PHARMAGENE. Optimization of Formulation Variables of Ranolazine Extended-Release Tablets by 3 2 Full Factorial Design
|
|
|
- Peter Nelson
- 5 years ago
- Views:
Transcription
1 ISSN (Print) ISSN (Online) PHARMAGENE Vol: 1 Issue: 2 Research Article Optimization of Formulation Variables of Ranolazine Extended-Release Tablets by 3 2 Full Factorial Design Shah Pranav*, Naik Bhargavi, Zalak Chandarana Maliba Pharmacy College, Bardoli, Gujarat ABSTRACT Ranolazine is antianginal drug, approved by US FDA in It is marketed as extended release tablets (Ranexa 500mg/1gm). Ranolazine is extensively metabolized in the liver and its absorption is highly variable. The present study was aimed to apply experimental design in the development and optimization of drug release from extended release matrix tablets of Ranolazine (antianginal drug) using two factor three level (3 2 ) full factorial design. The extended release matrix tablets of Ranolazine were formulated using ph dependent polymer (Eudragit L ), Sodium hydroxide, MCC, HPMC 5 cps and Magnesium stearate. The amount of independent variables, Eudragit L (X1) and Sodium Hydroxide (X2) were optimized on the basis of drug release profiles at 0.5, 4, 12, 24 hours (dependent variables) of different tablets batches as per 3 2 full factorial design. Tablets were prepared by wet granulation technique and evaluated for various physicochemical parameters and in vitro drug release. Polynomial equations and contour plots derived from the data obtained from 13 batches were used to predict the values of independent variables and their effect on dependent variables for the formulation of optimized tablets with desired properties. Optimized formulation from DOE had identical dissolution profile (f2 = and f1 = 2.29) with innovator s tablet. Stability studies of optimized batch were conducted at accelerated conditions for three months and tablets were found to be stable. Thus the study revealed that experimental design could efficiently be applied for optimization of amount of excipients affecting drug release. Also, it is an economical way of obtaining the maximum amount of information in a short period of time and with the few experiments. KEY WORDS: Ranolazine extended release tablet, Experimental design, Full Factorial Design, Received on Modified on Accepted on INTRODUCTION Oral administration of drugs is strongly preferred because of its convenience, relatively low production cost and the high level of patient safety. However there are some problems associated with the oral drug delivery such as poor bioavailability, high first pass metabolism, frequent drug administration etc. Extended-release systems allow the drug to be released over prolonged time periods. By extending the release profile of a drug, the frequency of dosing can be reduced. Extended release can be achieved using sustained or controlled release dosage forms [1]. *Address for correspondence: Dr. Pranav Shah, Professor, Maliba Pharmacy College, Bardoli, Gujarat, India pranav.shah@utu.ac.in The oral extended release system shows a typical pattern of drug release in which the drug concentration can be maintained in the therapeutic window for a prolonged period of time (extended release), thereby ensuring controlled therapeutic action. Ranolazine is antianginal drug, approved by US FDA in It is marketed as extended release tablets (Ranexa 500mg/1gm). Ranolazine is extensively metabolized in the liver and its absorption is highly variable. The apparent terminal half-life of Ranolazine is 7 hrs. Ranolazine has relatively high solubility (42.08 mg/ml in 0.1 N HCl) at low ph in the stomach (ph 1.2 3). The high acid soluble property of ranolazine results in rapid drug absorption and clearance, causing large and undesirable fluctuations in 1
