Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals
|
|
|
- Kelly Whitehead
- 5 years ago
- Views:
Transcription




1 ORIENTATION FOR NEW CLINICAL RESEARCH COORDINATORS Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals
2 Overall Agenda for Orientation Module 1: Introduction to Clinical Research, Education, and IRB Module 2: Informed Consent, Documentation and GCP, and Study Start up Module 3: Contracting, ClinicalTrials.gov, and Drug/ Device Policies, Conflict of Interest Policy Module 4: Recruitment, Preparing Industry & NIH Grant Budgets, Accounting, Billing Coverage Analysis Module 5: Essential Documents, From CDA to Study Close Out
3 ESSENTIAL (REGULATORY) Valorie Buchholz DOCUMENTS Associate Director for Clinical Trial Quality Assurance Office of Clinical Trials
4 Mornings in the Regulatory World
5 What Types of Trials Should Meet GCP Guidelines? FDA Regulated Trials Federally funded (NIH) Trials Requirement as of January, 2017 A quality research site complies with the ICH Good Clinical Practice guidelines, the accepted international ethical and scientific quality standards for designing, conducting, recording, and reporting trials involving human participants. American Society of Clinical Oncology Statement on Minimum Standards and Exemplary Attributes of Clinical Trial Sites; R. Zion et al; Journal of Clinical Oncology; April 7, 2008
6 What are Essential Documents? ICH defines Essential Documents as: the documents which individually and collectively permit the evaluation of the conduct of a trial and the quality of the data produced (ICH GCP E6 (R2) 8.1)
7 GCP Essential Documents Guidance Categorizes documents into 3 sections Defines whether the sponsor, site or both are responsible for maintenance of the documents Reminder If your investigator holds and IND or IDE, he/she also has sponsor responsibilities
8 BEFORE STUDY IS INITIATED: Investigative Site Sponsor/Funding Agency

9 IND trials 1572 Statement of Investigator IDE Trials Investigator Agreement
10 Documentation for Investigators and Sub- Investigators Curriculum Vitae Licensure Certifications Documents qualifications and eligibility to conduct trial Documents eligibility to provide medical supervision of subjects Documents qualifications to perform tasks as delegated GCP 2.8, 4.1.1, 4.1.5
11 AND MORE APPROVAL DOCUMENTS AGREEMENTS Institutional Review Board Regulatory Authorities Protocol Review Committee Institutional Biosafety Committee Scientific Review Committee Site and Sponsor Site and CRO Financial Aspects Disclosure Forms (FDA) GCP 3.4, 4.5.1, 8.2.4, 8.2.9
12 AND MORE Normal Ranges Lab Info CLIA CAP CV of Medical Director GCP
13 JUST A FEW MORE Investigational Product Shipping and Accountability Records Applicable for both Drugs and Devices Procedures for Decoding Blinded Trials used in case of emergency doesn t break blind for others Study Initiation Report Document questions asked during SIV! GCP , , , ,
14 READY, SET, GO! Documents updated during the study UPDATE Laboratory Accreditations (CLIA/CAP) 1572 /Investigator Agreement Only required by FDA if a new protocol is added to the IND and the Investigator is participating OR a new investigator is added Investigator s Brochures Normal values to lab ranges GCP , 8.3.1, 8.3.6
15 Update IRB renewals / modifications Regulatory Authorities Biosafety Committee Protocol Review Committee CV s / Licensures / Certifications for New Investigators GCP
16 ADD LOGS DELEGATIO N OF AUTHORITY LOG / SIGNATURE SHEET SCREENING LOG SUBJECT ENROLL- MENT LOG SUBJECT ID CODE LIST TRAINI NG LOGS Protocol IB Equipment CRF Completio n / Correction RETAI NED BODY FLUID / TISSU E SAMP LES GCP 4.1.5, 4.2.4, ,
17 INCLUDE RELEVANT COMMUNICATIO NS Letters Memos to File Meeting Notes Telephone Calls GCP
