriptome Analysis of Illumina RNA- Seq Data. Ryan Peters Field Application Specialist
 Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
Whole Transcriptome Analysis of Illumina RNA- Seq Data Ryan Peters Field Application Specialist Partek GS in your NGS Pipeline Your Start-to-Finish Solution for Analysis of Next Generation Sequencing Data
MODULE 1: INTRODUCTION TO THE GENOME BROWSER: WHAT IS A GENE?
 MODULE 1: INTRODUCTION TO THE GENOME BROWSER: WHAT IS A GENE? Lesson Plan: Title Introduction to the Genome Browser: what is a gene? JOYCE STAMM Objectives Demonstrate basic skills in using the UCSC Genome
MODULE 1: INTRODUCTION TO THE GENOME BROWSER: WHAT IS A GENE? Lesson Plan: Title Introduction to the Genome Browser: what is a gene? JOYCE STAMM Objectives Demonstrate basic skills in using the UCSC Genome
Lecture 2: Biology Basics Continued
 Lecture 2: Biology Basics Continued Central Dogma DNA: The Code of Life The structure and the four genomic letters code for all living organisms Adenine, Guanine, Thymine, and Cytosine which pair A-T and
Lecture 2: Biology Basics Continued Central Dogma DNA: The Code of Life The structure and the four genomic letters code for all living organisms Adenine, Guanine, Thymine, and Cytosine which pair A-T and
RNA Seq: Methods and Applica6ons. Prat Thiru
 RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
RNA Seq: Methods and Applica6ons Prat Thiru 1 Outline Intro to RNA Seq Biological Ques6ons Comparison with Other Methods RNA Seq Protocol RNA Seq Applica6ons Annota6on Quan6fica6on Other Applica6ons Expression
Variation detection based on second generation sequencing data. Xin LIU Department of Science and Technology, BGI
 Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Variation detection based on second generation sequencing data Xin LIU Department of Science and Technology, BGI liuxin@genomics.org.cn 2013.11.21 Outline Summary of sequencing techniques Data quality
Sequence Annotation & Designing Gene-specific qpcr Primers (computational)
") James Madison University From the SelectedWorks of Ray Enke Ph.D. Fall October 31, 2016 Sequence Annotation & Designing Gene-specific qpcr Primers (computational) Raymond A Enke This work is licensed under
James Madison University From the SelectedWorks of Ray Enke Ph.D. Fall October 31, 2016 Sequence Annotation & Designing Gene-specific qpcr Primers (computational) Raymond A Enke This work is licensed under
Course Presentation. Ignacio Medina Presentation
 Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Course Index Introduction Agenda Analysis pipeline Some considerations Introduction Who we are Teachers: Marta Bleda: Computational Biologist and Data Analyst at Department of Medicine, Addenbrooke's Hospital
Read Mapping and Variant Calling. Johannes Starlinger
 Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
Read Mapping and Variant Calling Johannes Starlinger Application Scenario: Personalized Cancer Therapy Different mutations require different therapy Collins, Meredith A., and Marina Pasca di Magliano.
