Department of Microbiology
|
|
|
- Rodney Hardy
- 5 years ago
- Views:
Transcription
 UGC FUNDED")
1 0 Department of Microbiology St Aloysius College, Mangalore (Autonomous) Re -accredited by NAAC A Grade CGPA3.62 College with Potential for Excellence LABORATORY MANUAL CERTIFICATE COURSE IN FOOD SAFETY AND ADULTERATION DETECTION (COP/FS) UGC FUNDED UNDER CAREER ORIENTED PROGRAMES
2 1 LIST OF EXPERIMENTS 1. Microscopic observation of microorganisms commonly found in food: Gram smear preparation and Tease mount preparation of fungi. 2. Observation and identification of bacterial types and fungi. 3. Preparation of culture media used in food examination. 4. Microbial examinations of foods: Isolation of bacteria and Fungi from different food types. 5. Tests for milk: Dye Reduction tests-methylene blue reduction and Resazurin test. 6. Tests for milk: Lactose and Lactic acid estimation in milk. 7. Direct Microscopic count of bacteria. 8. Water examination: Multiple tubes Method. Water examination: PA method. 9. Detection of adulterants in Food: Tests for milk adulterants and Ghee. 10. Detection of adulterants: Tests for honey and jaggery, sweetmeats and ice cream.
3 2 EXPERIMENT NO1: GRAM STAINING Materials Required: 1. Clean glass slides 2. Inoculating loop 3. Bunsen burner 4. Bibulous paper 5. Microscope 6. Lens paper and lens cleaner 7. Immersion oil 8. Distilled water to 24 hour cultures of organisms Reagents: 1. Primary Stain - Crystal Violet 2. Mordant - Grams Iodine 3. Decolourizer - Ethyl Alcohol 4. Secondary Stain - Safranin Procedure: Part 1: Preparation of the glass microscopic slide Grease or oil free slides are essential for the preparation of microbial smears. Grease or oil from the fingers on the slides is removed by washing the slides with soap and water. Wipe the slides
4 3 with spirit or alcohol. After cleaning, dry the slides and place them on laboratory towels until ready for use. Part 2: Labeling of the slides Drawing a circle on the underside of the slide using a glassware-marking pen may be helpful to clearly designate the area in which you will prepare the smear. You may also label the slide with the initials of the name of the organism on the edge of the slide. Care should be taken that the label should not be in contact with the staining reagents. Part 3: Preparation of the smear Bacterial suspensions in broth: With a sterile cooled loop, place a loopful of the broth culture on the slide. Spread by means of circular motion of the inoculating loop to about one centimeter in diameter. Excessive spreading may result in disruption of cellular arrangement. A satisfactory smear will allow examination of the typical cellular arrangement and isolated cells. Bacterial plate cultures: With a sterile cooled loop, place a drop of sterile water or saline solution on the slide. Sterilize and cool the loop again and pick up a very small sample of a bacterial colony and gently stir into the drop of water/saline on the slide to create an emulsion. Swab Samples: Roll the swab over the cleaned surface of a glass slide. Please note: It is very important to prevent preparing thick, dense smears which contain an excess of the bacterial sample. A very thick smear diminishes the amount of light that can pass through, thus making it difficult to visualize the morphology of single cells. Smears typically require only a small amount of bacterial culture. An effective smear appears as a thin whitish layer or film after heat-fixing. Part 4: Heat Fixing
5 4 Heat fixing kills the bacteria in the smear, firmly adheres the smear to the slide, and allows the sample to more readily take up stains. Allow the smear to air dry. After the smear has air-dried, hold the slide at one end and pass the entire slide through the flame of a Bunsen burner two to three times with the smear-side up. Now the smear is ready to be stained. Please Note: Take care to prevent overheating the slide because proteins in the specimen can coagulate causing cellular morphology to appear distorted.
6 5 Part 5: Gram Stain Procedure 1. Place slide with heat fixed smear on staining tray.
7 2. Gently flood smear with crystal violet and let stand for 1 minute Tilt the slide slightly and gently rinse with tap water or distilled water using a wash bottle. 4. Gently flood the smear with Gram s iodine and let stand for 1 minute. 5. Tilt the slide slightly and gently rinse with tap water or distilled water using a wash bottle. The smear will appear as a purple circle on the slide. 6. Decolorize using 95% ethyl alcohol or acetone. Tilt the slide slightly and apply the alcohol drop by drop for 5 to 10 seconds until the alcohol runs almost clear. Be careful not to overdecolorize. 7. Immediately rinse with water. 8. Gently flood with safranin to counter-stain and let stand for 45 seconds. 9. Tilt the slide slightly and gently rinse with tap water or distilled water using a wash bottle. 10. Blot dry the slide with bibulous paper. 11. View the smear using a light-microscope under oil-immersion.

8 7 &&&&&&&&&&&&&&& EXPERIMENT NO:2 TEASE MOUNT PREPARATION OF FUNGI The lactophenol cotton blue (LPCB) wet mount preparation is the most widely used method of staining and observing fungi and is simple to prepare. The preparation has three components: 1. Phenol: kills any live organisms; 2. Lactic acid : It preserves fungal structures, and 3. Cotton blue : It stains the chitin in the fungal cell walls. Lactophenol Cotton Blue Solution is a mounting medium and staining agent used in the preparation of slides for microscopic examination of fungi. Fungal elements are stained intensely
9 8 blue. MATERIALS REQUIRED: Glass slide Coverslips Needle REAGENTS REQUIRED: Lactophenol Cotton blue Preparation of lactophenol cotton blue (LPCB) slide mounts 1. Place a drop of seventy percent alcohol on a clean microscope slide. 2. Material from cultures of filamentous fungi should be removed using a stiff inoculating wire not the loop used for manipulations with bacteria or yeasts. 3. Flame the wire by holding it upright in the hottest part of the Bunsen flame, just above the blue cone, until the whole length of the wire glows red hot. 4. You must ensure that the inoculating wire has cooled before placing it in a fungal culture it should have cooled sufficiently after approximately ten seconds. 5. Remove the cap from the tube but do not put it on the bench. Kill any contaminating microorganisms by flaming the neck of the tube. 6. Remove a small amount of the culture. For fungal cultures, it is often useful to take a little of the agar medium together with the fungus. In any case, the material should be disturbed as little as possible when being transferred to the slide. 7. Flame the neck of the tube once more and replace the cap. 8. Immerse the fungal material in the drop of seventy percent alcohol. This drives out the air trapped between the hyphae. 9. Tease out the material very gently with mounted needles. 10. Do not forget to sterilise the inoculating wire and the needles after use by heating to red heat in a Bunsen flame 11. Fungal structures are readily visualised after staining with a lactophenol cotton blue dye preparation.
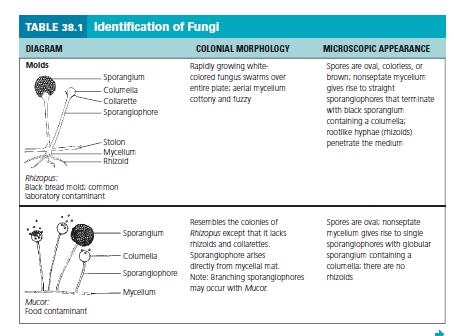
10 9 12. Before the alcohol dries out add one or at most two drops of the stain. A common fault is to add too much to the preparation. Holding the coverslip between your index finger and thumb, touch one edge of the drop of stain with the edge of the coverslip. 13. Lower the coverslip gently onto the slide, trying to avoid air bubbles. Your preparation is now ready for examination. 14. Make the initial examination using a low power objective lens. The thinner parts of the preparation, generally around the edges of the mounted material, will yield the best images. 15. Switch to a higher power 40X objective for more detailed examination of spores and other structures. &&&&&&&&&&&&&&&&&&&&&&&&& EXPERIMENT 3: IDENTIFICATION OF UNKNOWN FUNGI FUNGI Learning Objective Identify a fungal unknown based on colonial morphology and microscopic appearance. Principle In this experiment, you will be provided with a number-coded pure culture of a representative fungal organism for cultivation and subsequent identification. Materials Cultures Number-coded, 7-day Sabouraud broth spore suspensions of Aspergillus, Mucor, Penicillium, Alternaria, Rhizopus, Cladosporium, Fusarium, and Yeast. Media One Sabouraud agar plate per student. Reagent Lactophenol cotton-blue solution. Equipment Bunsen burner, dissecting microscope, hand lens,sterile cotton swabs, glass slides, coverslips, inoculating
11 10 loop, and glassware marking pencil. Procedure Lab One 1. With a sterile inoculating loop, inoculate an appropriately labeled Sabouraud agar plate with one of the provided unknown cultures by placing one loopful in the center of the plate. Note: Do not spread culture. 2. Incubate the plates in a noninverted position for 1 week at 25 C in a moist incubator. Procedure Lab Two 1. Observe mold cultures with a hand lens, noting and recording their colonial morphologies. 2. Prepare a wet mount by suspending some of the culture in a few drops of lactophenol cotton-blue solution. Be gentle to avoid damaging the fungal structures. 3. Examine the preparation under high-power and low-power magnifications with the aid of a dissecting microscope and record your observations in the Lab Report. Observations and Results 1. Draw representative microscopic fields of your culture below. 2. Using Figure given below, identify your unknown fungal organism. a. Color pigmentation b. Diameter (mm) c. Texture (cottony, smooth, etc.) d. Margin (entire, undulating, lobular, etc.) e. Aerial hyphae (septate, nonseptate)
12 11
13 12 &&&&&&&&&&&&&&&&&&
14 13 EXPERIMENT NO:4.OBSERVATION OF YEAST MORPHOLOGY Yeasts are non filamentous unicellular fungi. Yeast cultures resemble bacteria when grown on the surface of artificial laboratory media; however, they are 5 to 10 times larger than bacteria. Microscopically, yeast cells may be ellipsoidal, spherical, or in some cases, cylindrical. Unlike molds, yeast do not have aerial hyphae and supporting sporangia. Yeast reproduce asexually by budding or by fission. In budding, an outgrowth from the parent cell (a bud) pinches off, producing a daughter cell. Fission occurs in certain species of yeast, such as those in the genus Schizosaccharomyces. During fission, the parent cell elongates, its nucleus divides, and it splits evenly into two daughter cells. Some yeast may also undergo sexual reproduction when two sexual spores conjugate, giving rise to a zygote, or diploid cell. The nucleus of this cell divides by meiosis, producing four new haploid nuclei (sexual spores), called ascospores, contained within a structure called the ascus When the ascus ruptures, the ascospores are released and conjugate, starting the cycle again. Yeasts are important for many reasons. Saccharomyces cerevisiae is referred to as baker s yeast and is used as the leavening agent in dough. Two major strains of yeast, Saccharomyces carlsbergensis and Saccharomyces cerevisiae, are used for brewing. The wine industry relies on wild yeast (present on the grape) for the fermentation of grape juice, which is supplemented with Saccharomyces ellipsoideus to begin the fermentation. Also, the high vitamin content of yeasts makes them particularly valuable as food supplements. As useful as some yeasts are, there are a few species that can create problems in the food industry or are harmful to humans. Undesired yeast must be excluded from the manufacture of fruit juices, such as grape juice or apple cider, to prevent the fermentation of fruit sugars to alcohol. The contamination of soft cheese by some forms of yeast will destroy the product. Materials Cultures


15 14 7-day Sabouraud agar cultures of Saccharomyces cerevisiae. Reagents Water-iodine solution, lactophenol cotton-blue solution. Equipment Bunsen burner, inoculating loop and needle, 10 glass slides, 10 coverslips, 5 sterile Pasteur pipettes, glassware marking pencil, and microscope. Procedure Morphological Characteristics Prepare a wet mount of each yeast culture in the following manner: 1. Suspend a loopful of yeast culture in a few drops of lacto phenol cotton-blue solution on a microscope slide and cover with a cover slip. 2. Examine all yeast wet-mount slide preparations under low and high power, noting the shape and the presence or absence of budding. Record your observations in the Lab Report. &&&&&&&&&&&&&& EXPERIMENT NO:5- PREPARATION OF MICROBIOLOGICAL CULTURE MEDIA The survival and growth of microorganisms depend on available and a favourable growth environment. Culture media are nutrient solutions used in laboratories to grow microorganisms. For the successful cultivation of a given microorganism, it is necessary to understand its nutritional requirements and then supply the essential nutrients in the proper form and proportion in a culture medium. The general composition of a medium is as follows:
16 15 H-donors and acceptors (approximately 1-15 g/l) C-source (approximately 1-20 g/l) N-source (approximately g/l) Other inorganic nutrients e.g. S, P (50 mg/l) Trace elements (0.1-1 µg/l) Growth factors (amino acids, purines, pyrimidines, occasionally 50 mg/l, vitamins occasionally mg/l) Solidifying agent (e.g. agar g/l) Solvent (usually distilled water) Buffer chemicals Microbiological culture media could be classified according to: 1. Consistency, which could be adjusted by changing the concentration of solidifying or gelling agents, e.g. agar, gelatine (liquid media do not contain such materials) Cultures in liquid media (or broth) are usually handled in tubes or flasks and incubated under static or shaken conditions. This way, homogenous conditions are generated for growth and metabolism studies,(e.g. with the control of optical density and allowing sampling for the analysis of metabolic products). Semisolid media are usually used in fermentation and cell mobility studies, and are also suitable for promoting anaerobic growth. Solid media are prepared in test tubes or in Petri dishes, in the latter case, the solid medium is called agar plate. In the case of tubes, medium is solidified in a slanted position, which is called agar slant, or in an upright position, which is called agar deep tube. Solid media are used to determine colony morphology, isolate cultures, enumerate and isolate bacteria (e.g. using dilutions from a mixed bacterial population in combination with spreading), and for the detection of specific biochemical reactions (e.g. metabolic activities connected with diffusing extracellular enzymes that act with insoluble substrates of the agar medium).
