Functional Annotation - Faction 2 Background and Strategy
|
|
|
- Regina Webster
- 5 years ago
- Views:
Transcription

1 Functional Annotation - Faction 2 Background and Strategy March 8, 2017 Khushbu Patel Karan Kapuria Angela Mo Harrison Kim David Lu Christian Colon Nolan English Bowen Yang Cong Gao

2 RECAP. WE ARE HERE!!
3 Outline Objectives Review: Functional Annotation Tools Protein-Coding Non Protein-Coding Ontology Highly Automated Pipelines Annotation Comparisons Proposed Workflow
4 Objectives Annotation of Salmonella heidelberg sporadic genomes using the information obtained by the gene prediction group. Test individual tools for specific prediction and compare them with results of fully automated pipelines. Compilation of relevant annotation results into a refined GFF file for use by the comparative genomics group.
5 Functional Annotation Annotation may be defined as the part of genome analysis that is customarily performed before a genome sequence is deposited in GenBank and described in a published paper. The unit of genome annotation is the description of an individual gene and its protein (or RNA) product, and the focal point of each such record is the function assigned to the gene product.


6 Features to be annotated Protein-coding: TransMembrane, Signal Peptides, Domains and Motifs Non protein-coding: CRISPRs, ribosomes, ncrnas, trna Ontology: Pathways Operons Plasmids potentially distinguishing isolates
7 Tools Selected for Evaluation 1. Protein-Coding Tools
8 InterProScan Classification of proteins with signatures based on 14 InterPro databases Latest release 61.0: 29,774 entries
9
10 Advantages: Well-curated, wide selection of databases Compaction reduces redundancy while emphasizing strengths Database regularly updated every ~8 weeks Disadvantages: Results are completely dependent on the quality of the database Scanning through large volume of databases takes time
11 TMHMM Detection of transmembrane helices in proteins based on a hidden Markov model Combines information on hydrophobicity, charge bias, helix lengths, and grammatical constraints into one model

12 Advantages: Web and standalone version Accuracy: 97-98% of transmembrane helices Distinguish soluble and membrane proteins Disadvantages: Decreased accuracy with presence of signal peptides due to hydrophobic region, though not as inaccurate for Gram-negative bacteria
13 SignalP 4.1 Characterizes signal peptides based on two neural networks Negative training sets: Transmembrane-containing dataset and non transmembrane-containing dataset If transmembrane-trained network predicts 4+ transmembrane proteins, it will be used for final prediction
14 Advantages: More recently developed and updated Performed the best for Gram-negative bacteria compared with other tools Disadvantages: Sometimes doesn't perform as well as other tools when no transmembrane proteins present
15 Comparison of performance
16 OperonDB - - Many of the gene in bacteria are organized in operons. Component: Promoter, Operator, Structural gene and Regulatory gene. A database containing the results of a computational algorithm for locating operon in microbial genome. The algorithm is based on the concept of conserved gene pairs, and its probability of both gene belong in the same operon. ( gene pair is defined as genes that is adjacent to each other) Database size: 1059 bacterial and archaeal genomes.
17 DOOR2 - - Database of Prokaryotic Operons. Developed by computational system biology lab in University of Georgia A open database include the predictions of algorithm (data mining classifier). Algorithm feature: Intergenic distance, Neighborhood conservation, Phylogenetic distance, Information from short DNA motifs, Similarity score between GO terms of gene pairs, and Length ratio between a pair of genes. Database size: 2072 prokaryotic genomes. No command line tools. The process will have to be implemented using blast+ from NCBI.
18 Tools Selected for Evaluation 2. Non Protein-Coding Tools
.")
19 RNAcon It uses SVM-based model for the discrimination between coding and ncrnas and RandomForest-based prediction model for the classification of ncrnas into different classes. Prediction step: Discriminate coding or non-coding transcripts and thereafter classify ncrnas into respective classes by SVM using a single feature (tri-nucleotide composition). Classification step: Graph properties based feature set along with RandomForest algorithm to classify different ncrna classes.
20 Pros: 1.Much quicker and computationally less expensive 2.Provides predicted structure of ncrna and classification Cons: Machine learning algorithm dependents on many factors Prediction accuracy is only as good as the learning testing data Over Optimization issues
21 Rfam Database containing information on noncoding RNA families Divides ncrnas into families based on evolution from a common ancestor Uses multiple sequence alignments specifically to study this homology Users can view and download multiple sequence alignments for each ncrna family Stochastic context-free grammar (SCFG) used as probabilistic model INFERNAL software (developed by Eddy Lab) implementation Precaution: Search time is long, so BLAST is used to reduce the search space to speed things up. ( Rfam, 2016)
22 Cmsearch (Infernal) Finds more ncrna homologs in multiple sequence alignments Searches a covariance model against a sequence database Uses Cmbuild to build a covariance model Cmsearch will take the cmfile as the query and a database of your choosing Takes in an alignment file as input and outputs cmfile for the search step European Nucleotide Archive is a good starting point Align sequences with good scores greater than set threshold Align sequences back to the model which are now used to identify true homologs
23 PILER-CR Designed to identify the characteristic signature of CRISPR repeats Finds a chain of local alignments where the repeats and spacers are within the expected ranges of length and contain sequence conservation Implementation: a. b. c. d. e. f. g. Find local alignments of the genome to itself Constructs piles (set of bases which cover at least one local alignment) Graph construction Draft array identification Array refinement Adjacent array merge Clustering and alignment
24 CRISPR Recognition tool (CRT) Search for exact k-mer match separated by similar distance using small sliding window. (left to write) If exact match is found, it extends left and right to find the right repeat length (or words). If no exact match is found, the sliding window advances to the right to search for the next word. Filter out the unwanted repetitive region Check the right and left flank of CRISPR for missed repetitive sequence due to the interval calculation or mutation of last few repeats. Fast. RAM efficient.
25 RNAmmer - rrna is highly conserved in both sequential level and structural level due to ribosome s dependence on ribosomal RNA. Computational predictor for major rrna species across all kingdoms Based on hidden Markov model (HMM) trained by 5S ribosomal RNA database and European ribosomal RNA database project. A pre-screening procedure speeds up the process at the cost of little sensitivity.