Basic Bioinformatics: Homology, Sequence Alignment,
 Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
Basic Bioinformatics: Homology, Sequence Alignment, and BLAST William S. Sanders Institute for Genomics, Biocomputing, and Biotechnology (IGBB) High Performance Computing Collaboratory (HPC 2 ) Mississippi
UCSC Genome Browser. Introduction to ab initio and evidence-based gene finding
 UCSC Genome Browser Introduction to ab initio and evidence-based gene finding Wilson Leung 06/2006 Outline Introduction to annotation ab initio gene finding Basics of the UCSC Browser Evidence-based gene
UCSC Genome Browser Introduction to ab initio and evidence-based gene finding Wilson Leung 06/2006 Outline Introduction to annotation ab initio gene finding Basics of the UCSC Browser Evidence-based gene
RNAseq Differential Gene Expression Analysis Report
 RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
RNAseq Differential Gene Expression Analysis Report Customer Name: Institute/Company: Project: NGS Data: Bioinformatics Service: IlluminaHiSeq2500 2x126bp PE Differential gene expression analysis Sample
Introduction to the UCSC genome browser
 Introduction to the UCSC genome browser Dominik Beck NHMRC Peter Doherty and CINSW ECR Fellow, Senior Lecturer Lowy Cancer Research Centre, UNSW and Centre for Health Technology, UTS SYDNEY NSW AUSTRALIA
Introduction to the UCSC genome browser Dominik Beck NHMRC Peter Doherty and CINSW ECR Fellow, Senior Lecturer Lowy Cancer Research Centre, UNSW and Centre for Health Technology, UTS SYDNEY NSW AUSTRALIA
CNV and variant detection for human genome resequencing data - for biomedical researchers (II)
") CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
CNV and variant detection for human genome resequencing data - for biomedical researchers (II) Chuan-Kun Liu 劉傳崑 Senior Maneger National Center for Genome Medican bioit@ncgm.sinica.edu.tw Abstract Common
Next Generation Sequencing Lecture Saarbrücken, 19. March Sequencing Platforms
 Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
Next Generation Sequencing Lecture Saarbrücken, 19. March 2012 Sequencing Platforms Contents Introduction Sequencing Workflow Platforms Roche 454 ABI SOLiD Illumina Genome Anlayzer / HiSeq Problems Quality
L3: Short Read Alignment to a Reference Genome
 L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
L3: Short Read Alignment to a Reference Genome Shamith Samarajiwa CRUK Autumn School in Bioinformatics Cambridge, September 2017 Where to get help! http://seqanswers.com http://www.biostars.org http://www.bioconductor.org/help/mailing-list
RNA-Seq with the Tuxedo Suite
 RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
RNA-Seq with the Tuxedo Suite Monica Britton, Ph.D. Sr. Bioinformatics Analyst September 2015 Workshop The Basic Tuxedo Suite References Trapnell C, et al. 2009 TopHat: discovering splice junctions with
Data Analysis with CASAVA v1.8 and the MiSeq Reporter
 Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
Data Analysis with CASAVA v1.8 and the MiSeq Reporter Eric Smith, PhD Bioinformatics Scientist September 15 th, 2011 2010 Illumina, Inc. All rights reserved. Illumina, illuminadx, Solexa, Making Sense
BCHM 6280 Tutorial: Gene specific information using NCBI, Ensembl and genome viewers
 BCHM 6280 Tutorial: Gene specific information using NCBI, Ensembl and genome viewers Web resources: NCBI database: http://www.ncbi.nlm.nih.gov/ Ensembl database: http://useast.ensembl.org/index.html UCSC
BCHM 6280 Tutorial: Gene specific information using NCBI, Ensembl and genome viewers Web resources: NCBI database: http://www.ncbi.nlm.nih.gov/ Ensembl database: http://useast.ensembl.org/index.html UCSC
Sequence Assembly and Alignment. Jim Noonan Department of Genetics
 Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Sequence Assembly and Alignment Jim Noonan Department of Genetics james.noonan@yale.edu www.yale.edu/noonanlab The assembly problem >>10 9 sequencing reads 36 bp - 1 kb 3 Gb Outline Basic concepts in genome
Gap Filling for a Human MHC Haplotype Sequence
 American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
American Journal of Life Sciences 2016; 4(6): 146-151 http://www.sciencepublishinggroup.com/j/ajls doi: 10.11648/j.ajls.20160406.12 ISSN: 2328-5702 (Print); ISSN: 2328-5737 (Online) Gap Filling for a Human
