CBI s 15 th Annual Product Complaints Congress for Life Sciences
|
|
|
- Meagan Bradley
- 5 years ago
- Views:
Transcription
1 CBI s 15 th Annual Product Complaints Congress for Life Sciences June 14, 2017 Steven Niedelman Lead Quality Systems & Compliance Consultant King & Spalding LLP 1700 Pennsylvania Ave., NW Washington, DC sniedelman@kslaw.com
2 Potpourri of FDA Activities Program Alignment Combination Products update Final MDR Guidance changes ISO 13485:2016 and MDSAP Concerns to consider and potential impact 2
3 Program Alignment 3
4 Program Alignment Most significant reorganization of FDA s field organization (Office of Regulatory Affairs) since it s creation; operational as of May 15, 2017 Realigns current field staff investigators, compliance officers, Supervisors, District Directors into a matrix based organization based on specialty The goal is for investigators to be better trained in the areas they inspect by specializing their focus only in those areas District Offices will be identified by product specialty investigators may sit at a brick and mortar district different that of their supervisor; and both at yet another District Office as their District Director Brick and mortar offices will exist and will be staffed by employees who may report to managers at other offices Expect to see new investigators inspecting your sites! 4
5 Goal of Program Alignment Modernize and strengthen the FDA workforce to Improve Public Health 2013 FDA Program Alignment Charge 5
6 The Imperative for Change Acceleration of scientific innovation Global expansion of markets Improve public health response Enact modern legal authorities: 2009 Family Smoking Prevention and Tobacco Control Act 2011 FDA Food Safety Modernization Act (FSMA) 2012 FDA Safety and Innovation Act (FDASIA) 2013 Drug Quality and Security Act (DQSA) PUBLIC HEALTH NEED: FDA must move toward a more collaborative program-based model to continue to effectively address the demands of the modern regulatory environment. 6






7 Office of Medical Products and Tobacco Operations (OMPTO) Ellen Morrison, Assistant Commissioner Anne Reid, Director Tobacco Staff [acting] Ginette Michaud, Director Biological Products Chrissy Cochran, Director Bioresearch Monitoring Jan Welch, Director Medical Device and Radiological Health Alonza Cruse, Director Pharmaceutical Quality 7
8 OMPTO Advances FDA s Mission New ORA Offices FDA Center Counterpart Office of Biological Products Center for Biologics Evaluation Research Operations Office of Bioresearch Monitoring Center Offices of Bioresearch Monitoring Operations Office of Medical Device and Center for Devices and Radiological Health Radiological Health Operations Office of Pharmaceutical Quality Center for Drug Evaluation and Research Operations Center for Veterinary Medicine Tobacco Operations Center for Tobacco Products 8