18 SAFETY SERIOUS ADVERSE EVENTS Report to sponsor and IRB UNANTICIPATED / UNEXPECTED ADVERSE EVENTS Report to Sponsor and IRB SAFETY INFORMATION / REPORTS Sponsors Notify Sites
19 STUDY COMPLETION Logs Investigational Product Accountability Subject Identification Code Final Close-out monitoring report Includes where site documents will be stored Termination of study with IRB Clinical Study Report
20 Inspections and Essential Documents Sponsors and other regulatory agencies look to these documents as part of their processes to confirm the validity of the conduct of the trial and the data collected
21 Record Retention Requirements Depends on the Type of Trial FDA: 21CFR (IND) OR 21CFR (IDE) Contracts with Funding Source Institutional Policies: (see Section 6 Grants and Research Records) Pediatric Studies UNC Healthcare Policy? GCP
22 TAKE AWAY POINTS Organized Consistent
23 For Additional Information article/efficacy-guidelines.html guidancecomplianceregulatoryinformation/guidances/ucm pdf
24
25 FROM CDA TO STUDY CLOSURE Panel Members: Christine Nelson Val Buchholz
26 Objectives Review the steps for successful clinical trial implementation beginning with first sponsor contact through study closure
27 So it begins
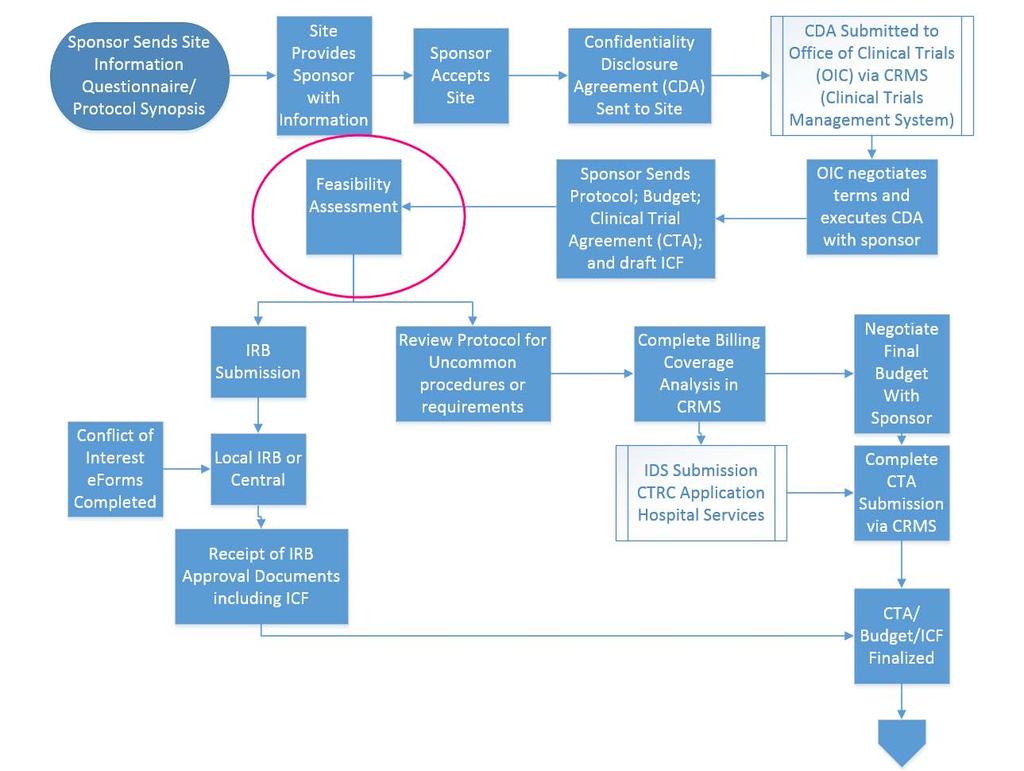
28 Site Survey Site information form Site Qualification form Site Feasibility form Sponsors and CROs track turn around times
29
30 CDA CDA must be submitted via CRMS Cannot be signed by PI Quick turn around Not every CDA results in receiving a protocol Sponsors and CROs track turn around times
31
32 CRMS Protocol final or draft? May just send protocol until you are selected as a site May send someone out to do site qualification visit Draft ICF Draft CTA Draft Budget Investigator brochure Pharmacy manual Lab manual
33
34 Feasibility Conduct a preliminary feasibility assessment Read the draft ICF Read the protocol Potential enrollment Study schedule (practical, reasonable) Study duration Non-routine care items Imaging Pharmacy Lab/specimens Resources (study coordinator, data manager, 24/7) Adequate staffing Training requirements Special vendor requirements
35
36 Budget Billing Coverage Analysis Spreadsheet from CRMS Deemed and Qualified Epic Billing calendar Funding source (federal or industry) Consistent approach Ensure start up fees are sufficient and invoiced Standardized fees Screen fails Monitoring visits Monthly invoicing IDS CTRC
37
38 CTA Submit CTA to OIC via CRMS, direct link to ALICE Complete review request form (RRF) Contract manager assigned Only the assigned contract manager negotiates the CTA Open communication with your contract manager The CTA can be negotiated while you negotiate your budget Once budget has been finalized with sponsor we can execute the CTA
39
40 IRB Submit when you are sure the PI wants to participate UNC local IRB or Central IRB ICF and contract must be consistent in respect to subject injury, stipends and what has been promised for free to the subjects
41
42 CTA and IRB IRB Approval ICF and CTA must be consistent OCT will check but you should also check If inconsistent the ICF will need to be revised
43
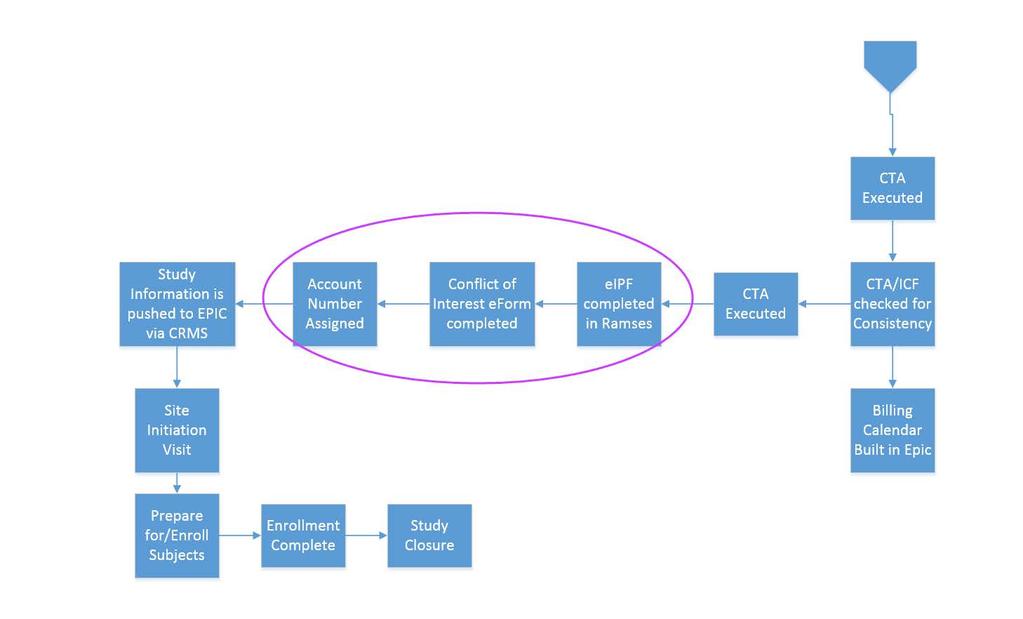
44 Ramses eipf Internal budget Need an account Set up after all compliance checks are completed by OCT Sent to OSR for final PS project ID assignment COI Individual Institutional

45 CTA Executed Study Information is pushed to EPIC via CRMS Account Number Assigned Conflict of Interest eform completed eipf completed in Ramses CTA Executed CTA/ICF checked for Consistency Site Initiation Visit Billing Calendar Built in Epic Prepare for/enroll Subjects Enrollment Complete Study Closure