measuring gene expression December 5, 2017
 measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
measuring gene expression December 5, 2017 transcription a usually short-lived RNA copy of the DNA is created through transcription RNA is exported to the cytoplasm to encode proteins some types of RNA
Release Notes for Genomes Processed Using Complete Genomics Software
 Release Notes for Genomes Processed Using Complete Genomics Software Version 1.11.0 Related Documents... 1 Changes to Version 1.11.0... 2 Changes to Version 1.10.0... 6 Changes to Version 1.9.0... 10 Changes
Release Notes for Genomes Processed Using Complete Genomics Software Version 1.11.0 Related Documents... 1 Changes to Version 1.11.0... 2 Changes to Version 1.10.0... 6 Changes to Version 1.9.0... 10 Changes
BIOINFORMATICS. Lacking alignments? The next-generation sequencing mapper segemehl revisited
 BIOINFORMATICS Vol. 00 no. 00 2014 Pages 1 7 Lacking alignments? The next-generation sequencing mapper segemehl revisited Christian Otto 1,2, Peter F. Stadler 2 8, Steve Hoffmann 1,2 1 Transcriptome Bioinformatics
BIOINFORMATICS Vol. 00 no. 00 2014 Pages 1 7 Lacking alignments? The next-generation sequencing mapper segemehl revisited Christian Otto 1,2, Peter F. Stadler 2 8, Steve Hoffmann 1,2 1 Transcriptome Bioinformatics
Identifying copy number alterations and genotype with Control-FREEC
 Identifying copy number alterations and genotype with Control-FREEC Valentina Boeva contact: freec@curie.fr Most approaches for predicting copy number alterations (CNAs) require you to have whole exomesequencing
Identifying copy number alterations and genotype with Control-FREEC Valentina Boeva contact: freec@curie.fr Most approaches for predicting copy number alterations (CNAs) require you to have whole exomesequencing
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow
 From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
From Variants to Pathways: Agilent GeneSpring GX s Variant Analysis Workflow Technical Overview Import VCF Introduction Next-generation sequencing (NGS) studies have created unanticipated challenges with
Training materials.
 Training materials Ensembl training materials are protected by a CC BY license http://creativecommons.org/licenses/by/4.0/ If you wish to re-use these materials, please credit Ensembl for their creation
Training materials Ensembl training materials are protected by a CC BY license http://creativecommons.org/licenses/by/4.0/ If you wish to re-use these materials, please credit Ensembl for their creation
Variant calling in NGS experiments
 Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
Variant calling in NGS experiments Jorge Jiménez jjimeneza@cipf.es BIER CIBERER Genomics Department Centro de Investigacion Principe Felipe (CIPF) (Valencia, Spain) 1 Index 1. NGS workflow 2. Variant calling
A step-by-step guide to ChIP-seq data analysis
 A step-by-step guide to ChIP-seq data analysis December 03, 2014 Xi Chen, Ph.D. EMBL-European Bioinformatics Institute Wellcome Trust Sanger Institute Target audience Wet-lab biologists with no experience
A step-by-step guide to ChIP-seq data analysis December 03, 2014 Xi Chen, Ph.D. EMBL-European Bioinformatics Institute Wellcome Trust Sanger Institute Target audience Wet-lab biologists with no experience
Intermediate RNA-Seq Tips, Tricks and Non-Human Organisms
 Intermediate RNA-Seq Tips, Tricks and Non-Human Organisms Kevin Silverstein PhD, John Garbe PhD and Ying Zhang PhD, Research Informatics Support System (RISS) MSI September 25, 2014 Slides available at
Intermediate RNA-Seq Tips, Tricks and Non-Human Organisms Kevin Silverstein PhD, John Garbe PhD and Ying Zhang PhD, Research Informatics Support System (RISS) MSI September 25, 2014 Slides available at
Exploring structural variation in the tomato genome with JBrowse
 Exploring structural variation in the tomato genome with JBrowse Richard Finkers, Wageningen UR Plant Breeding Richard.Finkers@wur.nl; @rfinkers Version 1.0, December 2013 This work is licensed under the
Exploring structural variation in the tomato genome with JBrowse Richard Finkers, Wageningen UR Plant Breeding Richard.Finkers@wur.nl; @rfinkers Version 1.0, December 2013 This work is licensed under the