26 trnascan-se Identifies % of transfer RNA genes in DNA sequence while giving less than one false positive per 15 gigabases Input consists of DNA or RNA sequences in FASTA format trnascan-se does no trna detection itself, but instead negotiates the flow of information between three independent trna prediction programs, performs some post-processing and outputs the results trnascan-se works in THREE phases: Analysis of Accuracy of Sub-Tools with trnascan-se
27 Tools Selected for Pathway/Notation 3. Gene Ontology (GO)
28 PANTHER Input: Gene List Output: GO Cellular Component Biological Processes Molecular Function Gene Ontology Consortium BioModels Database
, with functional categories (i.")
29 EggNOG EggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups) is a database of orthologous groups of genes. The orthologous groups are annotated with functional description lines (derived by identifying a common denominator for the genes based on their various annotations), with functional categories (i.e derived from the original COG categories). Two Parts: Functional annotation Phylogenetic reconstruction
30
31 Tools Selected for Evaluation 4. Highly Automated Pipelines
32 Prokka a command line software tool. Input: a set of scaffold sequences (in fasta format) produced by de novo assembly software. Advantage: High speed (a typical bacterial genome can be annotated in 10 min on a quad core desktop computer). Prokka compares genes with database of known sequences, but in a hierarchical manner, starting with a smaller trustworthy database, moving to medium-sized but domain-specific databases, and finally to curated models of protein families. Prokka uses BLAST+ to find similarity searching against protein sequence libraries. Coding sequences (CDS) can also be searched against user-provided annotations. hmmscan from HMMER 3.1 package is used to search sequences against profile databases, including Pfam and TIGRFAMs.

33 Prokka Pipeline
34 Description of Prokka output files Suffix Description of file contents.fna FASTA file of original input contigs (nucleotide).faa FASTA file of translated coding genes (protein).ffn FASTA file of all genomic features (nucleotide).fsa Contig sequences for submission (nucleotide).tbl Feature table for submission.sqn Sequin editable file for submission.gbk Genbank file containing sequences and annotations.gff GFF v3 file containing sequences and annotations.log Log file of Prokka processing output.txt Annotation summary statistics
35 ConsPred prokaryotic genome annotation framework. Workflow of annotation includes: prediction of protein-coding genes prediction of non protein-coding elements functional annotation of proteins prediction of conserved motifs mapping of protein-coding genes onto eggnog functional categories mapping of protein-coding genes onto KEGG metabolic pathways integration of the various types of annotations Predicts coding sequences (CDS) by the ab initio CDS prediction and the homology-based prediction. Prodigal, GeneMark, Critica and Glimmer are used for ab initio CDS prediction. BLAST is used for homology-based CDS prediction.
36 Non-protein-coding elements (NCEs) are predicted, using the prediction tools RNAmmer, trnascan-se, PilerCR and Infernal. For annotating the gene name, protein product and EC number, the CDS are compared to the manually curated UniProt/SwissProt database. Hits with e-value better or equal to 1.0e-5 and a minimal coverage of both query and subject by the alignment of 70% are used for annotation transfer. Conserved domains are annotated by comparing CDS to the profiles in the InterPro signature databases using InterProScan. CDS are also compared to the Kegg and eggnog databases and assignments to Kegg KOs, Kegg EC numbers, Kegg pathways and eggnog functional categories are exported. Can be run locally as well as on cloud: ConsPred annotation framework (Amazon Machine Image (AMI)). Output file formats:.gff, csv, gbk,.embl

37 DeNoGAP: De-Novo Genome Analysis Pipeline Shalabh Thakur and David S. Guttman (Last Update: December 2016) Written in Perl, BioPerl and SQLite on Ubuntu Linux version LTS This software package accompanies script for automated installation of necessary external programs on Ubuntu Linux The pipeline performs four primary analysis tasks: 1. Gene prediction 2. Functional annotation 3. Ortholog prediction 4. Pan-genome analysis.