46 Study Start Up When can I enroll! Its been months and I am already tired
47 Mornings in the Regulatory World
48 What Types of Trials Should Meet GCP Guidelines? FDA Regulated Trials Federally funded (NIH) Trials Requirement as of January, 2017 A quality research site complies with the ICH Good Clinical Practice guidelines, the accepted international ethical and scientific quality standards for designing, conducting, recording, and reporting trials involving human participants. American Society of Clinical Oncology Statement on Minimum Standards and Exemplary Attributes of Clinical Trial Sites; R. Zion et al; Journal of Clinical Oncology; April 7, 2008
49 What are Essential Documents? ICH defines Essential Documents as: The documents which individually and collectively permit the evaluation of the conduct of a trial and the quality of the data produced (ICH GCP E6 (R2) 8.1)


50 BEFORE STUDY IS INITIATED: Investigative Site Sponsor/Funding Agency
51 Documentation for Investigators and Sub- Investigators Curriculum Vitae Licensure Certifications Documents qualifications and eligibility to conduct trial Documents eligibility to provide medical supervision of subjects Documents qualifications to perform tasks as delegated GCP 2.8, 4.1.1, 4.1.5
52 AND MORE APPROVAL DOCUMENTS AGREEMENTS Institutional Review Board Regulatory Authorities Protocol Review Committee Institutional Biosafety Committee Scientific Review Committee Site and Sponsor/Funding Agency Site and CRO Financial Aspects Disclosure Forms (FDA) GCP 3.4, 4.5.1, 8.2.4, 8.2.9

53 AND MORE Normal Ranges Lab Info CLIA CAP CV of Medical Director GCP


54 JUST A FEW MORE Investigational Product Shipping and Accountability Records Applicable for both Drugs and Devices Procedures for Decoding Blinded Trials used in case of emergency doesn t break blind for others Study Initiation Report Document questions asked during SIV! GCP , , , ,
55 Include: LOGS DELEGATION OF AUTHORITY LOG / SIGNATURE SHEET SCREENING LOG SUBJECT ENROLL- MENT LOG SUBJECT ID CODE LIST TRAINING LOGS Protocol IB Equipment CRF Completion / Correction OTHER LOGS GCP 4.1.5, 4.2.4, ,
56 Study Start Up SIV Site Initiation Visit Study supplies CRMS Clinical Research Management System (OnCore for Oncology studies) Epic IDS Investigational Drug Services at UNC Healthcare Subject binders Source documents Study visit checklist
57 Study Conduct Enroll your first subject Inclusion/exclusion criteria ICF Documentation of the informed consent process Maintenance of Essential Documents


58 Documents updated during the study UPDATE Laboratory Accreditations (CLIA/CAP) 1572 /Investigator Agreement Only required by FDA if a new protocol is added to the IND and the Investigator is participating OR a new investigator is added Investigator s Brochures Normal values to lab ranges GCP , 8.3.1, 8.3.6

59 Update IRB renewals / modifications Regulatory Authorities Biosafety Committee Protocol Review Committee CV s / Licensures / Certifications for New Investigators GCP


60 INCLUDE RELEVANT COMMUNICATIO NS Letters Memos to File Meeting Notes Telephone Calls GCP
61 Ongoing conduct of study Study visit checklists Case report forms Epic Billing review Investigational product accountability SAE/AE reporting Monitor Access Annual IRB renewal Amendments Modifications Deviations

62 STUDY COMPLETION / CLOSE OUT All study subjects complete Data lock / IRB closure Logs Investigational Product Accountability Subject Identification Code Final Close-out monitoring report Includes where site documents will be stored Clinical Study Report Pack up the records Pat yourself on the back
 Contracts with Funding Source Institutional Policies: http://library.unc.edu/wpcontent/uploads/2016/06/unc_ret_sched.")