9 Program Alignment: Key Changes From To Geographic management of operations Program management of operations, management teams based on staff: Bioresearch Monitoring 2 management teams 2 management teams Human and Animal Food 12 management teams Medical Devices and Rad Health 3 management teams Pharmaceutical Quality 4 management teams Tobacco Plus Imports as a program 5 management teams Biologics Program Alignment: Key Changes SES Regional Food & Drug Directors SES Office Directors Degrees of program specialization for investigations, compliance and operational managers 20 District Directors who manage the geographic district and all programs operations within the district One import district and a range of import operations embedded within the 16 other districts Exclusive specialization in one program for investigations, compliance and operational managers 20 District Directors who manage the geographic district and only one program for operations. Plus eight new program division directors who manage program operations only total 28 management teams Five import divisions (four new import divisions) covering all borders, managing import operations nationally as a program 9
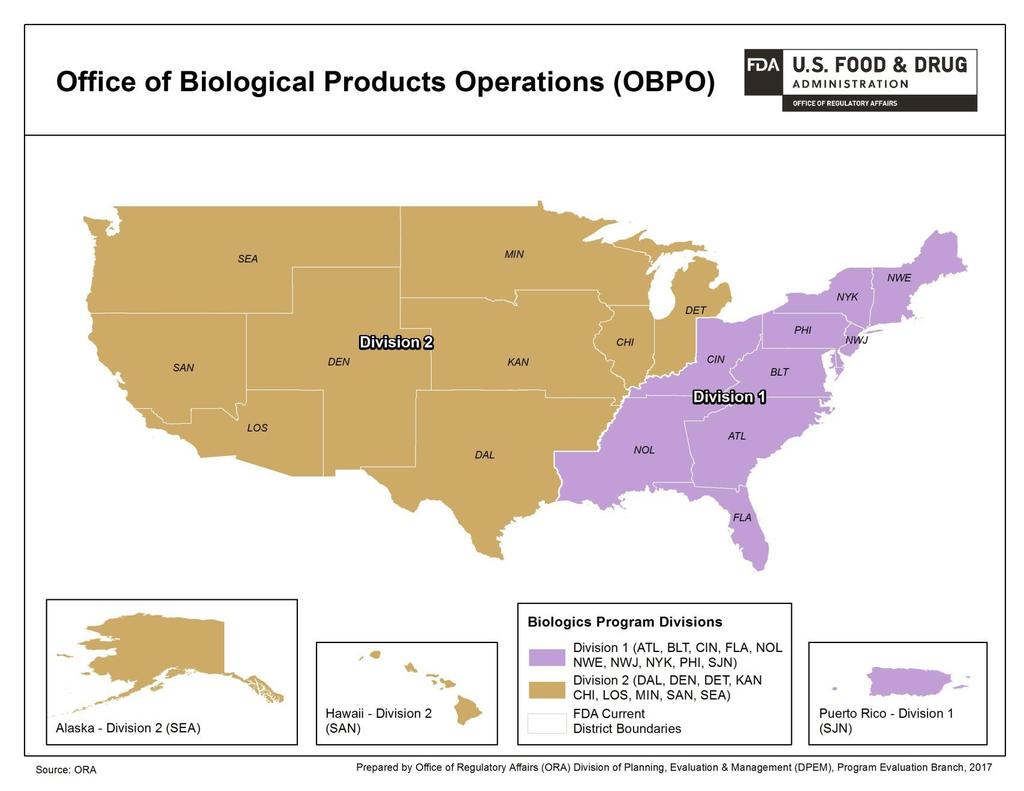
10 10 3
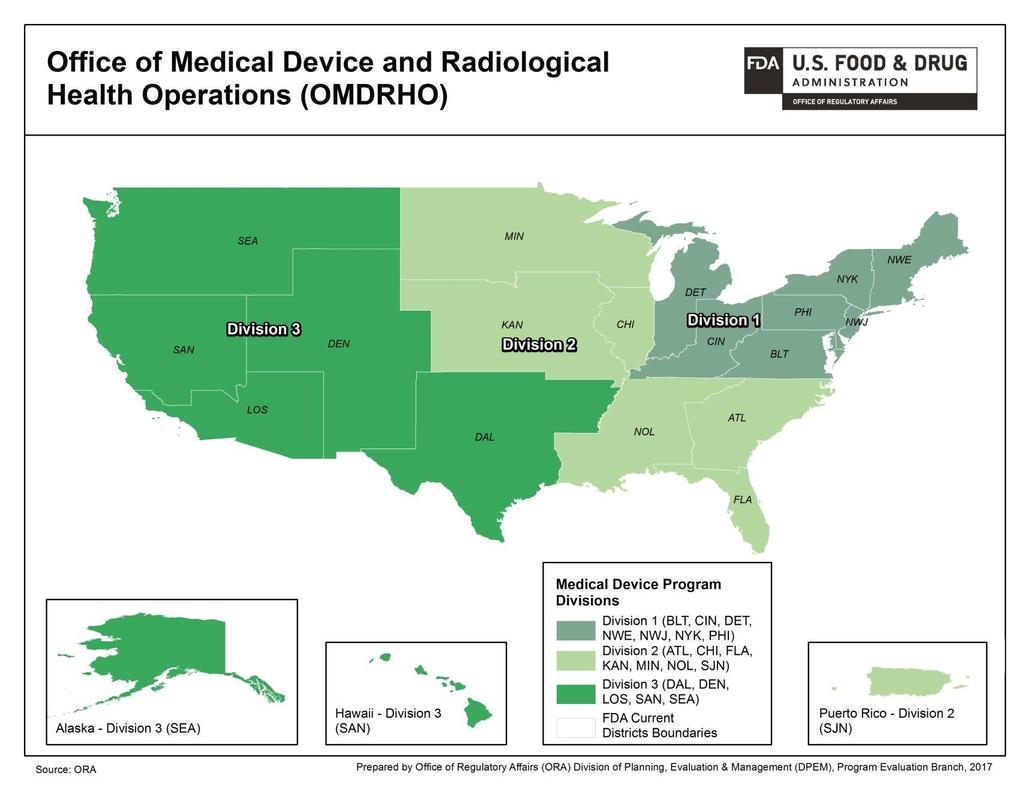
11 11 4
12 12 5

13 13 7
14 Interfacing with Program Alignment Unless instructed otherwise, continue to communicate with your existing local FDA District Office Recalls, Warning Letter and FDA 483 responses should be sent to your local District who will assure they are distributed to the correct contact within the new organization. At that point you may be directed to continue communications with an appropriate individual in the assigned program area (i.e., Medical devices, biologics, etc.) Program alignment does not affect your Center contacts Any questions on point of contact should contact Jessica Zeller, ORA Ombudsman at 14
15 Recent Postmarket Activities Affecting Combination Products 15
16 Overview of Recent Activity FDA recently published long awaited guidance and regulation affecting the growing combination products industry In January 2017, FDA finalized the January 2015 Draft Guidance implementing the 2013 Final Rule for applicability of Good Manufacturing Practices for Combination Products In December, 2016, FDA issued the long awaited final rule describing postmarket reporting requirements for Combination Products which was based on a draft that dated back to October 2009 If you manufacture Combination Products it is critical you understand your obligation and pathway you will follow to ensure compliance for reporting postmarket activities 16
 Proposed Rule (September 2009) Public")
 Final guidance (January")
17 Regulatory History GMP requirements Draft guidance (October 2004) Proposed Rule (September 2009) Public workshop (January 2010) Final rule (January 2013) Draft guidance (January 2015) Final guidance (January 2017) 17
18 Final Rule Issued January 2013 Added 21 CFR Part 4 Does not impose any new substantive manufacturing requirements; clarifying in nature Overarching principle: constituent parts retain their regulatory status after they are combined Focuses on how to demonstrate compliance with CGMP Provides two options to combination product manufacturers for demonstrating compliance with CGMP requirements 18