17 16 2. Composition 3. Function Chemically-defined (or synthetic) media are composed only of pure chemicals with defined quantity and quality. Complex (or non-synthetic) media are composed of complex materials, e.g. yeast extract, beef extract and peptone (partially digested protein), therefore their chemical composition is poorly defined. On the other hand, these materials are rich in nutrients and vitamins. All-purpose media do not contain any special additives and they aim to support the growth of most bacteria. Selective media enhance the growth of certain organisms while inhibit others due to the inclusion of particular substrate(s). Differential media allow identification of microorganisms usually through their unique (and visible) physiological reactions. In the detection of common pathogens, most practical media are both selective and differential. Enrichment media contain specific growth factors that allow the growth of metabolically fastidious microorganisms. An enrichment culture is obtained with selected media and incubation conditions to isolate the microorganisms of interest. PREPARATION OF AGAR SLANTS Object of study: preparation of agar slants Materials and equipment: distilled water measuring cylinder
18 17 flask bacteriological chemicals laboratory scales chemical spoons 1N NaOH solution 1N HCl solution ph indicator paper or ph meter cotton gloves dispenser test tubes test tube caps test tube basket slanting stage autoclave incubator PROCEDURE 1. Measure the components of the medium (e.g. TSA or nutrient, see Appendix) into a flask containing 9/10 volume of the solvent. Use a clean chemical spoon for every measurement. Dissolve the solid components and fill with the remaining solvent up to final volume. If the medium contains heat sensitive components (like sugars), they must be separately sterilised in solution (e.g. by filter sterilisation), and then mixed with the already sterilised and cooled agar medium. 2. Close the flask with cotton plug and cover with aluminium foil, put into the autoclave and start a sterilisation cycle. This cycle could be intermitted when the internal temperature has reached 121 C, at that temperature every component (e.g. agar-agar) will be dissolved correctly. 3. Check the ph of the medium with an indicator paper or with a ph meter and adjust to the proper value with NaOH or HCl solution.
19 18 4. Pour the C medium into the dispenser. Add 5-6 ml medium to each test tube, close them with caps and place them into a test tube basket. 5. Place the tubes into the autoclave and complete a whole sterilisation cycle for 20 min at 121 C. 6. Put the test tubes onto a slanting stage to let the medium solidify in the test tubes. 7. Label the slants according to the type of the medium and perform a sterility test: incubate the test tubes at 28 C for 24 hours, and check for sterility. 8. The prepared media can be stored for 1-2 weeks at C, or longer in a refrigerator. (Do not store medium containing agar-agar under 4-5 C as it destroys its structure!) PREPARATION OF AGAR PLATES Object of study: Preparation of agar plates Materials and equipment: distilled water measuring cylinder flask bacteriological chemicals laboratory scales chemical spoons 1N NaOH solution 1N HCl solution ph indicator paper or ph meter cotton gloves sterile, empty Petri dishes Bunsen burner autoclave incubator
20 19 Practise: 1. Prepare a medium as instructed in previous procedure. 2. Cool the sterilised medium to 55 C. 3. Take out the cotton plug and flame the mouth of the flask over a Bunsen burner, and then pour the medium into sterile, empty Petri dishes (15-20 ml into each Petri dish). 4. Keep the Petri dishes horizontally until the medium completely solidifies. Turn dishes upside-down and stack them up for storage. 5. Label the plates according to the type of the medium and perform a sterility test. 6. In case of longer storage, Petri plates must be placed into plastic bags or boxes to avoid drying out. &&&&&&&&&&&&&&&&&&&&&&&&&&&& EXPERIMENT NO:6 - MICROBIAL EXAMINATIONS OF FOODS: ISOLATION OF BACTERIA STANDARD PLATE COUNT METHOD OBJECTIVES To: undertake enumeration of viable microorganisms by SPC; perform the technique of serial dilution; do pour plating; and count colonies using colony counter. INTRODUCTION Often it is necessary to know the number of bacteria in a specimen, for example, to ensure that water, milk or other foods are safe to consume. Enumerating microbial populations is also important for evaluating products such as antibiotics, vitamins, and preservatives. Several methods can be used to determine bacterial concentrations. These include direct counts, plate counts, filtration, and turbidimetric measurements. The plate count is one of the most accurate means of enumeration of viable microbes because you get a visual indicator for every cell in the specimen. The technique stems from Robert Koch's insight gained from viewing colonies growing on the surface of a spoiling slice of potato. In practice, a small aliquot of a liquid suspension of microbes is spread on the surface of solidified nutrient medium, which when incubated, leads to each cell 'developing' into a visible colony through repeated fission.
21 20 The pour plate technique can be used to determine the number of microbes/ml or microbes/gram in a specimen. It has the advantage of not requiring previously prepared plates, and is often used to assay bacterial contamination of foodstuffs. Each colony represents a "colony forming unit" (CFU). For optimum accuracy of a count, the preferred range for total CFU/plate is between 30 to 300 colonies/plate. PRINCIPLE The number of bacteria in a given sample is usually too great to be counted directly. However, if the sample is serially diluted and then plated out on an agar surface in such a manner that single isolated bacteria form visible isolated colonies, the number of colonies can be used as a measure of the number of viable (living) cells in that known dilution. However, keep in mind that if the organism normally forms multiple cell arrangements, such as chains, the colony-forming unit may consist of a chain of bacteria rather than a single bacterium. In addition, some of the bacteria may be clumped together. Therefore, when doing the plate count technique, we generally say we are determining the number of Colony-Forming Units (CFUs) in that known dilution. By extrapolation, this number can in turn be used to calculate the number of CFUs in the original sample. Normally, the bacterial sample is diluted by factors of 10 and plated on agar. After incubation, the number of colonies on a dilution plate showing between 30 and 300 colonies are determined. Series of dilution used in SPC. A plate having colonies is chosen because this range is considered statistically significant. If there are less than 30 colonies on the plate, small errors in dilution technique or the presence of a few contaminants will have a drastic effect on the final count. Likewise, if there are more than 300 colonies on the plate, there will be poor isolation and colonies will have grown together. Generally, one wants to determine the number of CFUs per milliliter (ml) of sample. To find this, the number of colonies (on a plate having colonies) is multiplied by the number of times the original ml of bacteria was diluted (the dilution factor of the plate counted). For
22 21 example, if a plate containing a 1/1,000,000 dilution of the original ml of sample shows 150 colonies, then 150 represents 1/1,000,000 the number of CFUs present in the original ml. Therefore the number of CFUs per ml in the original sample is found by multiplying 150 x 1,000,000 as shown in the formula below: Number of CFUs per ml of sample =Number of colonies Dilution factor of the plate counted ml of sample plated In the case of the example above,150 x 1,000,000 = 150,000,000 CFUs per ml. For a more accurate count it is advisable to plate each dilution in duplicate or triplicate and then find an average count. One disadvantage of pour plates is that embedded colonies will be much smaller than those which happen to be on the surface, and must be carefully enumerated so that none are overlooked. Also, obligate aerobes may grow poorly if deeply embedded in the agar. MATERIALS REQUIRED Cultures: hour old nutrient agar slant or nutrient broth cultures of E.coli Reagents: Sterile Dilution blanks (containing 9ml of 0.9% NaCl2 ), Plate count agar (standard methods) Equipment and glassware: Petri dishes, glass or plastic (at least 15 x 90 mm), Pipettes with pipette aids, Pipette and petri dish containers, Incubator, Colony counter, blender or stomacher (for food sample), autoclave PROCEDURE E. Coli Culture 1. Label the bottom of six petri plates 1-6. Label four tubes of saline 10-2,10-4, 10-6 and Using aseptic technique, the initial dilution is made by transferring 1 mlof E. coli sample to a 9ml sterile saline blank (Figure 5.1). This is a1/100 or 10-2 dilution. 3. The 10-2 blank is then shaken by grasping the tube between the palms of both hands and rotating quicklyto create a vortex. This serves to distribute the bacteria and break up any clumps. 4. Immediately after the 10-2 blank has been shaken, uncap it and aseptically transfer 1ml to a second 9ml saline blank. Since this is a 10-2 dilution, this second blank represents a 10-4 dilution of the original sample.
23 22 5. Shake the 10-4 blank vigorously and transfer 1ml to the third 9ml blank. This third blank represents a 10-6 dilution of the original sample. Repeat the process once more to produce a 10-8 dilution. 6. Shake the 10-4 blank again and aseptically transfer 1.0 ml to one petri plate and 0.1 ml to another petriplate. Do the same for the 10-6 and the 10-8 blanks. 7. Remove one agar pour tube from the 48 to 50 o C water bath. Carefully remove the cover from the 10-4 petri plate and aseptically pour the agar into it. The agar and sample are immediately mixed gently moving the plate in a figure-eight motion or a circular motion while it rests on the tabletop. Repeat this process for the remaining five plates. 8. After the pour plates have cooled and the agar has hardened, they are inverted and incubated at 37oC for 24 hours. 9. At the end of the incubation period, select all of the petri plates containing between 30 and 300 colonies. Plates with more than 300 colonies cannot be counted and are designated too many to count (TMTC). Plates with fewer than 30 colonies are designated too few to count (TFTC). Count the colonies on each plate. A Quebec colony counter should be used. 10. Calculate the number of bacteria (CFU) per milliliter or gram of sample by dividing the number of colonies by the dilution factor multiplied by the amount of specimen added to liquified agar. Number of bacteria per ml = Number of colonies dilution amount plated 11. Record your results. Food Samples 1. Using separate sterile pipettes, prepare decimal dilutions of 10-2, 10-3, 10-4, and others as appropriate, of food homogenate. (For food homogenate. Add 450 ml phosphate-buffered dilution water to blender jar or stomacher sterile bag containing 50 g analytical food sample and blend for 2 min. This results in a dilution of 10-1.) 2. Make dilutions of original homogenate promptly, using pipettes that deliver required volume accurately.
24 23 3. Prepare all decimal dilutions with 9 ml of sterile diluent plus 1 ml of previous dilution, unless otherwise specified, by transferring 1 ml of previous dilution to 9 ml of diluent. 4. Pipette 1 ml of each dilution into separate, duplicate, appropriately marked petri dishes. 5. Add ml plate count agar (cooled to 45 ± 1 C) to each plate within 15 min of original dilution.. Pour agar and dilution water control plates for each series of samples. 6. Immediately mix sample dilutions and agar medium thoroughly and uniformly by alternate rotation and back-and-forth motion of plates on flat level surface. 7. Let agar solidify. Invert solidified petri dishes, and incubate promptly for 24 ± 2 h at 37 C. 8. Repeat step 9, 10 and 11 from culture procedure above. OBSERVATIONS 1. Choose a plate that appears to have between 30 and 300 colonies. 2. Count the exact number of colonies on that plate using the colony counter (as demonstrated by your instructor). 3. Calculate the number of CFUs per ml of original sample as follows: The number of CFUs per ml of sample = The number of colonies ( plate) ml of dilute sample plated = Number of colonies = Dilution factor of plate counted = Number of CFUs per ml RESULTS Record your results and find the average number of cfus/ ml by adding the results from all of your plates and dividing by the number of plates. PRECAUTIONS 1. Avoid sampling foam. 2. Do not deliver less than 10% of total volume of pipette. For example, do not use pipette with capacity greater than 10 ml to deliver 1 ml volumes; for delivering 0.1 ml volumes, do not use pipette with capacity higher than 1.0 ml. 3. Do not stack plates when pouring agar or when agar is solidifying.