Introduction to RNA-Seq
 Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Introduction to RNA-Seq Monica Britton, Ph.D. Sr. Bioinformatics Analyst March 2015 Workshop Overview of RNA-Seq Activities RNA-Seq Concepts, Terminology, and Work Flows Using Single-End Reads and a Reference
Green Center Computational Core ChIP- Seq Pipeline, Just a Click Away
 Green Center Computational Core ChIP- Seq Pipeline, Just a Click Away Venkat Malladi Computational Biologist Computational Core Cecil H. and Ida Green Center for Reproductive Biology Science Introduc
Green Center Computational Core ChIP- Seq Pipeline, Just a Click Away Venkat Malladi Computational Biologist Computational Core Cecil H. and Ida Green Center for Reproductive Biology Science Introduc
RNA-seq data analysis with Chipster. Eija Korpelainen CSC IT Center for Science, Finland
 RNA-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? 1. What you can do with Chipster and how to operate it 2. What RNA-seq can be
RNA-seq data analysis with Chipster Eija Korpelainen CSC IT Center for Science, Finland chipster@csc.fi What will I learn? 1. What you can do with Chipster and how to operate it 2. What RNA-seq can be
COMPUTER RESOURCES II:
 COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
COMPUTER RESOURCES II: Using the computer to analyze data, using the internet, and accessing online databases Bio 210, Fall 2006 Linda S. Huang, Ph.D. University of Massachusetts Boston In the first computer
Jenny Gu, PhD Strategic Business Development Manager, PacBio
 IDT and PacBio joint presentation Characterizing Alzheimer s Disease candidate genes and transcripts with targeted, long-read, single-molecule sequencing Jenny Gu, PhD Strategic Business Development Manager,
IDT and PacBio joint presentation Characterizing Alzheimer s Disease candidate genes and transcripts with targeted, long-read, single-molecule sequencing Jenny Gu, PhD Strategic Business Development Manager,
Outline. Annotation of Drosophila Primer. Gene structure nomenclature. Muller element nomenclature. GEP Drosophila annotation projects 01/04/2018
 Outline Overview of the GEP annotation projects Annotation of Drosophila Primer January 2018 GEP annotation workflow Practice applying the GEP annotation strategy Wilson Leung and Chris Shaffer AAACAACAATCATAAATAGAGGAAGTTTTCGGAATATACGATAAGTGAAATATCGTTCT
Outline Overview of the GEP annotation projects Annotation of Drosophila Primer January 2018 GEP annotation workflow Practice applying the GEP annotation strategy Wilson Leung and Chris Shaffer AAACAACAATCATAAATAGAGGAAGTTTTCGGAATATACGATAAGTGAAATATCGTTCT
Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction
 Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction Gunnar Rätsch Friedrich Miescher Laboratory Max Planck Society, Tübingen, Germany NGS Bioinformatics Meeting, Paris (March 24, 2010)
Machine Learning Methods for RNA-seq-based Transcriptome Reconstruction Gunnar Rätsch Friedrich Miescher Laboratory Max Planck Society, Tübingen, Germany NGS Bioinformatics Meeting, Paris (March 24, 2010)
Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017
 Andrew Parrish Exeter, 2 nd November 2017") Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017 Topics to cover today What is Next Generation Sequencing (NGS)? Why do we need NGS? Common approaches to NGS NGS
Introduction to Next Generation Sequencing (NGS) Andrew Parrish Exeter, 2 nd November 2017 Topics to cover today What is Next Generation Sequencing (NGS)? Why do we need NGS? Common approaches to NGS NGS
RNA Expression Time Course Analysis
 RNA Expression Time Course Analysis April 23, 2014 Introduction The increased efficiency and reduced per-read cost of next generation sequencing (NGS) has opened new and exciting opportunities for data
RNA Expression Time Course Analysis April 23, 2014 Introduction The increased efficiency and reduced per-read cost of next generation sequencing (NGS) has opened new and exciting opportunities for data
Annotating 7G24-63 Justin Richner May 4, Figure 1: Map of my sequence
 Annotating 7G24-63 Justin Richner May 4, 2005 Zfh2 exons Thd1 exons Pur-alpha exons 0 40 kb 8 = 1 kb = LINE, Penelope = DNA/Transib, Transib1 = DINE = Novel Repeat = LTR/PAO, Diver2 I = LTR/Gypsy, Invader