19 Regulatory Framework for Compliance Combination product manufacturers can demonstrate compliance with either: All CGMP regulations applicable to each constituent part; or Streamlined approach : Either drug GMP or device QSR; and specified provisions from the other of the two sets of requirements (GMP or QSR); and requirements specific to biological products and HCT/Ps Constituent parts that are manufactured at a separate facility from the other constituent parts to be included in a combination product must comply with CGMP applicable to that constituent part You must comply with the complaint handling requirements of the quality system you follow! 19
20 Option 1: Drug CGMP-based streamlined approach: CGMP + select provisions of QSR If the combination product includes device and drug constituent parts, and the combo product manufacturer chooses to comply with drug CGMP, the following provisions of the device QSR must be satisfied: Section (management responsibility) Section (design controls) Section (purchasing controls) Section (corrective action and preventive action) Section (installation) Section (servicing) 20
21 Option 2: Device based streamlined approach: QSR + select provisions of drug CGMP If the combination product includes both device and drug constituent parts, and the combo product manufacturer chooses to comply with QSR, the following provisions of the drug CGMP regulations must be satisfied: Section (testing and approval or rejection of components, drug product containers, and closures) Section (calculation of yield) Section (tamper-evident packaging requirements for OTC human drug products) Section (expiration dating) Section (testing and release for distribution) Section (stability testing) Section (special testing requirements) Section (reserve samples) 21
22 Streamlined approach: Biological products and HCT/Ps If the combination product includes: A biological product constituent part, must also comply with requirements applicable to that biological product if it were not part of a combination product; An HCT/P, must also comply with all current good tissue practice requirements (including donor eligibility requirements in part 1271) if it were not part of a combination product 22
23 What s Next? Section 3038 of 21st Century Cures Act requires FDA to publish a proposed list of combination products and manufacturing processes for which: GMP requirements may vary from 21 CFR Section 4.4; or the requirements of Section 4.4 can be satisfied through alternative or streamlined mechanisms. Proposed list must be published within 18 months After public comment period, FDA must publish a final list in the Federal Register and periodically review it 23