63 Record Retention Requirements Depends on the Type of Trial FDA: 21CFR (IND) OR 21CFR (IDE) Contracts with Funding Source Institutional Policies: (see Section 6 Grants and Research Records) Pediatric Studies UNC Healthcare Policy? GCP

64 Audits FDA or Sponsor Who do you call? Hint its not Ghost Busters!
65 Inspections and Essential Documents Sponsors and other regulatory agencies look to these documents as part of their processes to confirm the validity of the conduct of the trial and the data collected
66 TAKE AWAY POINTS Organized Consistent
67



68 ENTER NEXT Training November XX, 2017, 12 noon 1:30pm Brinkhous-Bullitt Bldg, Room 219
Human Research Protection Program Good Clinical Practice Guidance for Investigators Regulatory File Essential Documents
 Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL
 Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
Regulatory Binder Guidance
 Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
STANDARD OPERATING PROCEDURE FOR RESEARCH. 17. Study Close Down
 Basildon and Thurrock University Hospitals NHS FT Research & Development APPROVED STANDARD OPERATING PROCEDURE FOR RESEARCH 1. BACKGROUND 17. Study Close Down According to ICH Good Clinical Practice (GCP)
Basildon and Thurrock University Hospitals NHS FT Research & Development APPROVED STANDARD OPERATING PROCEDURE FOR RESEARCH 1. BACKGROUND 17. Study Close Down According to ICH Good Clinical Practice (GCP)
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)
 SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)") POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
Office Of Clinical Trials. Christine Nelson, RN, BSN, MBA, CCRC
 Office Of Clinical Trials Christine Nelson, RN, BSN, MBA, CCRC OCT Mission Statement The mission of the Office of Clinical Trials (OCT) is to serve the Carolina research community by improving the quality
Office Of Clinical Trials Christine Nelson, RN, BSN, MBA, CCRC OCT Mission Statement The mission of the Office of Clinical Trials (OCT) is to serve the Carolina research community by improving the quality
Compliance and Quality Monitoring: What, Why, When, and How
 Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
Regulatory Binder: Set-up and Maintenance
 Regulatory Binder: Set-up and Maintenance Introduction Federal and state regulations, institutional policy, and good clinical and research practices require investigators to maintain documents related
Regulatory Binder: Set-up and Maintenance Introduction Federal and state regulations, institutional policy, and good clinical and research practices require investigators to maintain documents related
Source Documents and Regulatory Binders October 6, 2016
 Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1
 Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
FDA Sponsor and Investigator Responsibility Checklist
 FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
Regulatory Document Guidelines for DMID Clinical Studies. Version Oct-2005
 Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
Clinical Trial Basics:
 Clinical Trial Basics: Components and Responsibilities Pre-Award Aida Nana Ama Manu, Project Coordinator Four Main Components Non-Disclosure Agreements Institutional Review Board Application Clinical Trial
Clinical Trial Basics: Components and Responsibilities Pre-Award Aida Nana Ama Manu, Project Coordinator Four Main Components Non-Disclosure Agreements Institutional Review Board Application Clinical Trial
Multi-Site Coordination Process. Drafted by: Ester Dimayuga Page 1 of 18
 Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Human Research Protection Program Policy
 Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
ESSENTIAL DOCUMENTS DURING THE CLINICAL CONDUCT OF THE TRIAL
 ESSENTIAL DOCUMENTS DURING THE CLINICAL CONDUCT OF THE TRIAL THIS INFORMATION SHEET (ISR-RG-014) HAS BEEN TAKEN DIRECTLY FROM THE ICH- GUIDELINE* WITH ADDITIONAL COMMENTS ADDED BY THE RESEARCH GOVERNANCE
ESSENTIAL DOCUMENTS DURING THE CLINICAL CONDUCT OF THE TRIAL THIS INFORMATION SHEET (ISR-RG-014) HAS BEEN TAKEN DIRECTLY FROM THE ICH- GUIDELINE* WITH ADDITIONAL COMMENTS ADDED BY THE RESEARCH GOVERNANCE
Guidelines for Setting Up a Regulatory Binder
 Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Investigational New Drug Development Steps for CRCs
 Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
STANDARD OPERATING PROCEDURE. STH Researcher. Investigator Site File
 Research Department STANDARD OPERATING PROCEDURE STH Researcher SOP History CSUH 00/016 SOP Number A116 Created STH Research Department (TL) Reviewed by STH Research Department (AL) 06 August 2009 Superseded
Research Department STANDARD OPERATING PROCEDURE STH Researcher SOP History CSUH 00/016 SOP Number A116 Created STH Research Department (TL) Reviewed by STH Research Department (AL) 06 August 2009 Superseded
VCU Faculty Held IND and IDE Procedure Handbook
 VCU Faculty Held IND and IDE Procedure Handbook Contents A. Introduction... 3 B. Purpose of Institutional Oversight... 3 C. Applicability... 4 D. University Oversight of Clinical Investigations Being Conducted
VCU Faculty Held IND and IDE Procedure Handbook Contents A. Introduction... 3 B. Purpose of Institutional Oversight... 3 C. Applicability... 4 D. University Oversight of Clinical Investigations Being Conducted
Investigator Site File Index (CTIMP)
") Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol Current version plus all previous versions (or provide file note to detail location of previous
Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol Current version plus all previous versions (or provide file note to detail location of previous
ELEMENTS OF A DATA MONITORING PLAN
 ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
Objectives. The Regulatory Binder = Investigator Site File= Trial Center File 8/16/2010. Essential Documents: Maintaining the Site's Regulatory Binder
 Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
1. POLICY STATEMENT: 2. BACKGROUND:
 POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework
 1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
Source And Regulatory Documentation for DMID Clinical Studies
 Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
RESEARCH AUDIT Standard Operating Procedure
 Reference Number: UHB 236 Version Number: 2 Date of Next Review: 17 th Oct 2020 Previous Trust/LHB Reference Number: N/A RESEARCH AUDIT Standard Operating Procedure Introduction and Aim As a legal Sponsor
Reference Number: UHB 236 Version Number: 2 Date of Next Review: 17 th Oct 2020 Previous Trust/LHB Reference Number: N/A RESEARCH AUDIT Standard Operating Procedure Introduction and Aim As a legal Sponsor