2 plasma concentration of ranolazine and short duration of action, thus necessitating frequent oral administration for adequate treatment [2]. The present study was aimed to develop a matrix tablet using ph-dependent polymer which is insoluble at low ph and begins to dissolve at about ph > 5. The extended release tablets were formulated using ph dependent polymer (Eudragit L ), ph independent binder (HPMC 5 cps) and sodium hydroxide as neutralizing agent. Sodium hydroxide facilitates the conversion of the Eudragit L into the latex like film formed around the individual granules which controls the drug release from the formulation above ph 4.5 [3]. Statistical experimental design methodologies are powerful, efficient and systematic tools in the design of pharmaceutical dosage forms, allowing a rationalistic study of the influence of formulation and/or processing parameters on the selected responses with short experiment time and improvement in the research and development work [4-6]. The main objective of the experimental design strategies is to plan experiments in order to obtain the maximum information regarding the considered experimental domain with the lowest number of experiments [7]. Moreover, the multi-variant strategy of experimental design enables the simultaneous evaluation of the influence of the different variables involved in any process, being therefore particularly useful when, as in the case of pre-formulation studies, multiple factors have to be evaluated simultaneously. In particular, optimization by means of statistical experimental design methodologies has been successfully applied in the development of different kinds of modified release dosage forms, allowing a quick and efficient quantification and prediction of the effects of formulation changes on the considered crucial responses [8-13]. MATERIALS AND METHODS Materials Ranolazine was purchased from Virdev Intermediates, Surat, India. Eudragit L was obtained from Evonik Industries, Germany. Microcrystalline cellulose was purchased from FMC Biopolymer, India. HPMC 5cps was purchased from Colorcon, Goa, India. Sodium hydroxide was purchased from Cadila Pharmaceutical Ltd., Dholka, India and Magnesium stearate was purchased from Skant Healthcare Ltd., Mumbai, India. Experimental Design Two factor three level (3 2 ) full factorial design was employed for development of ranolazine extended release matrix tablets. A translation of coded values of independent variables and experimental design is executed as in table 1. Independent variables were as follow: X1 : Amount of Eudragit L (mg/tab) X2 : Amount of Sodium hydroxide (mg/tab) Dependent variables evaluated were as follows: Y1 = % drug released in 0.5 hours Y2 = % drug released in 4 hours Y3 = % drug released in 12 hours Y4 = % drug released in 24 hours Preformulation studies Preformulation studies were designed to identify physicochemical properties of Ranolazine and excipients that may influence formulation design and method of manufacture. Table 1: Execution of experimental design and coding of actual values of independent variables for factorial design Batch Level of Factor X1 Level of Factor X2 Amount of Amount of Eudragit L (mg/tablet) Sodium hydroxide (mg/tablet) F1-1(67) -1(2.6) F2 0(83.50) -1(2.6) F3 +1(100) -1(2.6) F4-1(67) 0(3.9) F5 0(83.50) 0(3.9) F6 +1(100) 0(3.9) F7-1(67) +1(5.2) F8 0(83.50) +1(5.2) F9 +1(100) +1(5.2) F10* 0(83.50) 0(3.9) F11* 0(83.50) 0(3.9) F12* 0(83.50) 0(3.9) F13* 0(83.50) 0(3.9) *Centre point batches Analytical method development HPLC method was developed to perform assay and analysis of dissolution samples of Ranolazine. HPLC analysis of samples were done using Phenomenex Luna ODS 250 mm 4.6 mm, 5 microns column with mobile phase flow rate 1.0 ml/min. Mobile phase consist of buffer ph 7.5 and acetonitrile in the ratio of 40:60. Sample and standards were dissolved in mobile phase and detected using UV detector at wavelength 225nm. Fig. 1 shows the HPLC chromatogram of standard ranolazine (100µg/ml) with retention time of minutes. 2