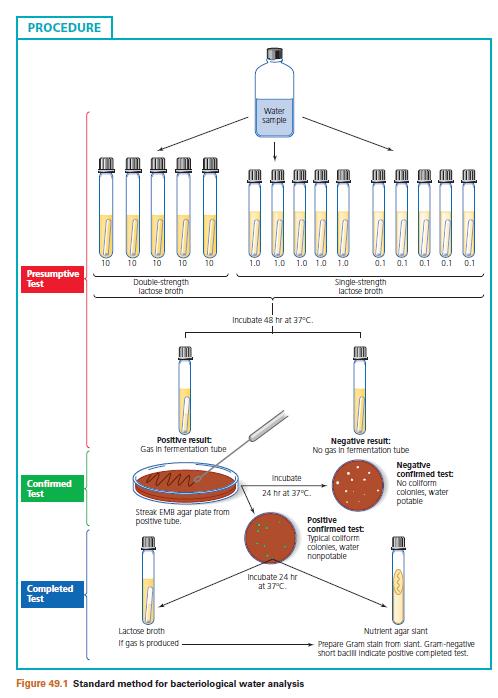
25 24 &&&&&&&&&&&&& EXPERIMENT NO:7-WATER EXAMINATION: MULTIPLE TUBES METHOD. WATER EXAMINATION EXPERIMENT : STANDARD QUALITATIVE MICROBIAL ANALYSIS OF WATER. Part A: Presumptive Test: Determination of the Most Probable Number of Coliform Bacteria. AIM:i.To determine the presence of coliform bacteria in a water sample,ii. To obtain some index as to the possible number of organisms present in the sample under analysis. PRINCIPLE: The presumptive test is specific for detection of coliform bacteria. Measured aliquots of the water to be tested are added to a lactose fermentative broth containing an inverted gas vial ( Durham tube). Because these bacteria are capable of using lactose as carbon source, their detection is facilitated by use of this medium. In this experiment the lactose fermentation broth also contains a surface tension depressant, a bile salt, which is used to suppress the growth of organisms other than coliform bacteria. Tubes of this lactose medium are inoculated with 10 ml, 1 ml and 0.1 ml aliquots of the water sample. The series consists of at least three groups, each composed of five tubes of specified medium. The tubes in each group are then inoculated with the designated volume of the water sample as desirable under procedure. The greater numbers of tubes per group, the greater the sensitivity of the test. Development of gas in any of the tubes is presumptive evidence of the presence of coliform bacteria in the sample. The presumptive test also enables the microbiologist to obtain some idea of the numbers of the coliform bacteria present by means of the most probable number test (MPN). The MPN is estimated by determining the number of tubes in each group that show gas following the incubation period. MATERIALS REQUIRED: Culture Water sample to be tested. Media: Lauryl Tryptose broth-double strength and single strength. Equipment: Bunsen burner, test tubes, test tube racks, sterile 10 ml, 1 ml and 0.1 ml pipettes, labels. PROCEDURE:
26 25 I. Set up three series consisting of three groups, a total of 15 tubes per series, in a test tube rack, for each tube label the water source and volume of sample to be inoculated. ii. Flame bottle and then, using a 10ml pipette, transfer 10 ml aliquots of water sample to the five tubes labeled LB2X-10 ml. iii. Flame bottle and then, using a 1ml pipette, transfer 1 ml aliquots of water sample to the five tubes labeled LB1X-1 ml. iv. Flame bottle and then, using a 0.1ml pipette, transfer 0.1 ml aliquots of water sample to the five tubes labeled LB1X-0.1 ml. v. Incubate all tubes for 48 hours at 37 ᴏ C. PART-B-CONFIRMED TEST. AIM: To confirm the presence of coliform bacteria in a water sample for which the presumptive test was positive. PRINCIPLE: The presence of a positive or doubtful presumptive test immediately suggests that the water sample is non potable. Confirmation of these results is necessary, since positive presumptive tests may be the result of organisms of non coliformorigin that are not recognized as indicators of faecal pollution. The confirmed test requires that selective and differential media such as Eosine Methylene Blue (EMB) or Endo agar be streaked from a positive lactose broth tube obtained from the presumptive test. Eosine Methylene Blue contains the dye methylene blue, which inhibits the growth of grampositive organisms. In the presence of an acid environment,emb forms a complex that precipitates out into the coliform colonies, producing dark centres and a green metallic sheen. This reaction is characteristic for Escherichia coli, the major indicator of faecal pollution. Endo agar is a nutrient medium containing the dye fuchsion, which is present in the decolorized state. In the presence of acid produced by the coliform bacteria, fuchsion forms a dark pink complex that turns the E.coli colonies and the surrounding medium pink. MATERIALS REQUIRED: Cultures
27 26 One 24 hour old positive lactose broth culture from each of the three series from the presumptive test. MEDIA: Eosine Methylene Blue agar plates or Endo agar plates. EQUIPMENT: Bunsen burner, label and inoculation loop. PROCEDURE: I. Label the covers of the three EMB plates or Endo agar plates with source of water sample. ii. Using a positive 24 hour lactose broth cultures from the presumptive test,streak the surface of one EMB agar or one Endo agar plate to obtain discrete colonies. iii. Incubate all plate cultures in an inverted position for 24 hours at 37 ᴏ C. PART-C-COMPLETE TEST AIM: To confirm the presence of coliform bacteria in a water or if necessary, to confirm a suspicious but doubtful result of the previous test. PRINCIPLE: The complete test is the final analysis of the water sample. It is used to examine the coliform colonies that appeared on the EMB or Endo agar plates used in the confirmed test..an isolated colony is picked from the confirmatory test plate and inoculated into a tube of lactose broth and streaked on a nutrient agar slant to perform a Gram stain. Following inoculation and incubation, tubes showing acid and gas in the lactose broth and presence of gram-negative bacilli on microscopic examination are further confirmation of the presence of E.coli and they are indicative of a positive completed test. MATERIALS REQUIRED: Cultures One 24 hours coliform positive EMB or Endo agar culture from each of the three series of confirmed test. MEDIA: Nutrient agar slants and Lactose fermentation broths. REAGENTS:
28 27 Crystal Violet, Grams iodine, decolorizer, Safranine. EQUIPMENT: Bunsen burner, staining tray, inoculation loop, blotting paper, microscope. PROCEDURE: I.Label each tube with the source of water sample. ii. Inoculate one broth and one nutrient agar slant from the same isolated E.coli colony obtained from an EMB agar or Endo agar plate. iii. Incubate all tubes for 24 hours at 37 ᴏ C.
29 28
30 29 OBSERVATION AND RESULT: PART-A PRESUMTIVE TEST-MPN TEST VOLUME OF SAMPLE A/G A/G A/G A/G A/G 1Oml 2X LTB 1ml 1XLTB 0.1ml 1X LTB MPN / 100 ml REFER MPN CHART. PART-B-CONFIRMED TEST COLONIES ON EMB PLATE FROM PRESUMTIVE POSITIVE TEST VOLUME OF SAMPLE E.COLI COLONIES 1Oml 2X LTB 1ml 1XLTB 0.1ml 1X LTB PART-C-COMPLETE TEST-GROWTH ON NUTRIENT AGAR SLANT AND GAS IN LACTOSE BROTH. VOLUME OF SAMPLE Oml 2X LTB GROWTH/ GAS
31 30 1ml 1XLTB 0.1ml 1X LTB &&&&&&&&&&&& EXPERIMENT NO:8 METHYLENE BLUE TEST AND RESAZURIN TEST FOR MILK Introduction This test is based on the work done by Wilson (1935) and Milk Regulations This test is used to check the contamination of bacteria in the sample of milk. It tells us about the viable count of bacteria that may be present in the milk. So by this particular test we can have a clue about the quality of the milk we have that whether it is invaded by bacteria or not and in which quantity these are present. Principle This test is based on the principle that if viable bacteria are present in the milk, they will reduce the metylene blue dye and decolourize the sample if kept for sometime in a dark place. The methylene blue is reduced due to depletion of oxygen in the milk as it is consumed by bacteria. Chemistry of Metylene Blue Methylene Blue is a dye and it is an aromatic chemical compound having the molecular formula C16H18N3SCl. Its molar mass is g/mol. It is a dark green powder at room temperature and when it is dissolved in water, it forms a blue solution.
32 31 The bacteria in the milk ferment lactose to lactic acid. During this process electrons are released, which are taken up by Methylene Blue and results in its reduction and ultimately decoloration. So, more bacteria will produce more electrons, in turn it would be faster reduction and rapid decolouration. Requirements Following apparatus and reagents are required for the Methylene Blue Test. Metylene Blue Solution Milk Sample Test Tube Pipette Rubber Stopper Water Bath Procedure Mix the sample well that has to be analyzed and then pour 10 ml of that milk into a clean and sterilized test tube. With the help of pipette add 1 ml of Methylene Blue Solution into the milk sample. The test tube is then closed with a rubber stopper and invered slowly so that all the contents have been mixed. Then this test tube is places in a water bath at 37 0 C for half an hour making sure that water level is upto sufficient height of the test tube. Covered with lid. Then the milk is examined after 30 minutes for checking the decoloration. Maintain the control tubes: one with milk boiled at 100 C for 3 minutes and another control with 1 ml tap water in place of methylene blue.
33 32 Interpretation If the viable bacteria decolourize the milk within 30 minutes then the test is positive and milk is of unsatisfactory quality. If the milk is not decolourized within 30 minutes then the test is negative or milk is of good quality. To interpret further, four levels of quality is used, 1. Quality of milk is Excellent if no reduction of blue color up to 8 hours. 2. Quality of milk is Good if decolouration occurs within hours.
34 3. Quality of milk is Fair if decolouration occurs within hours Quality of milk is Poor if decolouration occurs less than 2 hours. &&&&&&&&&&&&&& EXPERIMENT NO:9- RESAZURIN TEST FOR MILK Objectives: To determine extent of bacteriological quality of milk. Relevant Information: The majority of the organisms in milk are capable of reducing and decolorizing the resazurin dye. When bacteria grow in the milk they utilize oxygen, the rate of remove or reduction is proportional to the keeping quality. This test is also based on the same principles as M.B.R. (Methylene Blue Reduction) test, but dye is Resazurin which is much more sensitive that the Methylene blue. For this reason this test provides a rapid measure of the keeping quality of milk. During incubation, the dye undergoes reduction very largely through the metabolic activity of the organisms present. The greater the number of organisms present in milk, the more quickly the dye is reduced. The reduction takes place in two distinct stages. Resazurin is blue at the reaction of milk. In the first stage dye is changed to pink colour and in second stage pink colour is changed to colourness. The cells present in the milk may also influence the reduction of Resazurin and for that reason; the test may also measure physiologically or pathologically abnormal milk. Precaution: The test should be carried out aseptically. Material Required: i Milk sample ii. Resazurin colour/solution 0.05%.
35 34 Apparatus: i. Water path, thermostically controlled to maintain temperature of 37 0 C. ii. Test tube 10 ml. iii Pipette 10 ml. and 1 ml. Procedure: i. Milk the sample thoroughly by inverting from one to another container. ii. Pour 10 ml. of milk sample in to previously sterilized test tube. iii. Add quickly 1 ml. of Resazurin solution in the test tube. iv. Mix the milk and dye thoroughly by inverting 2-3 times. v. Place the tubes in the water bath at the temperature of C only for two minutes. vi. The tubes are then removed from the water bath. vii. Tubes are examined and classified at the end of an hour in the "one-hour test" or at the end of three successive hourly intervals in the "triple reading test.".