Annotating 7G24-63 Justin Richner May 4, 2005 Zfh2 exons Thd1 exons Pur-alpha exons 0 40 kb 8 = 1 kb = LINE, Penelope = DNA/Transib, Transib1 = DINE = Novel Repeat = LTR/PAO, Diver2 I = LTR/Gypsy, Invader
SCALABLE, REPRODUCIBLE RNA-Seq
 SCALABLE, REPRODUCIBLE RNA-Seq SCALABLE, REPRODUCIBLE RNA-Seq Advances in the RNA sequencing workflow, from sample preparation through data analysis, are enabling deeper and more accurate exploration
SCALABLE, REPRODUCIBLE RNA-Seq SCALABLE, REPRODUCIBLE RNA-Seq Advances in the RNA sequencing workflow, from sample preparation through data analysis, are enabling deeper and more accurate exploration
Hands-On Four Investigating Inherited Diseases
 Hands-On Four Investigating Inherited Diseases The purpose of these exercises is to introduce bioinformatics databases and tools. We investigate an important human gene and see how mutations give rise
Hands-On Four Investigating Inherited Diseases The purpose of these exercises is to introduce bioinformatics databases and tools. We investigate an important human gene and see how mutations give rise
Illumina (Solexa) Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme
 Throughput: 4 Tbp in one run (5 days) Cheapest sequencing technology. Mismatch errors dominate. Cost: ~$1000 per human genme") Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Illumina (Solexa) Current market leader Based on sequencing by synthesis Current read length 100-150bp Paired-end easy, longer matepairs harder Error ~0.1% Mismatch errors dominate Throughput: 4 Tbp in
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Supplementary Material
 Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions Joshua N. Burton 1, Andrew Adey 1, Rupali P. Patwardhan 1, Ruolan Qiu 1, Jacob O. Kitzman 1, Jay Shendure 1 1 Department
Introduction to genome biology
 Introduction to genome biology Lisa Stubbs We ve found most genes; but what about the rest of the genome? Genome size* 12 Mb 95 Mb 170 Mb 1500 Mb 2700 Mb 3200 Mb #coding genes ~7000 ~20000 ~14000 ~26000
Introduction to genome biology Lisa Stubbs We ve found most genes; but what about the rest of the genome? Genome size* 12 Mb 95 Mb 170 Mb 1500 Mb 2700 Mb 3200 Mb #coding genes ~7000 ~20000 ~14000 ~26000
Sequencing technologies. Jose Blanca COMAV institute bioinf.comav.upv.es
 Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Sequencing technologies Jose Blanca COMAV institute bioinf.comav.upv.es Outline Sequencing technologies: Sanger 2nd generation sequencing: 3er generation sequencing: 454 Illumina SOLiD Ion Torrent PacBio
Long and short/small RNA-seq data analysis
 Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
Long and short/small RNA-seq data analysis GEF5, 4.9.2015 Sami Heikkinen, PhD, Dos. Topics 1. RNA-seq in a nutshell 2. Long vs short/small RNA-seq 3. Bioinformatic analysis work flows GEF5 / Heikkinen
Genomics AGRY Michael Gribskov Hock 331
 Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
Genomics AGRY 60000 Michael Gribskov gribskov@purdue.edu Hock 331 Computing Essentials Resources In this course we will assemble and annotate both genomic and transcriptomic sequence assemblies We will
TSSpredator User Guide v 1.00
 TSSpredator User Guide v 1.00 Alexander Herbig alexander.herbig@uni-tuebingen.de Kay Nieselt kay.nieselt@uni-tuebingen.de June 3, 2013 1 Getting Started TSSpredator is a tool for the comparative detection
TSSpredator User Guide v 1.00 Alexander Herbig alexander.herbig@uni-tuebingen.de Kay Nieselt kay.nieselt@uni-tuebingen.de June 3, 2013 1 Getting Started TSSpredator is a tool for the comparative detection
Differential gene expression analysis using RNA-seq
 https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 1: Introduction into high-throughput
https://abc.med.cornell.edu/ Differential gene expression analysis using RNA-seq Applied Bioinformatics Core, August 2017 Friederike Dündar with Luce Skrabanek & Ceyda Durmaz Day 1: Introduction into high-throughput
Gene Identification in silico
 Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
Gene Identification in silico Nita Parekh, IIIT Hyderabad Presented at National Seminar on Bioinformatics and Functional Genomics, at Bioinformatics centre, Pondicherry University, Feb 15 17, 2006. Introduction
