Protocols for cell lines using CRISPR/CAS
|
|
|
- Isabel Blankenship
- 5 years ago
- Views:
Transcription

1 Protocols for cell lines using CRISPR/CAS Procedure overview Map
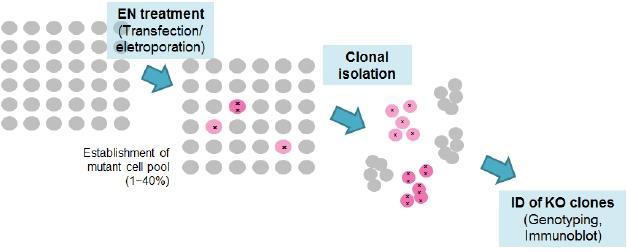
2 Preparation of CRISPR/CAS plasmids Expression vectors for guide RNA (U6-gRNA) and Cas9 gene (CMV-p-Cas9) are ampicillin-resist ant and stable in general E.coli strains such asdh5a or XL1. Surroate reporter vectors are knnamycin resistant. The yield of U6-gRNA plasmids purification could be lower than usual practice. Use less volume of suspension/elution buffers. Plasmid re-transformation: 1. Thaw 30 ul of E.coli DH5α on ice, add 1-50 ng of plasmid, vortex for 1 sec, incubate on ice for 15 min, heat shock at 42 for 45 sec, put it on ice for cooling down, add 1 ml of LB medium and shake at 37 for 1 hr. Dilute 50 ul of liquid culture in 150 ul of LB medium and plate it onto a LB agar plate with antibiotics, incubate at 37 for hr. 2. Mini-culture and preparation: pick colonies by 20ul-tip and put it into 2 ml of LB medium with 100 ug/ml antibiotics (for kanamycin: 50 ug/ml), incubate at 37 for hr. Mini-preparation protocol: EasyPrep Plasmid Mini kit, TOOLS, cat. no. DPT-BA03 3. Digest plasmids with restriction enzyme and run 1% agarose TBE gel to confirm the size of plasmids. 4. Maxi-culture and preparation: dilute 100X mini-culture in LB with antibiotics, incubate at 37 for hr. Maxi-preparation protocol: EasyPrep EndoFree Maxi Plasmid Extraction kit, TOOLS, cat. no. DPT-BA17 5. Determine the purity and amounts of plasmids by using NanoVue Spectrophotometer, dilute plasmids to a concentration of 0.5 ug/ul. 6. Repeat step 3 to confirm the correct plasmids. 7. Filtered the plasmids by 0.22 um filter in laminar flow, store the plasmids at -20. Transfer CRISPR/CAS to target cells Cell culture: 1. Thaw cell: thaw cells in water bath at 37, centrifuge at 300 xg for 1 min, resuspend cells in 10 ml of growth medium and plate in 10-cm dish 2. After 2-3 days, save 1 ml of medium for mycoplasma detection (protocol: EZ-PCR Mycoplasma Test Kit, Biological Industries, cat.# ) 3. One day before transfection, plate cells in 6 well-tc plates at % confluence in 2 ml growth medium w/o antibiotics. Cells will be 70-90% confluence at the time of transfection. 4. Transfect plasmids by using Lipofectamine LTX & PLUS Reagent (Life Technologies, cat.# ). Briefly, dilute 10ul of LTX Reagent in 100 ul Opti-MEM Medium, dilute 2.5ug of plasmid DNA (Cas9 : sgrna : surrogate = 1:1:0.5) in 100 ul Opti-MEM Medium, then add 2.5 ul Plus Reagent in diluted DNA, mix gently, combine LTX Reagent and diluted DNA, incubate at RT for 5 min. Replace with 1.8 ml fresh growth medium w/o antibiotics to cells, add DNA-reagent complex. Incubate at 37 for 48 hr. Remove the complex and replace medium at 48h. 5. Monitor the transfection efficiency (RFP+ population) and the activity of CRISPR (RFP+/ GFP+ population) at ~48 post-transfection by fluorescence microscopy 6. At 36~72h post-transfection, trypsinize cell population and plate APPROPRIATE number of cells into 10cm dish with APPROPRIATE hygromycin concentration. 1. APPROPRIATE number of cells: Each cell line has different colony forming efficiency. Appropriate number of cells to be plated should be determined empirically.