36 35 The following relationships of color and quality are generally accepted: Color of Sample: Quality of Milk 1. Blue (no color change): Excellent 2, Blue to deep mauve: Good
37 36 3. Deep mauve to deep pink: Fair 4. Deep pink to whitish pink: Poor 5. White: Bad &&&&&&&&&&&&&&&&&& EXPERIMENT NO:10- DIRECT MICROSCOPIC COUNT A. The Breed's Smear Method for Direct Microscopic Counts This is a method in which the number of bacteria or yeasts in a sample (e.g. broth culture, cell suspension, milk) may be determined by direct microscopic examination. Since the staining procedure does not differentiate between living and dead organisms, the bacterial count obtained is known as a "total count". Any suitable staining technique may be used (e.g. simple stain or Gram's method). In the case of samples of milk and other foods with a high fat content it is necessary first to defat (e.g. with xylene). Newman's stain conveniently combines both defatting and staining processes although not allowing the determination of the Gram's staining reaction. The Breed's smear method is also used for counting the animal cells - predominantly leucocytes -which are found in much larger numbers in mastitis milk (see page 179) than in milk from healthy animals and so can be diagnostic of mastitis. It is essential in this procedure that the microscope is first calibrated so that the area of the microscopic field is known. A known volume (0'01 ml) of the sample or an appropriate dilution is then spread over a known area (I cm2), on a glass slide. An alternative procedure often used which gives the same sample concentration on the slide is to draw two straight lines across the width of the slide to enclose a length of 2 cm. Since the standard microscope slide is 2.5 cm wide, this marks off an area of 5 cm2, over which 0.05 ml of sample is spread. The sample is allowed to dry, then stained and examined microscopically. The average number of bacteria or cells per field is determined and it is then possible to calculate the number of bacteria or cells per ml of original sample.
38 37 Determination of the area of the microscopic field 1. Set up the microscope and, using the X 10 eye-piece and low power objective, adjust the stage micrometer so that the graduated scale (I mm divided into 100 units of 10ILm each) is in the centre of the field. 2. Place a drop of immersion oil on the stage micrometer and focus with the oil-immersion objective. Determine the diameter of the microscopic field in ILm, using the micrometer scale, adjusting the tube length of the micro- scope slightly if necessary. In subsequent observations this same tube length must be maintained. 3. Calculate the area of the microscopic field in mm for the oil-immersion lens and X 10 eyepiece using the formula r 2 = Area of field r 2 where r = radius of field (0.08). Knowing the area of the microscopic field, it is then possible to determine the microscope factor (MF) by calculation: (a ) MF = Number of fields in I cm2 (100 mm2) organisms per ml of sample. Area of field in 1mm2 (b) Average number n of organisms or cells per field= Number counted Number of fields counted (c) MF x n = Number of organisms or cells present in 1 cm2 (d) Since 0.01 ml of sample was spread over 1 cm2, MF x n X 100 = Number of cells or organisms present in 1 ml of sample. For most microscopes as used above, one organism per field represents approximately 500,000 Preparation of smear I. Thoroughly mix the sample to be examined. Prepare dilutions as necessary -to produce smears with no more than 20 organisms per field. (If pure cultures are being examined a barely detectable turbidity will be given by a suspension containing about 106 cells per mi.) 2. Deliver 0.01 ml of sample onto a clean glass slide and spread over an area of I cm2 using either a guide card or a marked slide. The sample may be delivered either (0) by means of a capillary pipette calibrated to deliver 0.01 ml or (b) by using a wire loop. A closed loop of 4 mm
39 38 internal diameter will hold a drop of approximately 0.01 ml of sample, if withdrawn with the plane of the loop perpendicular to the surface. 3. Dry the smear immediately by placing on a warm level surface or in an incubator at 55 C. Drying should be complete within 5 min so as to prevent possible bacterial multiplication. 4. Stain by an appropriate method. 5. Examine, using the oil-immersion objective. 6. Determine the average number of organisms per field and hence calculate the number per ml.or per g of sample. &&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&& EXPERIMENT NO: 11 LACTIC ACID ESTIMATION IN MILK Determination of acidity The acidity in milk is measured, for example by titration with a 0.1 n NaOH solution, and indicates the consumption of NaOH necessary to shift the ph-value from 6.6 ± 0.1 (corresponding to fresh milk) to a ph-value of (phenolphthalein). Lactic acid is an organic acid with one carboxylic acid, CH3-CHOH-COOH, having a molecular weight of 90. One ml 0.1 n NaOH therefore corresponds to: 90 x 0.1 / 1000 = g of lactic acid If the titration requires e.g ml 0.1 n NaOH, the result is often expressed as: 14.5 x % lactic acid The titratable acidity is expressed as % lactic acid and is determined by titration of a known amount of reconstituted milk with 0.1 N NaOH using phenolphthalein as indicator. Acidity Expressed in SH (Soxhlet-Henkel Degrees) The titrable acidity test is employed to ascertain if milk is of such a high acidity as to reduce its keeping quality and heat stability. The acidity of milk is of two kinds. i. Natural acidity which is due to citrates and phosphates present in the milk and dissolved CO2 during the process of milking and thereafter.
40 39 ii. Developed acidity which is due to lactic acid produced by the action of bacteria on lactose in milk. Generally the acidity of milk means the total acidity (Natural + developed) or titrable acidity. It is determined by titrating a known volume of milk with standard alkali to the point of an indicator like phenolphthalein. The titrable acidity test measures the amount of alkali which is required to change the ph of milk from its initial value of about 6-6 to 6.8, to the ph of the colour change of phenolphthalein added to milk to indicate the end point (ph 8.3). In fact, the method measures the buffering capacity of milk and not the true acidity. Procedure: 1. Fill the burette with N/10 NaOH solution. 2. Mix the milk sample thoroughly by avoiding incorporation of air. 3. Transfer 10 ml milk with the pipette in conical flask. 5. Add equal quantity of glass distilled water. 6. Add 3-4 drops of phenolphthalein indicator solution* and stir with glass rod. 7. Take the initially reading of the alkali in the burette at the lowest point of meniscus. 8. Rapidly titrate the contents with N/10 NaOH solution continue to add alkali drop by the drop and stirring the content with glass rod till first definite change to pink colour which remains constant for 10 to 15 seconds. 9. Complete the titration within 20 seconds. 10. Note down the final burette reading. * Phenolphthalein Indicator Solution - Dissolve one gram of phenolphthalein in 100 ml of 95 % ethyl alcohol. Add 0 1 N sodium hydroxide solution until one drop gives a faint pink colouration.
41 40 Dilute with distilled water to 200 ml. Calculation: No of ml. of 0.1 N NaOH solutions required for neutralization x % Lactic acid = x 100 Weight of sample (Weight of sample = Volume of milk x specific gravity) &&&&&&&&&&&&&&& EXPERIMENT NO 12: ESTIMATION OF LACTOSE IN MILK PRINCIPLE: Sugars such as glucose, lactose maltose are oxidized in presence of sodium hydroxide and iodine is removed fom the solution as sodium iodide ( NaI). Excess of iodine can be titrated against 0.1 N sodium thiosulfate using starch as indicator. The difference in the titre value of blank titration and sample titration is used to calculate the percentage of lactose in the milk. PROCEDURE: a) Extraction of milk Lactose: i) 10ml of milk is transferred to a beaker diluted with 25ml of warm distilled water. ii) 10ml of Mayer s reagent is then added to coagulate proteins and fats present in the milk. iii) 2ml of dilute sulphuric acid(h2so4) is added and contents are mixed well. iv) The contents are then made upto 250ml in a standard volumetric flask. v) This filtered and the clear filtrate obtained is used for titration. b) Titration with sodium thiosulfate (Na2S2O3 ) solution: i) Pipette out 25ml of filtrate into a clean conical flask. ii) Add 20ml Iodine and 30ml NaOH solution.
42 41 iii) Mix the contents well and keep aside for 20 minutes. iv) Add 4ml of dilute H2SO4.The solution becomes dark brown in color. v) Titrate against standard sodium thiosulfate (Na2S2O3). vi) When the solution becomes pale yellow,add 1ml of freshly prepared starch as indicator. Solution turns to dark blue. vii) Titration is continued slowly till the blue colour disappears and solution turns colourless. viii) A blank titration is also carried out with 10 ml of water instead of filtrate. Difference in burette reading is used to calculate percentage of lactose. Calculations: Percentage of Lactose = (B-A) x N V1/V2 x 100/W Where: B=Burette Reading of blank titration A= Burette Reading of sample titration V1= 250ml V2 =25ml N=Normality of Na2S2O3 =0.1 W= Original volume of milk sample=10ml 1gm mol of Iodine = 1 gm mol of Lactose=342g 1gm eq.wt of Iodine = 171g 1000ml of 1N Iodine =171 g of lactose 1ml of 1N Iodine = 0.171g of lactose. &&&&&&&&&&&&&&&&&&&&&&&&&&&& EXPERIMENT NO:13-DETECTION OF STARCH IN MILK PRODUCTS LIKE KHOYA, BUTTER AND CHEESE GENERA[ Starch is a common adulterant in milk products as it gets easily mixed up with these products because of the similarity in the colour. Starch adds to the weight of the products, and is, therefore, a cheap source of adulteration. The adulteration of these products with starch can be detected by addition of iodine to these
43 42 products which results in the formation of a blue colour. The blue colour is due to the formation of an inclusion complex between iodine and the amylose. The amylase coils into the spiral and the iodine molecule aligns within the centre of this spiral and causes light absorption which gives a blue colour. Procedure Take 5 ml of milk in a test-tube and add 5 ml of p-dimethyl amino benza]dehyde reagent to it. The appearance of a distinct yellow colour indicates the presence of added urea. The appearance of slight yellow colour may be ignored which may be because of the presence of natural urea in milk. Detection of Sodium Chloride in Milk Lactometer is a simple device which can be used to detect adulteration of milk with water. Addition of water alters the density of milk which can be read on the Iactometer scale. Hence, to increase the density of milk, adulterant like sodium chloride is added. Procedure Take about 2 g of milk product in a test-tube and add Take 2 ml of milk in a test-tube and add O.I ml of about 5 ml water to it. Boil for a few minutes on the 5 percent potassium chromate solution and 2 ml of gas burner. COOI and add iodine solution to the test- 0.1 N silver nitrate to it. The appearance of a yellow tube. The formation of a blue coloration shows the colour indicates the presence of added sodium chloride in the milk while the appearance of a brick red precipitate indicates the absence of added sodium. Detection of Metanil Yellow in Sweets To make a sweet attractive, various colours are added to sweet preparations. As per the PFA Act, onlinatural
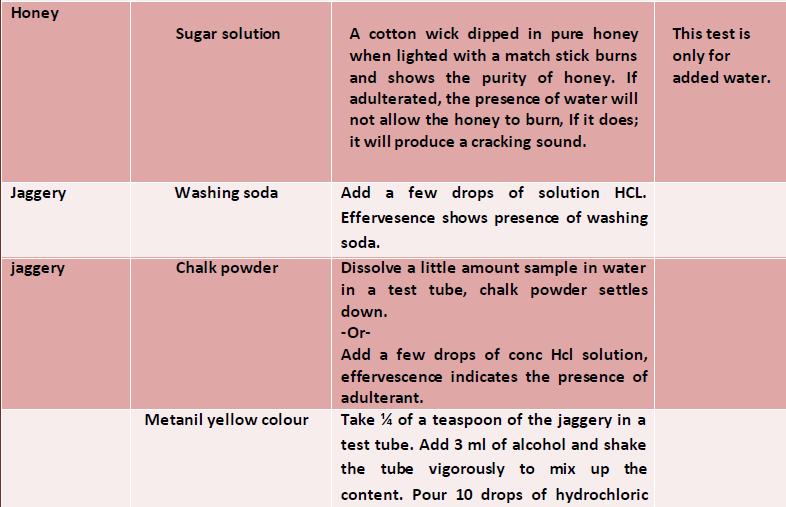
44 43 food colours or permitted artificial food colours can be used in sweets. Metanil yellow is a cheap textile dye which is commonly used in colouring sweets which amounts to adulteration. The metanil yellow is carcinogenic. Procedure Take about 5 g of the sweet sample in a test-tube and add hot water to bring the sample in pure solution. Separate the coloured matter by filtration and add few drops of concentrated hydrochloric acid. The appearance of a pinkish red coiour shows the presence of metanil yellow. Detection of Mashed Potatoes and Other Starches in Ghee/Butter General Mashed potatoes and edible starches are one of the adulterants in ghee/butter as these, without hampering the colour appeal of ghee/butter will add to the weight of ghee/butter. Procedure Take some ghee/butter in a test-tube and warm it. Add few drops of iodine solution to the sample. The appearance of a blue colour will indicate the presence of mashed potatoes and edible starches to gheelbutter. Detection of Chalk Powder in Bura Sugar/ Jaggery/Common Salt General Chalk powder is added to food items like bura sugar, jaggery and common salt as an adulterant to increase the weight. Chalk powder is carbonates of calcium which is insoluble in water and if consumed is injurious to health. Procedure
45 44 There are two methods for detection of chalk powder in adulterated foods like bura sugar(jaggery/common salt. Method 1 Dissolve about gof a sample in water in a beaker. Allow to settle for 5 min. The unadulterated samples will dissolve completely while the presence of any sediment at the bottom of the beaker will indicate the presence of chalk powder in the sample. Method II Take some sample of the food item to be examined in a test-tube and add few drops of 1:1 hydrochloric acid. The appearance of bubbles (effervescence) will indicate the presence of chalk powder in the sample.