4.2. Investigator Regulatory Binder: Files, usually a binder, maintained by the investigator at the investigative site.
 POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
Implementing Good Clinical Practice at an Academic Research Institution
 Implementing Good Clinical Practice at an Academic Research Institution Maintaining Essential Documents Partners Human Research Quality Improvement (QI) Program Stephen W. Hayes Outline Significance of
Implementing Good Clinical Practice at an Academic Research Institution Maintaining Essential Documents Partners Human Research Quality Improvement (QI) Program Stephen W. Hayes Outline Significance of
Investigator Site File Index (Medical Devices)
") Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol (Clinical Investigation Plan) Current version plus all previous versions (or provide file note
Study Title: Site Name/Number: REC Reference Number: Sponsor Reference Number: EudraCT Number: 1. Protocol (Clinical Investigation Plan) Current version plus all previous versions (or provide file note
Sponsor-Investigator Responsibilities In Clinical Trials
 In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd.
 Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES
 IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016
 UCLA CTSI WELCOME TO ONLINE TRAINING FOR CLINICAL RESEARCH COORDINATORS ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016 Objectives Define the sponsor of a clinical trial and learn
UCLA CTSI WELCOME TO ONLINE TRAINING FOR CLINICAL RESEARCH COORDINATORS ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016 Objectives Define the sponsor of a clinical trial and learn
Study Files and Filing
 Study Files and Filing The current version of all Hillingdon Hospital R&D Guidance Documents and Standard Operating Procedures are available from the R&D Intranet and Internet sites: www.ths.nhs.uk/departments/research/research.htm
Study Files and Filing The current version of all Hillingdon Hospital R&D Guidance Documents and Standard Operating Procedures are available from the R&D Intranet and Internet sites: www.ths.nhs.uk/departments/research/research.htm
SELF-ASSESSMENT CHECKLIST
 SELF-ASSESSMENT CHECKLIST SECTION 1: REGULATORY DOCUMENTATION Staff Documentation 1. Are all versions of the IRB approved protocol on file (including most recent)? 2. Are there CVs/biosketches of PI, Co-Is,
SELF-ASSESSMENT CHECKLIST SECTION 1: REGULATORY DOCUMENTATION Staff Documentation 1. Are all versions of the IRB approved protocol on file (including most recent)? 2. Are there CVs/biosketches of PI, Co-Is,
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices
 Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
ROLE OF THE RESEARCH COORDINATOR Investigational New Drug Application-Sponsor Responsibilities 21CFR Part , subpart D
 Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Demystifying Audits. Audits and Audit Preparation 5/23/2016. What is an Audit?
 Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
and Study Initiation
 10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
and Study Initiation
 10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
Quality Assurance QA STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits
 Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
Quality Assurance QA 601.01 STANDARD OPERATING PROCEDURE FOR FDA or Pharmaceutical Sponsored Audits Approval: Nancy Paris, MS, FACHE President and CEO 24 May 2017 (Signature and Date) Approval: Frederick
Compliance Program Guidance Manuals (CPGMs) -1-
 -1-") Inspector s Preparation for a CI Inspection: FDA Compliance Program & the Records Inventory Jean Toth-Allen, Ph.D. APEC GCP Inspection Workshop May 28, 2008 Compliance Program Guidance Manuals (CPGMs)
Inspector s Preparation for a CI Inspection: FDA Compliance Program & the Records Inventory Jean Toth-Allen, Ph.D. APEC GCP Inspection Workshop May 28, 2008 Compliance Program Guidance Manuals (CPGMs)
Clinical Trials and the Code of Federal Regulations. Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014
 Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol
 IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol Use this checklist to assemble your request for your institution s IBC to review a specific protocol. If WIRB does not already administer
IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol Use this checklist to assemble your request for your institution s IBC to review a specific protocol. If WIRB does not already administer
\\NAS1\George\Docs\SoCRA\CCRP communications\study guide management
 Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
LOUGHBOROUGH UNIVERSITY RESEARCH OFFICE STANDARD OPERATING PROCEDURE. Loughborough University (LU) Research Office
 Research Office") LOUGHBOROUGH UNIVERSITY RESEARCH OFFICE STANDARD OPERATING PROCEDURE Loughborough University (LU) Research Office SOP 1024 LU Study Closedown and End of Study Reporting for NHS Research Sponsored by Loughborough
LOUGHBOROUGH UNIVERSITY RESEARCH OFFICE STANDARD OPERATING PROCEDURE Loughborough University (LU) Research Office SOP 1024 LU Study Closedown and End of Study Reporting for NHS Research Sponsored by Loughborough
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities
 UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities BEVERLY ALGER, CCRP, CHRC Research Compliance Officer Office of Research Compliance November 2016
UC Irvine s Clinical Research Coordinator Certification Preparation Series PI Roles and Responsibilities BEVERLY ALGER, CCRP, CHRC Research Compliance Officer Office of Research Compliance November 2016
FDA Audit Preparation
 Duke University Office of Audit, Risk and Compliance (OARC) FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Associate Compliance Officer, Research Compliance Assurance (RCA) External audits
Duke University Office of Audit, Risk and Compliance (OARC) FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Associate Compliance Officer, Research Compliance Assurance (RCA) External audits
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
ORC Sponsor-Investigator IDE Checklist
 A sponsor-investigator assumes BOTH investigator and sponsor responsibilities as outlined in the FDA Code of Federal Regulations 21CFR812. This means that such investigators have additional responsibilities.