3 Preparation of tablets Tablets were prepared by wet granulation technique. Each batch of tablets (F1 F13) (Table 2) has varied amount of Eudragit L and sodium hydroxide. All ingredients were weighed accurately in required quantity. Ranolazine, Eudragit L , Avicel PH 101 and HPMC 5 cps were sifted through 20# sieve. The materials were mixed in rapid mixer granulator (RMG) at 75 rpm impeller speed. Binder solution was prepared by dissolving sodium hydroxide in sufficient amount of water. Granulation was done in RMG. The wet mass was dried in FBD at 60ºC for 20 minutes and the semi dried mass was passed through 16# and further dried at the same temperature till LOD value below 2% was obtained on moisture balance. The dried granules were passed through 16# sieve. Sized granules were then mixed with previously sifted magnesium stearate (60#) for 3 minutes. The tablets were compressed with mm capsule shaped, standard concave punches with break line on one side and plain on other side (D tooling). Table 2: Formulation of tablets batches Ingredients (mg) Batches F1 F2 F3 F4 F5 F6 F7 F8 F9 F10 F11 F12 F13 Ranolazine* Eudragit L HPMC 5 cps Sodium Hydroxide Magnesium Stearate MCC PH Total tablet weight * The potency of Ranolazine was found to be 506 mg for actual dose of 500 mg/tablet; LOD: 0.29%; Assay: 99.1% Figure 1: Chromatogram of standard Ranolazine (100µg/mL) (Retention time: min) Assay Assay of ranolazine was done using HPLC method. Standard solution was prepared by dissolving 50 mg of Ranolazine standard in 50 ml mobile phase in volumetric flask. Tablet powder equivalent to 50 mg of ranolazine was accurately weighed and transferred into 50 ml volumetric flask containing 25ml mobile phase (Buffer:Acetonitrile; 40:60), sonicated for 30 minutes, allowed to cool to room temperature and diluted upto 50ml volume with mobile phase and mixed. Resulting solution was filtered through 0.45 μm PVDF Millipore filter discarding first few ml of the filtrate. 5.0 ml of clear filtrate was diluted to 50.0 ml with mobile phase and mixed. 20µL of sample and standard preparation were injected into the column. Chromatogram was recorded and the response was measured at 225nm. (Figure: 1) Content of ranolazine per tablet was calculated. In vitro drug release study (In vitro dissolution study) [14] In vitro drug release study was performed as per the following specifications of OGD, 900 ml of 0.1 N HCl medium at 50 rpm for 24 hours at 37±0.5 C. A 10 ml of sample from dissolution medium was withdrawn at predetermined time intervals (0.5, 2, 4, 8, 12, 20, 24 hours) and replaced by an equal volume of dissolution medium. The samples were filtered through 0.45µm whatman filter paper and 5.0 ml of filtrate was diluted to 20.0ml with dissolution medium. Samples were analyzed using HPLC. Mechanism of drug release [15-17] To evaluate the mechanism of drug release from the dosage form, data for the first 60% of drug release were plotted in Korsmeyer s equation as log cumulative percentage of drug 3

4 released vs log time, and the exponent n was calculated from the slope of the straight line. / = Where, Mt/M is the fractional solute release, t is the release time, K is a kinetic constant characteristic of the drug/ polymer system, and n is an exponent that characterizes the mechanism of release. Stability Study RESULTS AND DISCUSSION Preformulation studies Ranolazine was found to have very poor compressibility and flow properties (Carr s index: 36.67, Hausner s ratio: ), hence wet granulation method was opted for better compression and good flow property for the preparation of the Ranolazine matrix tablet. The optimized formulation was subjected to short term accelerated stability study (40ºC/75% RH) for the period of three months as per ICH guidelines. Physical stability was analyzed by recording the change in appearance, hardness, friability and chemical stability was analyzed by the change in the assay and in vitro drug dissolution at the end of three months. Precompression evaluation of granules exhibited good flow property (Hausner s ratio < 1.25) and good compressibility (Carr s index < 20%). Post compression evaluation of tablets such as appearance, dimensions, weight variation, hardness, friability, and assay were within the specifications (Table 3). Table 3: In process quality control of Tablets Batch Average tablet weight (mg) n = 20 Hardness (kg/cm 2 ) n = 10 Length (mm) Dimensions n = 6 % Friability Assay n = 5 Width (mm) Thickness (mm) F ± ± ± ± ± ±0.10 F ± ± ± ± ± ±0.20 F ± ± ± ± ± ±0.23 F ± ± ± ± ± ±0.33 F ± ± ± ± ± ±0.19 F ± ± ± ± ± ±0.22 F ± ± ± ± ± ±0.30 F ± ± ± ± ± ±0.36 F ± ± ± ± ± ±0.20 F ± ± ± ± ± ±0.33 F ± ± ± ± ± ±0.19 F ± ± ± ± ± ±0.22 F ± ± ± ± ± ±0.30 In vitro drug release study (In vitro dissolution) In vitro drug release data (Figure 2) showed that the drug release from all the formulated batches (F1-F13) (n = 3) was extended upto 24 hours. Figure 2: Comparative % cumulative drug release profile of formulations (F1 F13) 4