46 45 &&&&&&&&&&
COUNT METHOD 5.0 OBJECTIVES 5.1 INTRODUCTION 5.2 PRINCIPLE. Structure
 Food Microbiology EXPERIMENT 5 STANDARD PLATE COUNT METHOD Structure 5.0 Objectives 5.1 Introduction 5.2 Principle 5.3 Materials Required 5.4 Procedure 5.4.1 E-coli Culture 5.4.2 Food Samples 5.5 Observations
Food Microbiology EXPERIMENT 5 STANDARD PLATE COUNT METHOD Structure 5.0 Objectives 5.1 Introduction 5.2 Principle 5.3 Materials Required 5.4 Procedure 5.4.1 E-coli Culture 5.4.2 Food Samples 5.5 Observations
Isolation & Characterization of Bacteria
 PR025 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Isolation & Characterization of Bacteria Teacher s Handbook (Cat. # BE 204) think proteins!
PR025 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Isolation & Characterization of Bacteria Teacher s Handbook (Cat. # BE 204) think proteins!
EXPERIMENT 4 STAINING TECHNIQUES
 Practical Manual Food Microbiology EXPERIMENT 4 STAINING TECHNIQUES Structure 4.0 Objectives 4.1 Introduction 4.2 Principle 4.3 Preparation of smear 4.4 Simple staining 4.4.1 Principle Involved 4.4.2 Materials
Practical Manual Food Microbiology EXPERIMENT 4 STAINING TECHNIQUES Structure 4.0 Objectives 4.1 Introduction 4.2 Principle 4.3 Preparation of smear 4.4 Simple staining 4.4.1 Principle Involved 4.4.2 Materials
Pathogenic Bacteria. culture media. Components of the Typical Culture Medium: Culture Media Importance:
 Level4 Lab2: Pathogenic Bacteria culture media Microorganisms, like all other living organisms, require basic nutrients for sustaining their life. All microorganisms have the same basic requirements but
Level4 Lab2: Pathogenic Bacteria culture media Microorganisms, like all other living organisms, require basic nutrients for sustaining their life. All microorganisms have the same basic requirements but
Microbiological Methods
 Microbiological Methods Making Media Pouring Culture Plates Sterile Technique Inoculating Plates and Culture Tubes Use of a Plate Counter to Estimate Microbial Population Densities Culturing Microorganisms
Microbiological Methods Making Media Pouring Culture Plates Sterile Technique Inoculating Plates and Culture Tubes Use of a Plate Counter to Estimate Microbial Population Densities Culturing Microorganisms
SELECTED QUESTIONS F ROM OLD MICRO 102 QUIZZES PART I EXPERIMENTS 1 THROUGH 7
 SELECTED QUESTIONS F ROM OLD MICRO 102 QUIZZES PART I EXPERIMENTS 1 THROUGH 7 Question numbers refer to the applicable experiment. Questions with blanks are multiple true-false questions unless otherwise
SELECTED QUESTIONS F ROM OLD MICRO 102 QUIZZES PART I EXPERIMENTS 1 THROUGH 7 Question numbers refer to the applicable experiment. Questions with blanks are multiple true-false questions unless otherwise
Adapted from Biology 15 Laboratory Manual Supplement: Wrightsman, Ininns and Cannon-Moloznic, Saddleback College, CA 92692
 Biology 4B Laboratory Bacteriological Examination of Water Adapted from Biology 15 Laboratory Manual Supplement: Wrightsman, Ininns and Cannon-Moloznic, Saddleback College, CA 92692 Objectives Carry out
Biology 4B Laboratory Bacteriological Examination of Water Adapted from Biology 15 Laboratory Manual Supplement: Wrightsman, Ininns and Cannon-Moloznic, Saddleback College, CA 92692 Objectives Carry out
Lab Activity #14 - Bacteriological Examination Of Water and Milk (Adapted from Lab manual by Dr. Diehl)
") Lab Activity #14 - Bacteriological Examination Of Water and Milk (Adapted from Lab manual by Dr. Diehl) Some of the diseases that humans can contract from drinking polluted water include typhoid, dysentery,
Lab Activity #14 - Bacteriological Examination Of Water and Milk (Adapted from Lab manual by Dr. Diehl) Some of the diseases that humans can contract from drinking polluted water include typhoid, dysentery,
INTRODUCTION water-soluble Figure 1.
 INTRODUCTION Natural waters contain bacteria. The aerobic gram negative bacillus of the genera Psedomonas, Alcalignes, and Flavobacterium are common in natural waters. Many of these bacteria are able to
INTRODUCTION Natural waters contain bacteria. The aerobic gram negative bacillus of the genera Psedomonas, Alcalignes, and Flavobacterium are common in natural waters. Many of these bacteria are able to
How to perform a Gram Stain. Jasleen Singh
 How to perform a Gram Stain Jasleen Singh Table of Contents iii Table of Contents Table of Contents... iii Introduction... 5 Terminology... 7 Terms to be familiar with... 7 Gram Staining... 8 What is
How to perform a Gram Stain Jasleen Singh Table of Contents iii Table of Contents Table of Contents... iii Introduction... 5 Terminology... 7 Terms to be familiar with... 7 Gram Staining... 8 What is
Aseptic Techniques. A. Objectives. B. Before coming to lab
 Aseptic Techniques A. Objectives Become familiar with 1. The ubiquity of microorganisms (see Note 1) 2. Aseptic techniques (see Note 2) 3. Standard methods for growing/observing microorganisms (see Note
Aseptic Techniques A. Objectives Become familiar with 1. The ubiquity of microorganisms (see Note 1) 2. Aseptic techniques (see Note 2) 3. Standard methods for growing/observing microorganisms (see Note
PURE CULTURE TECHNIQUES
 PURE CULTURE TECHNIQUES Most specimens (from animal tissue, plant tissue, or environmental samples) will be mixed, with a variety of bacteria (or other microorganisms). A single gram of feces, for example,
PURE CULTURE TECHNIQUES Most specimens (from animal tissue, plant tissue, or environmental samples) will be mixed, with a variety of bacteria (or other microorganisms). A single gram of feces, for example,
Microbiological Methods
 Microbiological Methods Making Media Pouring Culture Plates Sterile Technique Inoculating Plates and Culture Tubes Use of a Plate Counter to Estimate Microbial Population Densities Sterile Technique Sterile
Microbiological Methods Making Media Pouring Culture Plates Sterile Technique Inoculating Plates and Culture Tubes Use of a Plate Counter to Estimate Microbial Population Densities Sterile Technique Sterile
Appendix I Laboratory Procedures
 Canadian Shellfish Sanitation Program 02/11/2010 Version 1 Appendix I Laboratory Procedures This Appendix provides CSSP laboratories with information on: analytical methods and quality assurance procedures
Canadian Shellfish Sanitation Program 02/11/2010 Version 1 Appendix I Laboratory Procedures This Appendix provides CSSP laboratories with information on: analytical methods and quality assurance procedures
Exercise 13 DETERMINATION OF MICROBIAL NUMBERS
 Exercise 13 DETERMINATION OF MICROBIAL NUMBERS Introduction When biologists discuss the growth of microorganisms (microbial growth), they are actually referring to population size rather than to the size
Exercise 13 DETERMINATION OF MICROBIAL NUMBERS Introduction When biologists discuss the growth of microorganisms (microbial growth), they are actually referring to population size rather than to the size
Preparing and Gram-staining a bacteriological smear
 College of Life Sciences and Technology Medical Laboratory Science (Applied Learning) AP 84-205-00 (42) Module 4: Practical 2014-16 Preparing and Gram-staining a bacteriological smear Date Time Class Venue
College of Life Sciences and Technology Medical Laboratory Science (Applied Learning) AP 84-205-00 (42) Module 4: Practical 2014-16 Preparing and Gram-staining a bacteriological smear Date Time Class Venue
Most Probable Number (MPN) & Biological Oxygen Demand (BOD)
 & Biological Oxygen Demand (BOD)") Most Probable Number (MPN) & Biological Oxygen Demand (BOD) Part : Presumptive Coliform Test (MPN) Introduction This lab exercise will employ a commonly used multi-tube fermentation technique. The results
Most Probable Number (MPN) & Biological Oxygen Demand (BOD) Part : Presumptive Coliform Test (MPN) Introduction This lab exercise will employ a commonly used multi-tube fermentation technique. The results
Some Industrially Important Microbes and Their Products
 2 Some Industrially Important Microbes and Their Products 2.1. ENZYME PRODUCING MICROBES Type of enzyme Substrate Microorganism Amylase Starch Saccharomyces diastaticus Protease Proteins Bacillus sp. Lipase
2 Some Industrially Important Microbes and Their Products 2.1. ENZYME PRODUCING MICROBES Type of enzyme Substrate Microorganism Amylase Starch Saccharomyces diastaticus Protease Proteins Bacillus sp. Lipase
NATIONAL STANDARD OF THE PEOPLE S REPUBLIC OF CHINA. National Food Safety Standard
 GB NATIONAL STANDARD OF THE PEOPLE S REPUBLIC OF CHINA GB 4789.38-2012 National Food Safety Standard Microbiological Examination of Food Hygiene - Enumeration of Escherichia Coli 食品安全国家标准食品微生物学检验大肠埃希氏菌计数
GB NATIONAL STANDARD OF THE PEOPLE S REPUBLIC OF CHINA GB 4789.38-2012 National Food Safety Standard Microbiological Examination of Food Hygiene - Enumeration of Escherichia Coli 食品安全国家标准食品微生物学检验大肠埃希氏菌计数
ASEPTIC TRANSFER & PURE CULTURE TECHNIQUES
 ASEPTIC TRANSFER & PURE CULTURE TECHNIQUES GENERAL GUIDELINES & REMINDERS: SAFETY: NO EATING OR DRINKING IN THE LAB! Wash your hands with soap both BEFORE and AFTER lab, and, in addition, when you have
ASEPTIC TRANSFER & PURE CULTURE TECHNIQUES GENERAL GUIDELINES & REMINDERS: SAFETY: NO EATING OR DRINKING IN THE LAB! Wash your hands with soap both BEFORE and AFTER lab, and, in addition, when you have
Method 9.2 Drinking water and effluent: bacteria by the membrane filter method
 Section 9: Water and effluent p 1/5 Method 9.2 Drinking water and effluent: bacteria by the membrane filter method 1. Rationale The method is used for the determination of the total coliform bacteria,
Section 9: Water and effluent p 1/5 Method 9.2 Drinking water and effluent: bacteria by the membrane filter method 1. Rationale The method is used for the determination of the total coliform bacteria,
Determination of the microbial count
 Determination of the microbial count TEB Topics Microbial count, microorganisms, and analysis of drinking water Principle The microbial count is the number of viable microorganisms in one millilitre or
Determination of the microbial count TEB Topics Microbial count, microorganisms, and analysis of drinking water Principle The microbial count is the number of viable microorganisms in one millilitre or
DAIRY WATERS. (Coliform Group and Escherichia coli) [E. coli verification required only on source water] IMS #24
![DAIRY WATERS. (Coliform Group and Escherichia coli) [E. coli verification required only on source water] IMS #24](/thumbs/77/76598583.jpg "DAIRY WATERS. (Coliform Group and Escherichia coli) [E. coli verification required only on source water] IMS #24") DAIRY WATERS (Coliform Group and Escherichia coli) [E. coli verification required only on source water] IMS #24 [Unless otherwise stated all tolerances are ±5%] 1. Laboratory Requirements a. Cultural Procedures
DAIRY WATERS (Coliform Group and Escherichia coli) [E. coli verification required only on source water] IMS #24 [Unless otherwise stated all tolerances are ±5%] 1. Laboratory Requirements a. Cultural Procedures
Preparing and Gram-staining a bacteriological smear
 College of Life Sciences and Technology Medical Laboratory Science (Applied Learning) AP 84-205-00 (52) Module 4: Practical 2015-17 Preparing and Gram-staining a bacteriological smear Date Time Class Venue
College of Life Sciences and Technology Medical Laboratory Science (Applied Learning) AP 84-205-00 (52) Module 4: Practical 2015-17 Preparing and Gram-staining a bacteriological smear Date Time Class Venue
USEPA Membrane Filtration Method Method m-tec. Scope and Application: For potable water, nonpotable water, recreation water and wastewater.