38

39 PredictProtein An meta-service that searches up-to-date public sequence databases, creates alignments, and predicts aspects of protein structure and function. Input: protein sequence Output: MSAs, prediction of protein structural and functional features. Multiple sequence alignments, predicted aspects of structure (secondary structure, solvent accessibility, transmembrane helices (TMSEG) and strands, coiled-coil regions, disulfide bonds and disordered regions) and function

40 PredictProtein secondary structure, solvent accessibility, transmembrane helices, globular regions, coiled-coil regions, structural switch regions, B-values, disorder regions, intra-residue contacts, protein-protein binding sites protein-dna binding sites, sub-cellular localization, domain boundaries, beta-barrels, cysteine bonds, metal binding sites disulphide bridges.

41 RAST Fully automated annotation and assign functions to genes, built upon the framework provided by the SEED system. Offers both web-based version and standalone version(myrast). RASTtk- allows to build custom annotation pipelines(gene calling algorithms, annotation scripts, and output. Identifies protein-encoding, rrna, trna genes and seeks to produce an initial metabolic reconstruction. However, it takes about a single day after query file submission And limited throughput per day.
42 Annotation Comparison
")
43 Software tool that compares annotations of a particular genome from different Annotation Methods (AMs). Written in C++. Published in 2015 INPUT : GenBank format Consider two annotations X and Z Genes are considered IDENTICAL when.. Genes are considered UNIQUE when.. All other genes overlapping to some extent are divided into SIMILAR and UNIQUE based on a user-defined threshold (eg. 2%)


44 BEACON: Sample Results
45 Proposed Workflow
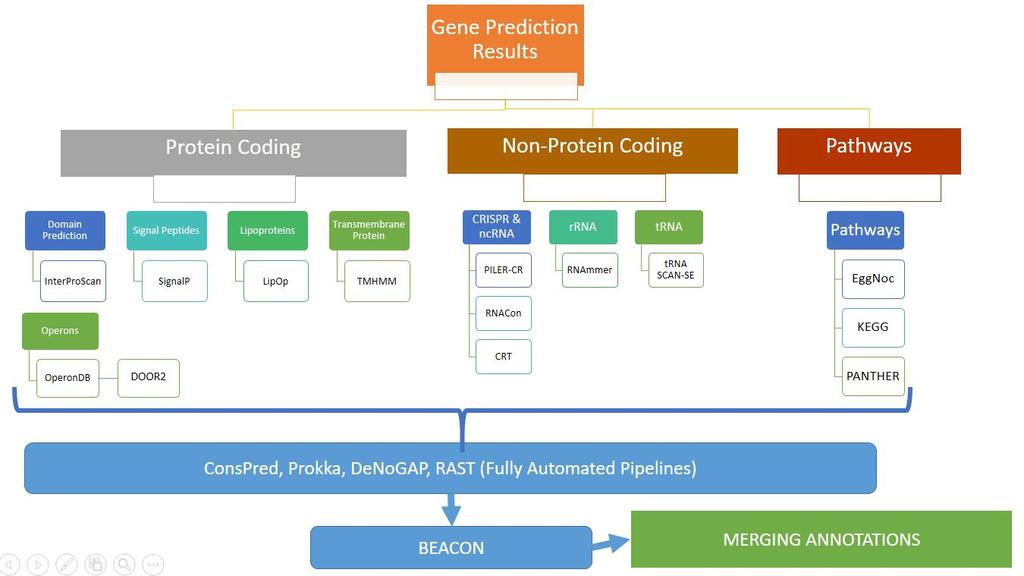
46
47 References Edgar, R.C. (2007) PILER-CR: fast and accurate identification of CRISPR repeats, BMC Bioinformatics, Jan 20;8:18. Torsten Seemann; Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30 (14): doi: /bioinformatics/btu153 Wikipedia contributors. "Rfam." Wikipedia, The Free Encyclopedia. Wikipedia, The Free Encyclopedia, 12 Nov Web. 8 Mar Thakur, Shalabh and David S. Guttman. "A De-Novo Genome Analysis Pipeline (DeNoGAP) For Large-Scale Comparative Prokaryotic Genomics Studies". BMC Bioinformatics 17.1 (2016): n. pag. Web. Guy Yachdav, Edda Kloppmann et al. PredictProtein an open resource for online prediction of protein structural and functional features. Nucleic Acids Research, 2014, Vol. 42. W337 W343 Ross Overbeek, Robert Olson et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Research, 2014, Vol. 42, Database issue. D206 D214. Lowe, T. "Trnascan-SE: A Program For Improved Detection Of Transfer RNA Genes In Genomic Sequence". Nucleic Acids Research 25.5 (1997): Web. Petersen, TN et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods Sep 29;8(10): doi: /nmeth Krogh, A et al. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J.Mol.Biol. (2001) 305, 567±580 Finn, R et al. InterPro in 2017 beyond protein family and domain annotations. Nucleic Acids Research, Jan 2017; doi: /nar/gkw1107