3 Recommended: Make multiple plates expected to produce 500/5000/50000/ (possibly more) each before selection. 2. APPROPRIATE hygromycin concentration: Each cell line has different hygromycin sensitivity. Appropriate hygromycin concentration that will kill 99% of cell population within 48 hours should be determined empirically. For 293T and HeLa cells, 2mg/ml of hygromycin was used. 3. Save part of transfected cells and prepare genomic DNA. Monitor if mutations we re introduced by CRISPR expression in your cell by genotyping assays like T7E1 assay hours after hygromycin selection, wash cells once and change media to a fresh me dia with no antibiotics. 8. Wait 2~4 weeks until monoclonal colonies are formed with occasional media change or addition. 9. Chose a plate with 10~200 colonies (if possible) to pick 20~100 colonies and move to 96- or 48-well plate. 10. Grow up each clones and identify mutant/ko cell colonies by genotyping and western b lot. Any DNA delivery method (transfection, electroporation) optimal for your cell line can be used for the delivery of CRISPR The recommended ratio of guide RNA expression plasmids and Cas9 gene expression plasmids is 1:1 ~ 5:1. (without surrogate reporter) 2~3 days after CRISPR treatment, plate appropriate density of cells to isolate monoclonal cell colonies The colony formation efficiency could vary among cell lines. Thus, optimal density of cell population needs to be determined empirically. Dish method : plate 50, 250, 1000, and 5000 cells / 10cm dish (2 plate / cell population) Limiting dilution method: plate 0.4 cell / well of 96 well plate (2~3 plate) Save part of the cell population treated with CRISPR and confirms the efficient mutation induction by CRISPR in your target cell by a T7E1 assay.

4 Validation of CRISPR activity Target PCR condition setup Detailed procedure of PCR condition setup and mutation detection assay can be found in the T7E1 assay protocol (separate file) The purpose of this step is to confirm and quantify the activity of CRISPR in cell. It also helps to have this step to practice and setup the T7E1 assay and other mutation analysis assay which are broadly used in the process of gene modification in cell lines. Common cell lines that have well-established DNA delivery protocol (such as 293T and HeLa) are good starting to validate the activity of CRISPR. 1. Day 1. Transfect CRISPR into target cell. The ratio of guide RNA expression plasmids and Cas9 expression plasmids can be 1:1 ~5:1 2. Day 3~4. Prepare genomic DNA from transfected cells. 3. Analyze the mutation at target locus by a T7E1 assay ~20 days after plating, isolate and expand monoclonal cell colonies (50~100 colonies are recommended) 5. Prepare genomic DNA from each clone at 48 well plate ~ 12 well plate. 6. Identify the knockout cell clones by genotyping and immunoblot analysis. Most mutation induced by CRISPR at target site is small deletions and insertions (-20bp~+10bp). When these mutations are causing the frameshift, it will function as knockout mutation Cell lines usually contains more than 2 alleles (polyploidy)
5 The complete knockout cell lines will have frameshift mutation on all alleles of target gene. These knockout cell lines can be identified by analyzing the mutation (genotyping) or/and by analyzing target gene expression Suggested: Fluorescence PCR (F-PCR) F-PCR is performed simply by amplifying target locus using fluorescence-labeled primer. The amplicons size for F-PCR is recommended to be smaller than 400bp to have good resolution. Most capillary sequencing service company provides the fluorescence-labeled DNA size analysis service. Because F-PCR result will show the size of all alleles in a cell population, this analysis identifies mutant cells with target modification (size change) in all alleles. Note that the size from the F-PCR analysis could vary by ~1bp. Fluorescence PCR: using 5 -carboxyfluorescein-labeled primers and ug of genomic DNA as templates, PCR protocol: High Fidelity PCR Enzyme Mix, TOOLS 2x Super Hi-Fi tag mastermix KTT-BB cycles. Analyze PCR products by using an ABI 3730xl DNA analyzer. The positions of peaks indicate the lengths of PCR products.