 , MF, m-tec, 8367 DOC316.53.001210 USEPA Membrane Filtration Method Method 8367 1 m-tec Scope and Application: For potable water, nonpotable water, recreation water and wastewater. 1 USEPA accepted. Test
, MF, m-tec, 8367 DOC316.53.001210 USEPA Membrane Filtration Method Method 8367 1 m-tec Scope and Application: For potable water, nonpotable water, recreation water and wastewater. 1 USEPA accepted. Test
SCHEDULE. Friday: Pet Investigations: Plate counts - how to know how many clones of your pet you have (pg. 9-10)
") SCHEDULE Wednesday: Pet Investigations: Phenol Red Broth with Durham tubes (pg. 3-4) Oxidation/Fermentation Agar (pg. 5-6) Anaerobic Growth (pg. 7) Growth in Liquid Culture (pg. 8-9) Friday: Pet Investigations:
SCHEDULE Wednesday: Pet Investigations: Phenol Red Broth with Durham tubes (pg. 3-4) Oxidation/Fermentation Agar (pg. 5-6) Anaerobic Growth (pg. 7) Growth in Liquid Culture (pg. 8-9) Friday: Pet Investigations:
Microbiology Chapter 2 Laboratory Equipment and Procedures 2:1 The Light Microscope MICROSCOPE: any tool with a lens to magnify and observe tiny
 Microbiology Chapter 2 Laboratory Equipment and Procedures 2:1 The Light Microscope MICROSCOPE: any tool with a lens to magnify and observe tiny details of specimens Micro tiny, small Scope to see SIMPLE
Microbiology Chapter 2 Laboratory Equipment and Procedures 2:1 The Light Microscope MICROSCOPE: any tool with a lens to magnify and observe tiny details of specimens Micro tiny, small Scope to see SIMPLE
MICROBIOLOGICAL TOOLS FOR QUALITY ASSURANCE IN HATCHERY: Laboratory Methods
 Issue No.29 / March 2010 MICROBIOLOGICAL TOOLS FOR QUALITY ASSURANCE IN HATCHERY: Laboratory Methods By Dr Vincent TURBLIN, Deputy Regional Market Manager Poultry - CEVA Animal Health Asia Pacific Most
Issue No.29 / March 2010 MICROBIOLOGICAL TOOLS FOR QUALITY ASSURANCE IN HATCHERY: Laboratory Methods By Dr Vincent TURBLIN, Deputy Regional Market Manager Poultry - CEVA Animal Health Asia Pacific Most
Section 8: Refined sugar p 1/5
 Section 8: Refined sugar p 1/5 1. Rationale Method 8.19 Refined sugar: total thermophilic organisms, flat sour spores, anaerobic organisms producing sulphide and anaerobic organisms producing gas The method
Section 8: Refined sugar p 1/5 1. Rationale Method 8.19 Refined sugar: total thermophilic organisms, flat sour spores, anaerobic organisms producing sulphide and anaerobic organisms producing gas The method
Serial dilution and colony count (Viable count) Pour plate. Spread plate Membrane filtration. Turbidity. Microscopic cell count
 Pour plate. Spread plate Membrane filtration. Turbidity. Microscopic cell count") Aljawharah Alabbad 2016 Serial dilution and colony count (Viable count) Pour plate Spread plate Membrane filtration Turbidity Microscopic cell count Many studies require the quantitative determination
Aljawharah Alabbad 2016 Serial dilution and colony count (Viable count) Pour plate Spread plate Membrane filtration Turbidity Microscopic cell count Many studies require the quantitative determination
Exercise 4 ASEPTIC TECHNIQUE & STREAK PLATE PREPARATION
 Introduction Exercise 4 ASEPTIC TECHNIQUE & STREAK PLATE PREPARATION In order to work with pure microbial cultures, microbiologists must start with sterile culture media and must be able to prevent contamination.
Introduction Exercise 4 ASEPTIC TECHNIQUE & STREAK PLATE PREPARATION In order to work with pure microbial cultures, microbiologists must start with sterile culture media and must be able to prevent contamination.
Exercise 19. Fungi: Molds and Yeasts F10 Or The Rotten World Around Us
 Exercise 19 119 Fungi: Molds and Yeasts F10 Or The Rotten World Around Us INTRODUCTION: Student Learning Objectives: After completing this exercise students will: a. Define the terms Saprophyte, Mycosis,
Exercise 19 119 Fungi: Molds and Yeasts F10 Or The Rotten World Around Us INTRODUCTION: Student Learning Objectives: After completing this exercise students will: a. Define the terms Saprophyte, Mycosis,
Laboratory Procedure October 1999 HEALTH PROTECTION BRANCH OTTAWA ANALYSIS OF SPROUTS FOR COLIFORMS, ESCHERICHIA COLI, AND KLEBSIELLA PNEUMONIAE..
 Government of Canada Gouvernement du Canada Laboratory Procedure MFLP-64 October 1999 HEALTH PROTECTION BRANCH OTTAWA ANALYSIS OF SPROUTS FOR COLIFORMS, ESCHERICHIA COLI, AND KLEBSIELLA PNEUMONIAE.. Don
Government of Canada Gouvernement du Canada Laboratory Procedure MFLP-64 October 1999 HEALTH PROTECTION BRANCH OTTAWA ANALYSIS OF SPROUTS FOR COLIFORMS, ESCHERICHIA COLI, AND KLEBSIELLA PNEUMONIAE.. Don
Determination of the microbial count (Item No.: P )
") Determination of the microbial count (Item No.: P4140400) Curricular Relevance Area of Expertise: Biology Education Level: University Topic: Microbiology Subtopic: Microbiology Experiment: Determination
Determination of the microbial count (Item No.: P4140400) Curricular Relevance Area of Expertise: Biology Education Level: University Topic: Microbiology Subtopic: Microbiology Experiment: Determination
DNA TRANSFORMATION OF BACTERIA RED COLONY REVISED 3/2003
 DNA TRANSFORMATION OF BACTERIA RED COLONY REVISED 3/2003 Prepared by the Office of Biotechnology, Iowa State University TEACHER PREPARATION AND INSTRUCTION GUIDE Preparation for the DNA transformation
DNA TRANSFORMATION OF BACTERIA RED COLONY REVISED 3/2003 Prepared by the Office of Biotechnology, Iowa State University TEACHER PREPARATION AND INSTRUCTION GUIDE Preparation for the DNA transformation
TRANSFER OF BACTERIA USING ASEPTIC TECHNIQUE
 TRANSFER OF BACTERIA USING ASEPTIC TECHNIQUE GENERAL GUIDELINES: Safety Wear a lab coat and have your goggles on! ALWAYS disinfect the tables BEFORE and AFTER lab. Wash your hands with soap both BEFORE
TRANSFER OF BACTERIA USING ASEPTIC TECHNIQUE GENERAL GUIDELINES: Safety Wear a lab coat and have your goggles on! ALWAYS disinfect the tables BEFORE and AFTER lab. Wash your hands with soap both BEFORE
Core practical 12: Investigate the rate of growth of microorganisms in liquid culture
 Core practical 12 Teacher sheet Core practical 12: Investigate the rate of growth of microorganisms in liquid Objectives To understand how microorganism growth rate in liquid can be measured To be able
Core practical 12 Teacher sheet Core practical 12: Investigate the rate of growth of microorganisms in liquid Objectives To understand how microorganism growth rate in liquid can be measured To be able
LESSON ASSIGNMENT. After completing this lesson, you should be able to:
 LESSON ASSIGNMENT LESSON 9 Media and Reagents TEXT ASSIGNMENT Paragraphs 9-1 through 9-13. TASK OBJECTIVES After completing this lesson, you should be able to: 9-1. Select the statement that correctly
LESSON ASSIGNMENT LESSON 9 Media and Reagents TEXT ASSIGNMENT Paragraphs 9-1 through 9-13. TASK OBJECTIVES After completing this lesson, you should be able to: 9-1. Select the statement that correctly
Biotechnology In Your Mouth
 PR005 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Biotechnology In Your Mouth Teacher s Guidebook (Cat. # BE 102) think proteins! think
PR005 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Biotechnology In Your Mouth Teacher s Guidebook (Cat. # BE 102) think proteins! think
GB Translated English of Chinese Standard: GB
 Translated English of Chinese Standard: GB4789.35-2016 www.chinesestandard.net Sales@ChineseStandard.net GB NATIONAL STANDARD OF THE PEOPLE S REPUBLIC OF CHINA GB 4789.35-2016 National food safety standard
Translated English of Chinese Standard: GB4789.35-2016 www.chinesestandard.net Sales@ChineseStandard.net GB NATIONAL STANDARD OF THE PEOPLE S REPUBLIC OF CHINA GB 4789.35-2016 National food safety standard
Bacterial Plate Preparation. ~ Using aseptic techniques ~
 Bacterial Plate Preparation ~ Using aseptic techniques ~ Bacterial Plates Laboratory and research scientists have to prepare nutrient media to grow specific strains of bacteria for their research. To do
Bacterial Plate Preparation ~ Using aseptic techniques ~ Bacterial Plates Laboratory and research scientists have to prepare nutrient media to grow specific strains of bacteria for their research. To do
PRESERVATIVE EFFICACY TEST FOR COSMETIC PRODUCT
 1 SCOPE AND FIELD OF APPLICATION To determine the efficacy of the antimicrobial activity of preservatives used in cosmetic products. The method covers the determination of the suitability of preservation
1 SCOPE AND FIELD OF APPLICATION To determine the efficacy of the antimicrobial activity of preservatives used in cosmetic products. The method covers the determination of the suitability of preservation
GB Translated English of Chinese Standard: GB NATIONAL STANDARD OF THE
 Translated English of Chinese Standard: GB4789.11-2014 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA GB 4789.11-2014 National Food Safety Standard
Translated English of Chinese Standard: GB4789.11-2014 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA GB 4789.11-2014 National Food Safety Standard
M I C R O B I O L O G Y
 ninth edition TORTORA FUNKE CASE M I C R O B I O L O G Y a n i n t r o d u c t i o n 6 Microbial Growth PowerPoint Lecture Slide Presentation prepared by Christine L. Case Microbial Growth Microbial growth
ninth edition TORTORA FUNKE CASE M I C R O B I O L O G Y a n i n t r o d u c t i o n 6 Microbial Growth PowerPoint Lecture Slide Presentation prepared by Christine L. Case Microbial Growth Microbial growth
--> Buy True-PDF --> Auto-delivered in 0~10 minutes. GB Translated English of Chinese Standard: GB4789.