ChIP-seq analysis. adapted from J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant
 ChIP-seq analysis adapted from J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant http://biow.sb-roscoff.fr/ecole_bioinfo/training_material/chip-seq/documents/presentation_chipseq.pdf A model
ChIP-seq analysis adapted from J. van Helden, M. Defrance, C. Herrmann, D. Puthier, N. Servant http://biow.sb-roscoff.fr/ecole_bioinfo/training_material/chip-seq/documents/presentation_chipseq.pdf A model
Next Generation Sequencing: An Overview
 Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Next Generation Sequencing: An Overview Cavan Reilly November 13, 2017 Table of contents Next generation sequencing NGS and microarrays Study design Quality assessment Burrows Wheeler transform Next generation
Alignment methods. Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics
 Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Alignment methods Martijn Vermaat Department of Human Genetics Center for Human and Clinical Genetics Alignment methods Sequence alignment Assembly vs alignment Alignment methods Common issues Platform
Haploid Assembly of Diploid Genomes
 Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Haploid Assembly of Diploid Genomes Challenges, Trials, Tribulations 13 October 2011 İnanç Birol Assembly By Short Sequencing IEEE InfoVis 2009 2 3 in Literature ~40 citations on tool comparisons ~20 citations
Cancer Genetics Solutions
 Cancer Genetics Solutions Cancer Genetics Solutions Pushing the Boundaries in Cancer Genetics Cancer is a formidable foe that presents significant challenges. The complexity of this disease can be daunting
Cancer Genetics Solutions Cancer Genetics Solutions Pushing the Boundaries in Cancer Genetics Cancer is a formidable foe that presents significant challenges. The complexity of this disease can be daunting
Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH. BIOL 7210 A Computational Genomics 2/18/2015
 Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH BIOL 7210 A Computational Genomics 2/18/2015 The $1,000 genome is here! http://www.illumina.com/systems/hiseq-x-sequencing-system.ilmn Bioinformatics bottleneck
Leonardo Mariño-Ramírez, PhD NCBI / NLM / NIH BIOL 7210 A Computational Genomics 2/18/2015 The $1,000 genome is here! http://www.illumina.com/systems/hiseq-x-sequencing-system.ilmn Bioinformatics bottleneck
Workflow of de novo assembly
 Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Workflow of de novo assembly Experimental Design Clean sequencing data (trim adapter and low quality sequences) Run assembly software for contiging and scaffolding Evaluation of assembly Several iterations:
Annotating Fosmid 14p24 of D. Virilis chromosome 4
 Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
Lo 1 Annotating Fosmid 14p24 of D. Virilis chromosome 4 Lo, Louis April 20, 2006 Annotation Report Introduction In the first half of Research Explorations in Genomics I finished a 38kb fragment of chromosome
Assessing De-Novo Transcriptome Assemblies
 Assessing De-Novo Transcriptome Assemblies Shawn T. O Neil Center for Genome Research and Biocomputing Oregon State University Scott J. Emrich University of Notre Dame 100K Contigs, Perfect 1M Contigs,
Assessing De-Novo Transcriptome Assemblies Shawn T. O Neil Center for Genome Research and Biocomputing Oregon State University Scott J. Emrich University of Notre Dame 100K Contigs, Perfect 1M Contigs,
SNP calling and VCF format
 SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
SNP calling and VCF format Laurent Falquet, Oct 12 SNP? What is this? A type of genetic variation, among others: Family of Single Nucleotide Aberrations Single Nucleotide Polymorphisms (SNPs) Single Nucleotide
Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition
 RESEARCH ARTICLE Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition Moses M. Muraya 1,2, Thomas Schmutzer 1 *, Chris Ulpinnis
RESEARCH ARTICLE Targeted Sequencing Reveals Large-Scale Sequence Polymorphism in Maize Candidate Genes for Biomass Production and Composition Moses M. Muraya 1,2, Thomas Schmutzer 1 *, Chris Ulpinnis
Section 10.3 Outline 10.3 How Is the Base Sequence of a Messenger RNA Molecule Translated into Protein?