 TA ligation: TOOLS Zback Faster Ligation Kit TGVTB04-2 3) Transformation: thaw 50-100 ul of E.")
6 TA-cloning and sequencing: 1) Target site PCR amplification: use ug of genomic DNA as templates, PCR protocol: High Fidelity PCR Enzyme Mix, TOOLS 2x Super Hi-Fi tag mastermix KTT-BB cycles. Clean up PCR products and run 1-2% agarose TBE gel. 2) TA ligation: TOOLS Zback Faster Ligation Kit TGVTB04-2 3) Transformation: thaw ul of E.coli DH5α on ice, add all TA ligation product, vortex for 1 sec, incubate on ice for 15 min, heat shock at 42 for 45 sec, put it on ice for cooling down, add 1 ml of LB medium and shake at 37 for 1 hr. Plate 100 ul of liquid culture onto LB LB/ampicillin/IPTG/X-Gal plates. Incubate at 37 for hr until white colonies growth. 4) Mini-culture and preparation: pick white colonies by 20ul-tip and put it into 2 ml of LB medium with 100 ug/ml antibiotics, incubate at 37 for hr. Mini-preparation protocol: EasyPrep Plasmid Mini kit, TOOLS, cat. no. DPT-BA03 5) Restriction enzyme digestion: Digest plasmids with restriction enzyme and run 1% agarose TBE gel to confirm insert fragments. 6) Determine the purity and amounts of plasmids by using NanoVue Spectrophotometer, send plasmid to sequencing. 7) analyze sequences by pairwise aligment with wt sequence. Immunoblot assay: Most direct way to confirm the gene knockout will be to determine the loss of target gene expression at protein level by immunoblot assay.
7
8 Enrichment of Knockout Cells induced by RGENs using Hygromycin-surrogate reporter References Magnetic separation and antibiotics selection enable enrichment of cells with ZFN/TALEN-induced mutations. (PLoS One. 2013;8(2):e56476) Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. (Nat Commun Feb 26;5:3378) Protocol 1. Transfer drgen plasmids (prgen-sgrna-u6 and prgen-cas9-cmv) into your cell line together with appropriate phrs plasmids (hygromycin-surrogate reporter) A. Any DNA delivery methods efficient for your cell line is compatible. Generally speaking, electroporation methods seems to be efficient for wide variety of cell lines. B. The recommended ratio of DNA mixture to be delivered is 2:2:1~5:5:1 (prgen-sgrna- U6 : prgen-cas9-cmv : phrs). 2. Monitor the transfection efficiency (RFP+ population) and the activity of RGENs (RFP+/GFP+ population) at ~48 post-transfection. 3. At 36~72h post-transfection, trypsinize cell population and plate APPROPRIATE number of cells into 10cm dish with APPROPRIATE hygromycin concentration. A. APPROPRIATE number of cells: Each cell line has different colony forming efficiency. Appropriate number of cells to be plated should be determined empirically. i. Recommended: Make multiple plates expected to produce 500/5000/50000/(possibly more) each before selection. B. APPROPRIATE hygromycin concentration: Each cell line has different hygromycin sensitivity. Appropriate hygromycin concentration that will kill 99% of cell population within 48 hours should be determined empirically. For 293T and HeLa cells, 2mg/ml of hygromycin was used. C. Save part of transfected cells and prepare genomic DNA. Monitor if mutations were introduced by RGEN expression in your cell by genotyping assays like T7E1 assay. (Control 1) hours after hygromycin selection, wash cells once and change media to a fresh media with no antibiotics. 5. Wait 2~4 weeks until monoclonal colonies are formed with occasional media change or addition. A. If there is plate with too many cells, and thus will not likely to support isolation of monoclonal colonies, it can be used to confirm the process is going well by performing genotyping assays to monitor high-content of genome-modified cells, especially compared to control 1 from above step 3.C. 6. Chose a plate with 10~200 colonies (if possible) to pick 20~100 colonies and move to 96- or 48-well plate. 7. Grow up each clones and identify mutant/ko cell colonies by genotyping and western blot.
9