 Translated English of Chinese Standard: GB4789.15-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA National Food Safety Standard Food Microbiological
Translated English of Chinese Standard: GB4789.15-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA National Food Safety Standard Food Microbiological
An Effective Use of Petri Dishes for Microcultures
 31 TURNER-GRAFF, RUHAMA (1952). J. gen. Microbiol. 7, 31-35 An Effective Use of Petri Dishes for Microcultures BY RUHAMA TURXER-GRAFF Dairy Research Laboratory, Agricultural Research Station, Rehovot,
31 TURNER-GRAFF, RUHAMA (1952). J. gen. Microbiol. 7, 31-35 An Effective Use of Petri Dishes for Microcultures BY RUHAMA TURXER-GRAFF Dairy Research Laboratory, Agricultural Research Station, Rehovot,
Bacterial Transformation: Unlocking the Mysteries of Genetic Material
 PR009 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Bacterial Transformation: Unlocking the Mysteries of Genetic Material Teacher s Guidebook
PR009 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Bacterial Transformation: Unlocking the Mysteries of Genetic Material Teacher s Guidebook
MATERIALS AND METHODS
 MATERIALS AND METHODS 2.1. Toys The toys that were used vary in material. This is particularly important when comparing and contrasting the types of bacteria that may grow on different surfaces and therefore
MATERIALS AND METHODS 2.1. Toys The toys that were used vary in material. This is particularly important when comparing and contrasting the types of bacteria that may grow on different surfaces and therefore
Coliform bacteria are quantitated by the fractional gram pour plate technique (Note 1). Test tubes containing gas collector tubes (Durham Tubes)
. Test tubes containing gas collector tubes (Durham Tubes)") Microbiological Methods IV-A- 1 (STANDARD PLATE COUNT METHOD) PRINCIPLE SCOPE Coliform bacteria are quantitated by the fractional gram pour plate technique (Note 1). The method is applicable to starches,
Microbiological Methods IV-A- 1 (STANDARD PLATE COUNT METHOD) PRINCIPLE SCOPE Coliform bacteria are quantitated by the fractional gram pour plate technique (Note 1). The method is applicable to starches,
ENVR 421 Laboratory #1: Basic Bacteriology Techniques
 ENVR 421 Laboratory #1: Basic Bacteriology Techniques Introduction The purpose of this laboratory exercise is to familiarize you with two fundamental bacteriology techniques: the streak plate and the spread
ENVR 421 Laboratory #1: Basic Bacteriology Techniques Introduction The purpose of this laboratory exercise is to familiarize you with two fundamental bacteriology techniques: the streak plate and the spread
Bacterial Transformation and Protein Purification
 Bacterial Transformation and Protein Purification Group 4 Natalie Beale Gregory A. Pate Justin Rousseau Dohee Won Introduction The purpose of this experiment is to perform a genetic transformation and
Bacterial Transformation and Protein Purification Group 4 Natalie Beale Gregory A. Pate Justin Rousseau Dohee Won Introduction The purpose of this experiment is to perform a genetic transformation and
á62ñ MICROBIOLOGICAL EXAMINATION OF NONSTERILE PRODUCTS: TESTS FOR SPECIFIED MICROORGANISMS
 USP 40 Microbiological Tests / á62ñ Microbiological Examination 1 á62ñ MICROBIOLOGICAL EXAMINATION OF NONSTERILE PRODUCTS: TESTS FOR SPECIFIED MICROORGANISMS INTRODUCTION The tests described hereafter
USP 40 Microbiological Tests / á62ñ Microbiological Examination 1 á62ñ MICROBIOLOGICAL EXAMINATION OF NONSTERILE PRODUCTS: TESTS FOR SPECIFIED MICROORGANISMS INTRODUCTION The tests described hereafter
Making Saline SOLUTION. Lab Number 2 Part 1
 Making Saline SOLUTION Lab Number 2 Part 1 Purpose The purpose of part 1 of this lab is to learn the proper way to make reagents that are needed for labs. Materials Need for the Lab are: Volumetric flasks
Making Saline SOLUTION Lab Number 2 Part 1 Purpose The purpose of part 1 of this lab is to learn the proper way to make reagents that are needed for labs. Materials Need for the Lab are: Volumetric flasks
genus is Sordaria. Sordaria can be found worldwide in the feces of herbivores (Maryland
 Solverson!1 Introduction The specific species of Sordaria being worked with in this lab is Sordaria fimicola. Sordaria fimicola is a species of microscopic fungus. It is from the family Sordariaceae, and
Solverson!1 Introduction The specific species of Sordaria being worked with in this lab is Sordaria fimicola. Sordaria fimicola is a species of microscopic fungus. It is from the family Sordariaceae, and
Bacterial Counts - Quantitative Analysis of Microbes
 Bacterial Counts - Quantitative Analysis of Microbes Introduction: It is often important to know not only what types of bacteria are in a sample but also how many of them are present. Food manufacturers
Bacterial Counts - Quantitative Analysis of Microbes Introduction: It is often important to know not only what types of bacteria are in a sample but also how many of them are present. Food manufacturers
MOLEBIO LAB #12: Bacterial Culture Techniques Part I
 MOLEBIO LAB #12: Bacterial Culture Techniques Part I Introduction: This lab introduces an introduction to plating and culturing E. coli on LB agar plates such that single cells can be isolated from one
MOLEBIO LAB #12: Bacterial Culture Techniques Part I Introduction: This lab introduces an introduction to plating and culturing E. coli on LB agar plates such that single cells can be isolated from one
LABORATORY #2 -- BIOL 111 BACTERIAL CULTIVATION & NORMAL FLORA
 LABORATORY #2 -- BIOL 111 BACTERIAL CULTIVATION & NORMAL FLORA OBJECTIVES After completing this exercise you should be able to: 1. Identify various types of media 2. Isolate bacteria using aseptic technique.
LABORATORY #2 -- BIOL 111 BACTERIAL CULTIVATION & NORMAL FLORA OBJECTIVES After completing this exercise you should be able to: 1. Identify various types of media 2. Isolate bacteria using aseptic technique.
Microorganisms In Our Environment
 PR015 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Microorganisms In Our Environment Teacher s Guidebook (Cat. # BE 106) think proteins!
PR015 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Microorganisms In Our Environment Teacher s Guidebook (Cat. # BE 106) think proteins!
TRYPTIC SOY AGAR (TSA) WITH LECITHIN AND TWEEN 80
 WITH LECITHIN AND TWEEN 80") TRYPTIC SOY AGAR (TSA) WITH LECITHIN AND TWEEN 80 Cat. no. P45 TSA with Lecithin and Tween 80, 15x60mm Contact Plate, 15ml Cat. no. Q13 TSA with Lecithin and Tween 80, 20x125mm Tube, 18ml Deep Cat. no.
TRYPTIC SOY AGAR (TSA) WITH LECITHIN AND TWEEN 80 Cat. no. P45 TSA with Lecithin and Tween 80, 15x60mm Contact Plate, 15ml Cat. no. Q13 TSA with Lecithin and Tween 80, 20x125mm Tube, 18ml Deep Cat. no.
Chapter 03 - Tools of the Laboratory: Methods for the Culturing of Microscopic Analysis of microorganisms
 Microbiology: A Systems Approach 4th Edition Cowan Test Bank Completed download: https://testbankreal.com/download/microbiology-systems-approach-4thedition-test-bank-cowan/ (Downloadable package TEST BANK
Microbiology: A Systems Approach 4th Edition Cowan Test Bank Completed download: https://testbankreal.com/download/microbiology-systems-approach-4thedition-test-bank-cowan/ (Downloadable package TEST BANK
Diagnostic Microbiology
 Diagnostic Microbiology Identification of Microbes Lecture: 1 Out lines What is expected out of this course??? At the end of this course, you will be able to apply Conventional/ Molecular diagnostic methods
Diagnostic Microbiology Identification of Microbes Lecture: 1 Out lines What is expected out of this course??? At the end of this course, you will be able to apply Conventional/ Molecular diagnostic methods
National food safety standard
 Translated English of Chinese Standard: GB4789.26-2013 Translated by: www.chinesestandard.net Wayne Zheng et al. Email: Sales@ChineseStandard.net ICS 65.020.30 GB B 40 NATIONAL STANDARD OF THE PEOPLE S
Translated English of Chinese Standard: GB4789.26-2013 Translated by: www.chinesestandard.net Wayne Zheng et al. Email: Sales@ChineseStandard.net ICS 65.020.30 GB B 40 NATIONAL STANDARD OF THE PEOPLE S
Biotechnology Science for the New Millennium by Ellyn Daugherty
 G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Biotechnology Science for the New Millennium by Ellyn Daugherty Characterizing Bacteria Using
G-Biosciences 1-800-628-7730 1-314-991-6034 technical@gbiosciences.com A Geno Technology, Inc. (USA) brand name Biotechnology Science for the New Millennium by Ellyn Daugherty Characterizing Bacteria Using
ENVIRONMENTAL PARAMETERS OF GROWTH
 ENVIRONMENTAL PARAMETERS OF GROWTH The growth and survival of microorganisms are affected by the chemical and physical conditions of the external environment. Environmental factors which have significant
ENVIRONMENTAL PARAMETERS OF GROWTH The growth and survival of microorganisms are affected by the chemical and physical conditions of the external environment. Environmental factors which have significant
MICROBIOLOGICAL EXAMINATION OF NON-STERILE PRODUCTS: TEST FOR SPECIFIED MICRO-ORGANISMS Test for specified micro-organisms
 5-2-3. Most-probable-number method Prepare and dilute the sample using a method that has been shown to be suitable as described in section 4. Incubate all tubes at 30-35 C for 3-5 days. Subculture if necessary,
5-2-3. Most-probable-number method Prepare and dilute the sample using a method that has been shown to be suitable as described in section 4. Incubate all tubes at 30-35 C for 3-5 days. Subculture if necessary,
Culture Media. Provide certain environmental conditions, nutrients & energy in order to grow and produce bacteria
 Culture Media Culture Media Provide certain environmental conditions, nutrients & energy in order to grow and produce bacteria Different categories of media can be made according to the type and combination
Culture Media Culture Media Provide certain environmental conditions, nutrients & energy in order to grow and produce bacteria Different categories of media can be made according to the type and combination
Exercise 23-C BACTERIOPHAGE REPRODUCTION AND PLAQUE FORMATION
 Exercise 23-C BACTERIOPHAGE REPRODUCTION AND PLAQUE FORMATION Introduction The reproductive cycle of a cytolytic bacteriophage called T2 begins with its adsorption onto a sensitive host cell. The phage
Exercise 23-C BACTERIOPHAGE REPRODUCTION AND PLAQUE FORMATION Introduction The reproductive cycle of a cytolytic bacteriophage called T2 begins with its adsorption onto a sensitive host cell. The phage
Requirements for Growth
 Requirements for Growth Definition: Bacterial growth defined as an increase in the number of cells. Physical Requirements: temperature, ph, tonicity Temperature: On the basis of growth range of temperature
Requirements for Growth Definition: Bacterial growth defined as an increase in the number of cells. Physical Requirements: temperature, ph, tonicity Temperature: On the basis of growth range of temperature
Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet
 Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet Overview In this lab, students will learn about ethanol and its important role in our world s ever-increasing demand for
Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet Overview In this lab, students will learn about ethanol and its important role in our world s ever-increasing demand for
2/25/2013. Psychrotrophs Grow between 0 C and C Cause food spoilage Food Preservation Temperatures
 3 4 5 6 7 8 9 0 Chapter 6 Microbial Growth Microbial Growth Increase in number of cells, not cell size Populations Colonies The Requirements for Growth Physical requirements Temperature ph Osmotic pressure
3 4 5 6 7 8 9 0 Chapter 6 Microbial Growth Microbial Growth Increase in number of cells, not cell size Populations Colonies The Requirements for Growth Physical requirements Temperature ph Osmotic pressure
Aerobic Count. Interpretation Guide. 3M Food Safety 3M Petrifilm Aerobic Count Plate
 3M Food Safety 3M Petrifilm Aerobic Count Plate Aerobic Count Interpretation Guide The 3M Petrifilm Aerobic Count (AC) Plate is a ready-made culture medium system that contains Standard Methods nutrients,
3M Food Safety 3M Petrifilm Aerobic Count Plate Aerobic Count Interpretation Guide The 3M Petrifilm Aerobic Count (AC) Plate is a ready-made culture medium system that contains Standard Methods nutrients,
Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet
 Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet Overview In this lab, students will learn about ethanol and its important role in our world s everincreasing demand for
Kansas Corn: Ethanol - Corn Mash and Distillation High School Student Lab Packet Overview In this lab, students will learn about ethanol and its important role in our world s everincreasing demand for
UNIVERSITEIT GENT. Laboratory of Microbiology K.L. Ledeganckstr. 35 B-9000 Gent (BELGIUM) SOP. Standard Operating Procedure.