5 (a) (c) (b) (d) Figure 3: Linear correlation plots of dependent variables Y1-Y4 (a-d) between actual and predicted value Drug Release kinetics Dissolution data were fitted to zero order, first order, Higuchi and Korsmeyer kinetic treatment for all the formulations and different kinetic equations were applied to interpret the release rate. The formulation with higher correlation coefficient R 2 was found with Higuchi s Model as shown in Table 4 indicating that release from gel forming system is based on diffusion mechanism for all formulations. The value of release exponent was more than 0.45 and less than indicating non-fickian anomalous release from all the formulations except F1. Statistical Analysis The two factor three level full factorial design allowed the development of mathematical equations, where predicted results (Y) were assessed as a function of amount of Eudragit (X1) and amount of Sodium hydroxide (X2) and calculated as the sum of a constant, two first-order effects (terms in X1 and X2), one interaction effect (X12) and two second-order effects (X1 2 and X2 2 ). The relationship between the two independent variables (amount of Eudragit L and sodium hydroxide) and the four dependent variables (% drug release at 0.5, 4, 12 and 24 hour) were analyzed using response surface methodology Data given in Table 5 depicts that all the models were significant at the 5% confidence level since P values were less than The large P values for lack of fit (>0.05) presented in Table 5 (PLOF) show that the F-statistic was insignificant, implying significant model correlation between the variables and process responses. Adequate Precision (AP) values higher than four (Table 5) for all the responses confirmed that all predicted models can be used to navigate the design space defined by the full factorial design. For all the models % CV were not greater than 10% (Table 5) which indicate that models are reproducible. Model equation for the all the variables showed the negative co-efficient terms for the first order effect, interaction term and second order effects which indicated the negative effect on the response with respect to the independent variable. Contour Plots and Response Surface Analysis Figure 4, 5, 6 and 7 (a and b) represents the response surface plot and contour plot of dependent variables Y1, Y2, 5
6 Y3 and Y4 respectively. For responses Y1, Y2, Y3 and Y4 drug release decreases rapidly with increase in amount of one variable while other at low level. This showed that both the variables (Eudragit L and sodium hydroxide) had prominent negative effect on Y1, Y2, Y3 and Y4. These results are in confirmation with mechanism. As the concentration of ph dependent binder increases in the formulation, there is decrease in the release rate of ranolazine at ph below 4.5 as enteric coating formed by the binder was less soluble in acidic ph. Partial neutralizing agent, sodium hydroxide facilitated the conversion of the binder into the latex like film formed around the individual granules which controled the drug release from the formulation above ph 4.5. Table 4: Release kinetic data for F1-F13 formulations Batch no. Zero order First order Higuchi Korsmeyer Peppas kinetic R 2 kinetic R 2 kinetic R 2 R 2 n F F F F F F F F F F F F F Table 5: ANOVA analysis of data ANOVA results for dependent variables Y Mathematical model Y1 Y1= X X X X X 2 Y2 Y2= X X X X X 2 Y3 Y3= X X X X Y4 Y4= X X X X X 2 P value PLOF R 2 Adjusted R 2 Predicted AP S.D CV R 2 % PRES S < < <
 for response Y1 Figure 6: Response")

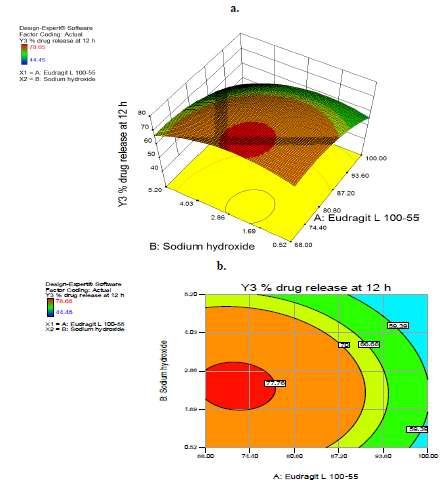
7 Figure 4: Response surface plot (a) and Contour plot (b) for response Y1 Figure 6: Response surface plot (a) and Contour plot (b) for response Y4 Figure 5: Response surface plot (a) and Contour plot (b) for response Y2 Figure 7: Response surface plot (a) and Contour plot (b) for response Y3 7
. The optimized batch F14 contains: Amount of Eudragit L 100-55 (X2) : 80 mg/tab Amount of NaOH : 3.")