A sponsor-investigator assumes BOTH investigator and sponsor responsibilities as outlined in the FDA Code of Federal Regulations 21CFR812. This means that such investigators have additional responsibilities.
Conducted Under an IND to Support a
 Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Clinical Trials Series Part II
 Clinical Trials Series Part II Agenda Recap December Presentation J. Schmelz Example: New CT from HSC Investigator Non Cancer Clinical Trial J. Bates, P. Miranda Example: New CT from External Entity Non
Clinical Trials Series Part II Agenda Recap December Presentation J. Schmelz Example: New CT from HSC Investigator Non Cancer Clinical Trial J. Bates, P. Miranda Example: New CT from External Entity Non
1 The Clinical Research Coordinator (CRC)... 1
... 1") TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
Objectives Discuss the importance of proper data collection. Identify the types of data collected for clinical trials. List potential source documents
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Introduction to Clinical Research
 Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Investigator-Initiated INDs
 Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Louise Brook Clinical Trials Quality Monitor. Date
 Details: Author: Louise Brook Clinical Trials Quality Monitor SOP Pages: 12 Version No. of replaced SOP: Effective date of replaced SOP: NA NA Approval: Version No: of the SOP being approved. Name of person
Details: Author: Louise Brook Clinical Trials Quality Monitor SOP Pages: 12 Version No. of replaced SOP: Effective date of replaced SOP: NA NA Approval: Version No: of the SOP being approved. Name of person
The Right Prescription for Working with Investigational Drug Service at BMC
 The Right Prescription for Working with Investigational Drug Service at BMC Andrew Schoch Pharmacy Intern Northeastern University Class of 2010 Hyeseon Hong, Pharm.D. IDS Pharmacy Manager What is Investigational
The Right Prescription for Working with Investigational Drug Service at BMC Andrew Schoch Pharmacy Intern Northeastern University Class of 2010 Hyeseon Hong, Pharm.D. IDS Pharmacy Manager What is Investigational
Investigator s Responsibility
 Investigator s Responsibility Introduction Investigator s Qualifications Clinical Trial Agreement Adequate Resources Medical Care of Trial Subjects Communication with IRB/IEC Study Initiation Patient Recruitment
Investigator s Responsibility Introduction Investigator s Qualifications Clinical Trial Agreement Adequate Resources Medical Care of Trial Subjects Communication with IRB/IEC Study Initiation Patient Recruitment
Sponsor/Investigator Responsibilities
 Sponsor/Investigator Responsibilities Marian Serge, RN Division of Bioresearch Monitoring Office of Compliance Center for Devices and Radiological Health Food and Drug Administration Marian.Serge@fda.hhs.gov
Sponsor/Investigator Responsibilities Marian Serge, RN Division of Bioresearch Monitoring Office of Compliance Center for Devices and Radiological Health Food and Drug Administration Marian.Serge@fda.hhs.gov
Preparing for Close-Out of Studies and Sites
 Preparing for Close-Out of Studies and Sites Presented by: Christie Thomas, MPH, CCRA Matthew Wright, BS, CCRP Dikla Blumberg, PhD CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
Preparing for Close-Out of Studies and Sites Presented by: Christie Thomas, MPH, CCRA Matthew Wright, BS, CCRP Dikla Blumberg, PhD CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced
Research Study Close-down and Archiving Procedures
 Title: Research Study Close-down and Archiving Procedures Outcome Statement: To inform researchers of the process for closing down research studies, retaining and storing research materials in the Trust.
Title: Research Study Close-down and Archiving Procedures Outcome Statement: To inform researchers of the process for closing down research studies, retaining and storing research materials in the Trust.
I. Purpose. II. Definitions. Last Approval Date
 Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
DANA-FARBER / HARVARD CANCER CENTER STANDARD OPERATING PROCEDURES FOR CLINICAL RESEARCH
 DANA-FARBER / HARVARD CANCER CENTER STANDARD OPERATING PROCEDURES FOR CLINICAL RESEARCH TITLE: SOP #: QA-706 Page: 1 of 7 Applicable Regulations & Guidelines: 21 CFR 312.60 General Responsibilities of
DANA-FARBER / HARVARD CANCER CENTER STANDARD OPERATING PROCEDURES FOR CLINICAL RESEARCH TITLE: SOP #: QA-706 Page: 1 of 7 Applicable Regulations & Guidelines: 21 CFR 312.60 General Responsibilities of
University of California, Irvine Human Research Protections Standard Operating Policies and Procedures
 University of California, Irvine Human Research Protections Standard Operating Policies and Procedures Policy Number: 41 Title: Investigational Drugs, Agents, and Biologics Date of Last of Revision: 07/28/2006;
University of California, Irvine Human Research Protections Standard Operating Policies and Procedures Policy Number: 41 Title: Investigational Drugs, Agents, and Biologics Date of Last of Revision: 07/28/2006;
FDA Audit Preparation
 Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services
 National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services The Division of Microbiology and Infectious Diseases (DMID), National Institute
National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services The Division of Microbiology and Infectious Diseases (DMID), National Institute
US Special Operations Command Human Research Protection Office