 SOP. Standard Operating Procedure.") SOP Standard Operating Procedure Author: Acronym: Date last modified: Geert Huys ASIARESIST-PRES 20-11-2002 Title: PRESERVATION OF BACTERIA USING COMMERCIAL CRYOPRESERVATION SYSTEMS References: Reviewed
SOP Standard Operating Procedure Author: Acronym: Date last modified: Geert Huys ASIARESIST-PRES 20-11-2002 Title: PRESERVATION OF BACTERIA USING COMMERCIAL CRYOPRESERVATION SYSTEMS References: Reviewed
ENVIRONMENTAL PARAMETERS OF GROWTH
 ENVIRONMENTAL PARAMETERS OF GROWTH The growth and survival of microorganisms are affected by the chemical and physical conditions of the external environment. Environmental factors which have significant
ENVIRONMENTAL PARAMETERS OF GROWTH The growth and survival of microorganisms are affected by the chemical and physical conditions of the external environment. Environmental factors which have significant
LAB NOTES FOR EXAM 1 SECTION
 LAB NOTES FOR EXAM 1 SECTION EX. 2-1: DIVERSITY AND UBIQUITY OF MICROOGANISMS Purpose: Microorganisms are found everywhere in the environment around us. To demonstrate this and to get a taste of the different
LAB NOTES FOR EXAM 1 SECTION EX. 2-1: DIVERSITY AND UBIQUITY OF MICROOGANISMS Purpose: Microorganisms are found everywhere in the environment around us. To demonstrate this and to get a taste of the different
Bacteria and other microbes have particular requirements for growth When they reside in and on our bodies or in the environment, they harvest their
 Bacteria and other microbes have particular requirements for growth When they reside in and on our bodies or in the environment, they harvest their food from us or from the environment When we grow bacteria
Bacteria and other microbes have particular requirements for growth When they reside in and on our bodies or in the environment, they harvest their food from us or from the environment When we grow bacteria
3.2 Test for sterility
 This text is based on the internationally-harmonized texts developed by the Pharmacopoeial Discussion Group (PDG). Some editorial modifications have been made in order to be in line with the style used
This text is based on the internationally-harmonized texts developed by the Pharmacopoeial Discussion Group (PDG). Some editorial modifications have been made in order to be in line with the style used
Project 7: Wound Cultures and Identification
 Project 7: Wound Cultures and Identification Readings: https://labtestsonline.org/understanding/analytes/wound-culture/tab/test Identification of Gram-Positive & Gram-Negative Bacteria Guide to laboratory
Project 7: Wound Cultures and Identification Readings: https://labtestsonline.org/understanding/analytes/wound-culture/tab/test Identification of Gram-Positive & Gram-Negative Bacteria Guide to laboratory
MICROBIAL GROWTH. Dr. Hala Al-Daghistani
 MICROBIAL GROWTH Dr. Hala Al-Daghistani Microbial Growth Microbial growth: Increase in cell number, not cell size! Physical Requirements for Growth: Temperature Minimum growth temperature Optimum growth
MICROBIAL GROWTH Dr. Hala Al-Daghistani Microbial Growth Microbial growth: Increase in cell number, not cell size! Physical Requirements for Growth: Temperature Minimum growth temperature Optimum growth
Heterotrophic Bacteria
 , m-hpc, 8242 DOC316.53.01225 Pour Plate Method Method 8242 m-hpc Scope and Application: For water and wastewater. Test preparation Introduction Before starting the test: The Pour Plate Method, also known
, m-hpc, 8242 DOC316.53.01225 Pour Plate Method Method 8242 m-hpc Scope and Application: For water and wastewater. Test preparation Introduction Before starting the test: The Pour Plate Method, also known
ANALYTICAL REPORT: Comparison of the Microbial Recovery Efficacy of QI Medical EnviroTest Paddles versus a Conventional Contact Plate
 FOCUS Scientific Services LLC ANALYTICAL REPORT: Comparison of the Microbial Recovery Efficacy of QI Medical EnviroTest Paddles versus a Conventional Contact Plate REPORT: FS-QI-GM-003 Study Manager: Anthony
FOCUS Scientific Services LLC ANALYTICAL REPORT: Comparison of the Microbial Recovery Efficacy of QI Medical EnviroTest Paddles versus a Conventional Contact Plate REPORT: FS-QI-GM-003 Study Manager: Anthony
--> Buy True-PDF --> Auto-delivered in 0~10 minutes. GB Translated English of Chinese Standard: GB4789.
 Translated English of Chinese Standard: GB4789.36-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA National Food Safety Standard - Microbiological
Translated English of Chinese Standard: GB4789.36-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA National Food Safety Standard - Microbiological
GB Translated English of Chinese Standard: GB NATIONAL STANDARD OF THE
 Translated English of Chinese Standard: GB4789.36-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA GB 4789.36-2016 National Food Safety Standard
Translated English of Chinese Standard: GB4789.36-2016 www.chinesestandard.net Sales@ChineseStandard.net NATIONAL STANDARD OF THE GB PEOPLE S REPUBLIC OF CHINA GB 4789.36-2016 National Food Safety Standard
Test Method of Specified Requirements of Antibacterial Textiles for Medical Use FTTS-FA-002
 Test Method of Specified Requirements of Antibacterial Textiles for Medical Use FTTS-FA-002 FTTS-FA-002 Antibacterial Textiles for Medical Use Antibacterial Textiles suppress and even kill harmful bacteria
Test Method of Specified Requirements of Antibacterial Textiles for Medical Use FTTS-FA-002 FTTS-FA-002 Antibacterial Textiles for Medical Use Antibacterial Textiles suppress and even kill harmful bacteria
2. 47 mm grid marked, white sterile 0.45 micron membranes (Millipore or equivalent) 4. Vacuum pump capable of inches of vacuum
 4. Vacuum pump capable of inches of vacuum") Microbiological Methods IX-B- 1 PRESUMPTIVE MEMBRANE FILTER METHOD AND CONFIRMATION OF PSEUDOMONAS AERUGINOSA PRINCIPLE SCOPE Presumptive Pseudomonas bacteria are quantitated by a membrane filter technique,
Microbiological Methods IX-B- 1 PRESUMPTIVE MEMBRANE FILTER METHOD AND CONFIRMATION OF PSEUDOMONAS AERUGINOSA PRINCIPLE SCOPE Presumptive Pseudomonas bacteria are quantitated by a membrane filter technique,
Microbiology sheet (6)
") Microbiology sheet (6) Made by marah marahleh corrected by : abd. Salman DATE :9/10/2016 Microbial growth / control of microbial growth 1 The method of counting bacteria is divided into: 1) direct 2) indirect
Microbiology sheet (6) Made by marah marahleh corrected by : abd. Salman DATE :9/10/2016 Microbial growth / control of microbial growth 1 The method of counting bacteria is divided into: 1) direct 2) indirect
Project 5: Urine Cultures and Identification
 Project 5: Urine Cultures and Identification Readings: http://www.webmd.com/a-to-z-guides/urine-culture http://www.medscape.com/viewarticle/558845 (Listen to the two lectures by Dr. Robert A. Weinstein.)
Project 5: Urine Cultures and Identification Readings: http://www.webmd.com/a-to-z-guides/urine-culture http://www.medscape.com/viewarticle/558845 (Listen to the two lectures by Dr. Robert A. Weinstein.)
LAB NOTES FOR EXAM 1 SECTION
 LAB NOTES FOR EXAM 1 SECTION EX. 2-1: DIVERSITY AND UBIQUITY OF MICROOGANISMS Purpose: Microorganisms are found everywhere in the environment around us. To demonstrate this and to get a taste of the different
LAB NOTES FOR EXAM 1 SECTION EX. 2-1: DIVERSITY AND UBIQUITY OF MICROOGANISMS Purpose: Microorganisms are found everywhere in the environment around us. To demonstrate this and to get a taste of the different
Inoculate: Media. Physical State of Media: Liquid. The Five I s: Basic Techniques to Culture Microbes Tools of the Microbiology Laboratory
 The Five I s: Basic Techniques to Culture Microbes Tools of the Microbiology Laboratory 1. Inoculate 2. Incubate 3. Isolate 4. Inspect 5. Identify The Five I s: Inoculate Inoculate: Media Classified according
The Five I s: Basic Techniques to Culture Microbes Tools of the Microbiology Laboratory 1. Inoculate 2. Incubate 3. Isolate 4. Inspect 5. Identify The Five I s: Inoculate Inoculate: Media Classified according
Petrifilm RYM. Interpretation Guide. Rapid Yeast and Mould Count Plate. Brand
 Petrifilm Brand Interpretation Guide The 3M Petrifilm Rapid Yeast and Mould Count Plate is a sample-ready culture medium system that contains nutrients supplemented with antibiotics, a cold-water-soluble
Petrifilm Brand Interpretation Guide The 3M Petrifilm Rapid Yeast and Mould Count Plate is a sample-ready culture medium system that contains nutrients supplemented with antibiotics, a cold-water-soluble
Kit Information 4 Introduction. 4 Kit Contents, Storage, and Testing Conditions. 4 Principle 4 Applicability. 5 Precautions Sample Preparation 6
 Contents 3. Kit Information 4 Introduction. 4 Kit Contents, Storage, and Testing Conditions. 4 Principle 4 Applicability. 5 Precautions.......................... 5 Sample Preparation 6 Peel Plate EB Test
Contents 3. Kit Information 4 Introduction. 4 Kit Contents, Storage, and Testing Conditions. 4 Principle 4 Applicability. 5 Precautions.......................... 5 Sample Preparation 6 Peel Plate EB Test
National food safety standard Food microbiological examination: Listeria monocytogenes
 National Standard of the People s Republic of China GB 4789.30-2010 National food safety standard Food microbiological examination: Listeria monocytogenes Issued on: 2010-03 - 26 Implemented on: 2010-06
National Standard of the People s Republic of China GB 4789.30-2010 National food safety standard Food microbiological examination: Listeria monocytogenes Issued on: 2010-03 - 26 Implemented on: 2010-06
Document No. FTTS-FA-001. Specified Requirements of Antibacterial Textiles for General Use
 1. Purpose and Scope This criterion is applicable to the evaluation and testing of antibacterial activity of textile for general use. The quantitative evaluation of antibacterial activity is judged by
1. Purpose and Scope This criterion is applicable to the evaluation and testing of antibacterial activity of textile for general use. The quantitative evaluation of antibacterial activity is judged by
CONTROL OF MICROBIAL GROWTH - DISINFECTANTS AND ANTISEPTICS
 CONTROL OF MICROBIAL GROWTH - DISINFECTANTS AND ANTISEPTICS Specific control measures can be used to kill or inhibit the growth of microorganisms. A procedure which leads to the death of cells is broadly
CONTROL OF MICROBIAL GROWTH - DISINFECTANTS AND ANTISEPTICS Specific control measures can be used to kill or inhibit the growth of microorganisms. A procedure which leads to the death of cells is broadly
ENV H 433 LABORATORY EXERCISE 1
 ENV H 433 LABORATORY EXERCISE 1 Multiple tube fermentation and presence/absence methods to detect total coliforms, fecal coliforms and Escherichia coli I. LABORATORY GOAL To determine concentration of
ENV H 433 LABORATORY EXERCISE 1 Multiple tube fermentation and presence/absence methods to detect total coliforms, fecal coliforms and Escherichia coli I. LABORATORY GOAL To determine concentration of
number Done by Corrected by Doctor
 L number Lab 2 Done by حسام أبو عوض Corrected by Mahdi sharawi Doctor In many cases we need to identify the type of bacteria causing an infection in order to be able to choose the right medication (antibiotic).
L number Lab 2 Done by حسام أبو عوض Corrected by Mahdi sharawi Doctor In many cases we need to identify the type of bacteria causing an infection in order to be able to choose the right medication (antibiotic).
M. Dalbey/Bio 105M Isolation of E. coli - Isolation of E. coli from an Environmental Sample
 Isolation of E. coli from an Environmental Sample We want to expand our horizons a bit beyond the domesticated lab strains of E. coli. In this exercise you will isolate "wild" E. coli strains from an environmental
Isolation of E. coli from an Environmental Sample We want to expand our horizons a bit beyond the domesticated lab strains of E. coli. In this exercise you will isolate "wild" E. coli strains from an environmental
NATURE OF MICROBES WORKBOOK
 NATURE OF MICROBES WORKBOOK Name: Tutor Group: 1 Microbes and Mankind 4. NATURE OF MICROBES 1. OBJECTIVES: What are microbes and are there different types? How are they seen? How can they be grown? How
NATURE OF MICROBES WORKBOOK Name: Tutor Group: 1 Microbes and Mankind 4. NATURE OF MICROBES 1. OBJECTIVES: What are microbes and are there different types? How are they seen? How can they be grown? How
MICROBIOLOGY #2 PREPERATION AND STERILIZATION OF CULTURE MEDIA
 MICROBIOLOGY #2 PREPERATION AND STERILIZATION OF CULTURE MEDIA When we receive a sample (ex. Urine sample) for detection, we cannot gram stain it right away if it requires to be inoculated because when
MICROBIOLOGY #2 PREPERATION AND STERILIZATION OF CULTURE MEDIA When we receive a sample (ex. Urine sample) for detection, we cannot gram stain it right away if it requires to be inoculated because when
New York State Department of Health - Wadsworth Center Laboratory of Environmental Biology NYS ELAP Laboratory ID 10765
 New York State Department of Health - Wadsworth Center Laboratory of Environmental Biology NYS ELAP Laboratory ID 10765 Division of Environmental Health Sciences Albany, New York NYS DOH LEB-608 Identification
New York State Department of Health - Wadsworth Center Laboratory of Environmental Biology NYS ELAP Laboratory ID 10765 Division of Environmental Health Sciences Albany, New York NYS DOH LEB-608 Identification