8 Optimization After generating the model polynomial equations to relate the dependant and independent variables, the process was optimized for all four responses. The final optimal experimental parameters were calculated using Design- Expert V8 (8.071). The optimized batch F14 contains: Amount of Eudragit L (X2) : 80 mg/tab Amount of NaOH : 3.25 mg/tab Dissolution profile of Ranexa (innovator s formulation) and optimized formulation F14 were compared using the FDA recommended similarity factor (f2) (figure 8). The value of f2 was found to be which was above the critical value (50) indicating an equivalence to the release profile of the optimum formulation and the innovator profile. Validation of Response Surface Methodology Five check point batches were formulated for the validation of response surface methodology. Actual experimental responses and predicted responses were then compared to validate design (Table 6). For all the 5 checkpoint formulations, the results of the dependent variables were found to be within limits. For validation of RSM results, the experimental values of the responses were compared with the anticipated values and the prediction error was found to vary between and These results demonstrate the reliability of the optimization procedure in predicting the effect of process variables on the dissolution behavior of the ranolazine extended release tablet profile. Table 6: Composition of check point batches and comparison of experimental and predicted values of response variables Figure 8: Dissolution profile comparison of optimized batch and innovator s product Check point Formulations X1 X Respon se Variabl es Experi mental values Predict ed values % predicti on Error Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Y Figure 9: Overlay plot of optimized batch Stability study Stability study of the optimized formulation proved the physical and chemical integrity of the developed ranolazine extended release matrix tablet with no significant change in the assay and dissolution profile. CONCLUSION Ranolazine extended release tablets were manufactured by wet granulation technique. The tablets exhibited drug release for a period of 24 hours and followed Higuchi kinetics. The amount of Eudragit L and Sodium hydroxide was optimized by 3 2 full factorial design based on the drug release. The contour plots represented the influence of the amount of the independent variable on the 8
9 drug release. The design was also validated by the check point batches. The optimized formulation exhibited drug release similar to the innovator (f2= 85.95). The accelerated stability studies suggested no significant change in the drug content, physical properties and drug release. REFERENCES 1. Perrie Y, Rades T. FASTtrack-Pharmaceutics-: Drug Delivery and Targeting: Pharmaceutical Press; Goodman LS. Goodman and Gilman's the pharmacological basis of therapeutics: Pergamon Press New York; Andrew A. Wolf, Sustained Release Ranolazine Formulation, United States Patent B2, 2005 March Lewis GA, Mathieu D, Phan RTL. Pharmaceutical experimental design: CRC Press; Schwartz JB, O'Connor RE. Optimization techniques in pharmaceutical formulation and processing. DRUGS AND THE PHARMACEUTICAL SCIENCES. 1996;72: Gabrielsson J, Lindberg NO, Lundstedt T. Multivariate methods in pharmaceutical applications. Journal of chemometrics. 2002;6(3): Lundstedt T, Seifert E, Abramo L, Thelin B, Nyström Å, Pettersen J, et al. Experimental design and optimization. Chemometrics and Intelligent Laboratory Systems. 1998;42(1): Renoux R, Demazieres J, Cardot J, Aiache J. Experimentally designed optimization of direct compression tablets. Drug development and industrial pharmacy. 1996;22(2): Sastry SV, Reddy IK, Khan MA. Atenolol gastrointestinal therapeutic system: optimization of formulation variables using response surface methodology. Journal of controlled release. 1997;45(2): Takahara J, Takayama K, Nagai T. Multi-objective simultaneous optimization technique based on an artificial neural network in sustained release formulations. Journal of controlled release. 1997;49(1): Geoffroy J-M, Fredrickson JK, Shelton JT. A mixture experiment approach for controlling the dissolution rate of a sustained-release tablet. Drug development and industrial pharmacy. 1998;24(9): Hamed E, Sakr A. Application of multiple response optimization technique to extended release formulations design. Journal of controlled release. 2001;73(2): Kramar A, Turk S, Vrečer F. Statistical optimisation of diclofenac sustained release pellets coated with polymethacrylic films. International journal of pharmaceutics. 2003;256(1): ion/index.cmf 15. Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. European journal of pharmaceutical sciences. 2001;13(2): Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. International Journal of Pharmaceutics. 1983;15(1): Korsmeyer R, Peppas N. Solute and penetrant diffusion in swellable polymers. III. Drug release from glassy poly (HEMA-co-NVP) copolymers. J Controlled Release. 1984;1(2):