 Section 10.3 Outline 10.3 How Is the Base Sequence of a Messenger RNA Molecule Translated into Protein? Messenger RNA Carries Information for Protein Synthesis from the DNA to Ribosomes Ribosomes Consist
Section 10.3 Outline 10.3 How Is the Base Sequence of a Messenger RNA Molecule Translated into Protein? Messenger RNA Carries Information for Protein Synthesis from the DNA to Ribosomes Ribosomes Consist
NOW GENERATION SEQUENCING. Monday, December 5, 11
 NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
NOW GENERATION SEQUENCING 1 SEQUENCING TIMELINE 1953: Structure of DNA 1975: Sanger method for sequencing 1985: Human Genome Sequencing Project begins 1990s: Clinical sequencing begins 1998: NHGRI $1000
Outline. Introduction to ab initio and evidence-based gene finding. Prokaryotic gene predictions
 Outline Introduction to ab initio and evidence-based gene finding Overview of computational gene predictions Different types of eukaryotic gene predictors Common types of gene prediction errors Wilson
Outline Introduction to ab initio and evidence-based gene finding Overview of computational gene predictions Different types of eukaryotic gene predictors Common types of gene prediction errors Wilson
QIAGEN s NGS Solutions for Biomarkers NGS & Bioinformatics team QIAGEN (Suzhou) Translational Medicine Co.,Ltd
 Translational Medicine Co.,Ltd") QIAGEN s NGS Solutions for Biomarkers NGS & Bioinformatics team QIAGEN (Suzhou) Translational Medicine Co.,Ltd 1 Our current NGS & Bioinformatics Platform 2 Our NGS workflow and applications 3 QIAGEN s
QIAGEN s NGS Solutions for Biomarkers NGS & Bioinformatics team QIAGEN (Suzhou) Translational Medicine Co.,Ltd 1 Our current NGS & Bioinformatics Platform 2 Our NGS workflow and applications 3 QIAGEN s
RNA-Seq Tutorial 1. Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016
 RNA-Seq Tutorial 1 Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016 Slides available at www.msi.umn.edu/tutorial-materials RNA-Seq Tutorials Lectures RNA-Seq experiment
RNA-Seq Tutorial 1 Kevin Silverstein, Ying Zhang Research Informatics Solutions, MSI October 18, 2016 Slides available at www.msi.umn.edu/tutorial-materials RNA-Seq Tutorials Lectures RNA-Seq experiment
MAKING WHOLE GENOME ALIGNMENTS USABLE FOR BIOLOGISTS. EXAMPLES AND SAMPLE ANALYSES.
 MAKING WHOLE GENOME ALIGNMENTS USABLE FOR BIOLOGISTS. EXAMPLES AND SAMPLE ANALYSES. Table of Contents Examples 1 Sample Analyses 5 Examples: Introduction to Examples While these examples can be followed
MAKING WHOLE GENOME ALIGNMENTS USABLE FOR BIOLOGISTS. EXAMPLES AND SAMPLE ANALYSES. Table of Contents Examples 1 Sample Analyses 5 Examples: Introduction to Examples While these examples can be followed
Next Generation Sequencing Technologies. Some slides are modified from Robi Mitra s lecture notes
 Next Generation Sequencing Technologies Some slides are modified from Robi Mitra s lecture notes What will you do to understand a disease? What will you do to understand a disease? Genotype Phenotype Hypothesis
Next Generation Sequencing Technologies Some slides are modified from Robi Mitra s lecture notes What will you do to understand a disease? What will you do to understand a disease? Genotype Phenotype Hypothesis
user s guide Question 3
 Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
Single Nucleotide Variant Analysis. H3ABioNet May 14, 2014
 Single Nucleotide Variant Analysis H3ABioNet May 14, 2014 Outline What are SNPs and SNVs? How do we identify them? How do we call them? SAMTools GATK VCF File Format Let s call variants! Single Nucleotide
Single Nucleotide Variant Analysis H3ABioNet May 14, 2014 Outline What are SNPs and SNVs? How do we identify them? How do we call them? SAMTools GATK VCF File Format Let s call variants! Single Nucleotide