 S- US Special Operations Command Human Research Protection Office Human Research Protocol Submission Form for Headquarters Level Administrative Review of Extramural* Research PURPOSE: All United States
S- US Special Operations Command Human Research Protection Office Human Research Protocol Submission Form for Headquarters Level Administrative Review of Extramural* Research PURPOSE: All United States
Clinical Evaluation Phases 1,2,3,4
 Clinical Evaluation Phases 1,2,3,4 Matt Laurens, MD MPH Associate Professor of Pediatrics Center for Vaccine Development Institute for Global Health University of Maryland School of Medicine February 1,
Clinical Evaluation Phases 1,2,3,4 Matt Laurens, MD MPH Associate Professor of Pediatrics Center for Vaccine Development Institute for Global Health University of Maryland School of Medicine February 1,
Obtaining Pre-Study Approvals for Clinical Trials K-30 Module 5 Navigating the Pre-Award Process
 Obtaining Pre-Study Approvals for Clinical Trials K-30 Module 5 Navigating the Pre-Award Process Helene Orescan, J.D. Bishoy Anastasi, MBA, CCRP David Geffen School of Medicine at UCLA Industry Sponsored
Obtaining Pre-Study Approvals for Clinical Trials K-30 Module 5 Navigating the Pre-Award Process Helene Orescan, J.D. Bishoy Anastasi, MBA, CCRP David Geffen School of Medicine at UCLA Industry Sponsored
STANDARD OPERATING PROCEDURE SOP 410. Set up and Initiation of an Investigator Site
 STANDARD OPERATING PROCEDURE SOP 410 Set up and Initiation of an Investigator Site Version 1.2 Version date 4.01.2017 Effective date 5.07.2017 Number of pages 13 Review date July 2019 Author Role NNUH
STANDARD OPERATING PROCEDURE SOP 410 Set up and Initiation of an Investigator Site Version 1.2 Version date 4.01.2017 Effective date 5.07.2017 Number of pages 13 Review date July 2019 Author Role NNUH
Risk-Based Monitoring: How Can It Be Implemented For More Effective Study Oversight
 Risk-Based Monitoring: How Can It Be Implemented For More Effective Study Oversight Lisa Marie Saldanha Senior Director & Head of Operations Real World Insights Asia Singapore Research & Ethics Conference
Risk-Based Monitoring: How Can It Be Implemented For More Effective Study Oversight Lisa Marie Saldanha Senior Director & Head of Operations Real World Insights Asia Singapore Research & Ethics Conference
GCP INSPECTION FINDINGS Singapore Research Ethics Conference 2018
 1 GCP INSPECTION FINDINGS Singapore Research Ethics Conference 2018 Sumitra Sachidanandan GCP Inspection Consultant Clinical Trials Branch Health Products Regulation Group Health Sciences Authority Singapore
1 GCP INSPECTION FINDINGS Singapore Research Ethics Conference 2018 Sumitra Sachidanandan GCP Inspection Consultant Clinical Trials Branch Health Products Regulation Group Health Sciences Authority Singapore
FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up?
 FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up? The following framework of characteristics focuses on attributes that are within the control of investigators and their
FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up? The following framework of characteristics focuses on attributes that are within the control of investigators and their
OCTC 2012 CRO Selection
 OCTC 2012 CRO Selection Colin Macaulay Viron Therapeutics Inc. 15 Nov 2012 Viron Therapeutics Inc. Virtual Biotech Company (6) Phase 2a (48 pt) clinical trial in acute coronary syndrome (ACS) completed
OCTC 2012 CRO Selection Colin Macaulay Viron Therapeutics Inc. 15 Nov 2012 Viron Therapeutics Inc. Virtual Biotech Company (6) Phase 2a (48 pt) clinical trial in acute coronary syndrome (ACS) completed
Compassionate Use Navigator Information for Physicians
 Compassionate Use Navigator Information for Physicians Contact: Elena Gerasimov, Program Director, Elena@kidsvcancer.org. As a physician, you must have wished there would be more treatment options for
Compassionate Use Navigator Information for Physicians Contact: Elena Gerasimov, Program Director, Elena@kidsvcancer.org. As a physician, you must have wished there would be more treatment options for
Annex IV to guidance for the conduct of good clinical practice inspections sponsor and CRO
 23 August 2017 EMA/431267/2016 Annex IV to guidance for the conduct of good clinical practice inspections sponsor Adopted by GCP Inspectors Working Group (GCP IWG) 29 November 2017 Keywords GCP inspection,
23 August 2017 EMA/431267/2016 Annex IV to guidance for the conduct of good clinical practice inspections sponsor Adopted by GCP Inspectors Working Group (GCP IWG) 29 November 2017 Keywords GCP inspection,
Good Clinical Practice Compliance
 Good Clinical Practice Compliance Pharmaceutical Regulatory & Compliance Congress Greg Levine LLP Agenda? GCP compliance rules What is the law? What other (non-binding) standards apply? What are the unwritten
Good Clinical Practice Compliance Pharmaceutical Regulatory & Compliance Congress Greg Levine LLP Agenda? GCP compliance rules What is the law? What other (non-binding) standards apply? What are the unwritten
Human Research Protection Program Policy
 Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
ClinicalTrials.gov REGISTRATION REQUIREMENTS
 ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
EVENT TYPE TIMING REQUIRED FORM
 Reportable Events EVENT TYPE TIMING REQUIRED FORM Unanticipated Problem or Adverse Event 10 days Event Reporting Form Death (related) 10 days Event Reporting Form Death (not related) Renewal Summary with
Reportable Events EVENT TYPE TIMING REQUIRED FORM Unanticipated Problem or Adverse Event 10 days Event Reporting Form Death (related) 10 days Event Reporting Form Death (not related) Renewal Summary with