Measuring transcriptomes with RNA-Seq
 Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2017 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC
Measuring transcriptomes with RNA-Seq BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2017 Anthony Gitter gitter@biostat.wisc.edu These slides, excluding third-party material, are licensed under CC BY-NC
Shuji Shigenobu. April 3, 2013 Illumina Webinar Series
 Shuji Shigenobu April 3, 2013 Illumina Webinar Series RNA-seq RNA-seq is a revolutionary tool for transcriptomics using deepsequencing technologies. genome HiSeq2000@NIBB (Wang 2009 with modifications)
Shuji Shigenobu April 3, 2013 Illumina Webinar Series RNA-seq RNA-seq is a revolutionary tool for transcriptomics using deepsequencing technologies. genome HiSeq2000@NIBB (Wang 2009 with modifications)
Welcome to the NGS webinar series
 Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
Welcome to the NGS webinar series Webinar 1 NGS: Introduction to technology, and applications NGS Technology Webinar 2 Targeted NGS for Cancer Research NGS in cancer Webinar 3 NGS: Data analysis for genetic
The dsrbp and inactive editor, ADR-1, utilizes dsrna binding to regulate A-to-I RNA editing across the C. elegans transcriptome
 The dsrbp and inactive editor, ADR-1, utilizes dsrna binding to regulate A-to-I RNA editing across the C. elegans transcriptome Michael C. Washburn 1,8, Boyko Kakaradov 2,3,8, Balaji Sundararaman 3, Emily
The dsrbp and inactive editor, ADR-1, utilizes dsrna binding to regulate A-to-I RNA editing across the C. elegans transcriptome Michael C. Washburn 1,8, Boyko Kakaradov 2,3,8, Balaji Sundararaman 3, Emily
user s guide Question 3
 Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
Question 3 During a positional cloning project aimed at finding a human disease gene, linkage data have been obtained suggesting that the gene of interest lies between two sequence-tagged site markers.
RNA-Seq analysis workshop
 RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
RNA-Seq analysis workshop Zhangjun Fei Boyce Thompson Institute for Plant Research USDA Robert W. Holley Center for Agriculture and Health Cornell University Outline Background of RNA-Seq Application of
Agenda. Web Databases for Drosophila. Gene annotation workflow. GEP Drosophila annotation projects 01/01/2018. Annotation adding labels to a sequence
 Agenda GEP annotation project overview Web Databases for Drosophila An introduction to web tools, databases and NCBI BLAST Web databases for Drosophila annotation UCSC Genome Browser NCBI / BLAST FlyBase
Agenda GEP annotation project overview Web Databases for Drosophila An introduction to web tools, databases and NCBI BLAST Web databases for Drosophila annotation UCSC Genome Browser NCBI / BLAST FlyBase
Atelier Chip-Seq. Stéphanie Le Gras, IGBMC Strasbourg Violaine Saint-André, Institut Curie Paris Morgane Thomas-Chollier, ENS Paris
 Atelier Chip-Seq Stéphanie Le Gras, IGBMC Strasbourg Violaine Saint-André, Institut Curie Paris Morgane Thomas-Chollier, ENS Paris École de bioinformatique AVIESAN-IFB 2017 Get connected to the server
Atelier Chip-Seq Stéphanie Le Gras, IGBMC Strasbourg Violaine Saint-André, Institut Curie Paris Morgane Thomas-Chollier, ENS Paris École de bioinformatique AVIESAN-IFB 2017 Get connected to the server
Interpreting RNA-seq data (Browser Exercise II)
") Interpreting RNA-seq data (Browser Exercise II) In previous exercises, you spent some time learning about gene pages and examining genes in the context of the GBrowse genome browser. It is important to
Interpreting RNA-seq data (Browser Exercise II) In previous exercises, you spent some time learning about gene pages and examining genes in the context of the GBrowse genome browser. It is important to