Hub Performance Assessment
 Hub Performance Assessment Hub complex performance assessment will be assessed with a standardized report card mechanism. The report card draws on data quality reports, enrollment reports, and monitoring
Hub Performance Assessment Hub complex performance assessment will be assessed with a standardized report card mechanism. The report card draws on data quality reports, enrollment reports, and monitoring
Standard Operating Procedures (SOPs)
") Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Standard Operating Procedure
 Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Managing a Research Team. Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services
 Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Good Clinical Practice. Martin Rose, MD, JD February 8, 2018 ASQ
 Good Clinical Practice Martin Rose, MD, JD February 8, 2018 ASQ Disclaimer The views expressed in this presentation are those of the presenter and do not necessarily represent the official position of
Good Clinical Practice Martin Rose, MD, JD February 8, 2018 ASQ Disclaimer The views expressed in this presentation are those of the presenter and do not necessarily represent the official position of
Trial Master File. SOP No. SOP 7
 Trial Master File SOP Title Trial Master File SOP No. SOP 7 Author Consultation Departments Date approved Julia Farmery Lincolnshire Clinical Research Facility, Research and Development, Trust consultants
Trial Master File SOP Title Trial Master File SOP No. SOP 7 Author Consultation Departments Date approved Julia Farmery Lincolnshire Clinical Research Facility, Research and Development, Trust consultants
Document Title: Site Recruitment and Initiation for Papworth Sponsored Studies
 Document Title: Site Recruitment and Initiation for Papworth Sponsored Studies Document Number: SOP015 Staff involved in development: Job titles only Document author/owner: Directorate: Department: For
Document Title: Site Recruitment and Initiation for Papworth Sponsored Studies Document Number: SOP015 Staff involved in development: Job titles only Document author/owner: Directorate: Department: For
Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success. Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist
 with the FDA: Keys for Success. Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist") Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist Agenda How an investigator should prepare and submit
Maintaining your Investigational Device Exemption (IDE) with the FDA: Keys for Success Jenna Stump, MS, CCRP Clinical Research Regulatory Specialist Agenda How an investigator should prepare and submit
1.0 INTRODUCTION AND OVERVIEW
 Table of Contents 1.0 INTRODUCTION AND OVERVIEW... 3 2.0 DEFINITIONS... 4 3.0 OVERSIGHT AND ORGANIZATION... 5 3.1 CLINICAL RESEARCH OVERSIGHT COMMITTEE (CROC)... 5 3.2 CLINICAL RESEARCH SUPPORT (CRS)...
Table of Contents 1.0 INTRODUCTION AND OVERVIEW... 3 2.0 DEFINITIONS... 4 3.0 OVERSIGHT AND ORGANIZATION... 5 3.1 CLINICAL RESEARCH OVERSIGHT COMMITTEE (CROC)... 5 3.2 CLINICAL RESEARCH SUPPORT (CRS)...
Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective
 1 Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective Kate-Louise Gottfried, JD, MSPH, Senior Director Stephanie C. Guzik, RN, BSN, MBA, Assistant Director Research
1 Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective Kate-Louise Gottfried, JD, MSPH, Senior Director Stephanie C. Guzik, RN, BSN, MBA, Assistant Director Research
Study Start-Up SS STANDARD OPERATING PROCEDURE FOR Investigator Selection
 Effective date of version: April 01, 2012 Study Start-Up SS 202.00 STANDARD OPERATING PROCEDURE FOR Investigator Selection Approval: Nancy M. Paris, MS, FACHE President and CEO 08 March 2012 (Signature
Effective date of version: April 01, 2012 Study Start-Up SS 202.00 STANDARD OPERATING PROCEDURE FOR Investigator Selection Approval: Nancy M. Paris, MS, FACHE President and CEO 08 March 2012 (Signature
Standard Operating Procedures (SOP) Research and Development Office
 Research and Development Office") Standard Operating Procedures (SOP) Research and Development Office Title of SOP: Essential Documentation and Creation and Maintenance of Trial Master File SOP Number: 13 Version Number: 2.0 Supercedes:
Standard Operating Procedures (SOP) Research and Development Office Title of SOP: Essential Documentation and Creation and Maintenance of Trial Master File SOP Number: 13 Version Number: 2.0 Supercedes:
GCP Refresher and GCP/GCDMP Trends. in the CTN. Denise King, MS, RD, CCRA & Lauren Yesko, BS. Presented by:
 GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
Regulatory Visits, Audits and Investigations
 Regulatory Visits, Audits and Investigations The Role of the Compliance Officer Carole A. Klove, JD, RN, CHRC Juliann Tenney, JD, CHRC Welcome! The content of this presentation does not necessarily represent
Regulatory Visits, Audits and Investigations The Role of the Compliance Officer Carole A. Klove, JD, RN, CHRC Juliann Tenney, JD, CHRC Welcome! The content of this presentation does not necessarily represent
Show Me Your Docs! A REVIEW OF UNIVERSITY OF PITTSBURGH GUIDELINE: STUDY DOCUMENTATION FOR FDA REGULATED RESEARCH
 Show Me Your Docs! A REVIEW OF UNIVERSITY OF PITTSBURGH GUIDELINE: STUDY DOCUMENTATION FOR FDA REGULATED RESEARCH Speakers Kelly Dornin-Koss, MPPM, RN, CCRC Director Education and Compliance for Human
Show Me Your Docs! A REVIEW OF UNIVERSITY OF PITTSBURGH GUIDELINE: STUDY DOCUMENTATION FOR FDA REGULATED RESEARCH Speakers Kelly Dornin-Koss, MPPM, RN, CCRC Director Education and Compliance for Human