24 Regulatory History: Postmarket Adverse Event Reporting On December 20, 2016, FDA published a final rule Postmarketing Safety Reporting for Combination Products that is codified at 21 CFR part 4 subpart B. (81 Fed. Reg ) Effective date: January 19, 2017 Some provisions have a compliance date later than effective date Accessible at products 24
25 Background No prior postmarketing safety reporting (PMSR) for combination products Public hearing (November 25, 2002) and public workshops (July 8, 2003 and January 10, 2010) Proposed Rule issued on October 1, 2009 Common themes for both FDA and industry Consistency and clarity for PMSR for combination products Reduction of unnecessary duplicative reporting 25
26 Purpose To describe PMSR requirements when two or more products (drugs, devices, and/or biological products), which are referred to as constituent parts, comprise a combination product and the combination product or its constituent parts have received FDA marketing authorization. Definition of combination product has not changed (See 21 CFR 3.1) Rule does not apply to investigational combination products 26
27 Required Reports - All Applicants Both combination product applicants and constituent part applicants must comply with all of the PMSR requirements based on the marketing authorization of the product. See 21 CFR 4.102(b) Device applications: 21 CFR parts 803 and 806 Drug (NDA or ANDA): 21 CFR part 314 Biological product (BLA): 21 CFR parts 600 and

28 Required Reports - All Applicants 28
29 Additional Reports Only Combination Product Applicants Combination products, which are authorized under a NDA, ANDA or BLA, that include a device constituent part must also submit: Correction and removal reports (21 CFR ), and keep related records (21 CFR ) 5-day reports (21 CFR and ) Malfunction MDR reports, including follow-up (21 CFR and ), See 21 CFR 4.102(c) 29
30 Additional Reports Only Combination Product Applicants Combination products, which are authorized under a device application or BLA, that include a drug constituent part must also submit Field alert reports (21 CFR ) 15-day reports (21 CFR ) If the combination product is authorized for marketing as a device, this report may be submitted within 30 calendar days rather than 15 calendar days See 21 CFR 4.102(c) 30
31 Additional Reports Only Combination Product Applicants Combination products, which are authorized under a device application, NDA or ANDA, that include a biological product constituent part must also submit Biological Product Deviation Reports (BPDR), (21 CFR and ) 15-day reports (21 CFR ) If the combination product is authorized for marketing as a device, this report may be submitted within 30 calendar days rather than 15 calendar days See 21 CFR 4.102(c) 31
32 When Can One Report Be Submitted? A combination product applicant may submit only one PMSR if the report Contains all of the information that would be required in another type of report, Is required to be submitted in the same manner under this rule as the other report, and Is submitted within the applicable deadlines See 21 CFR
33 Medical Device Reporting (MDR)Final Guidance 33
34 New MDR Guidance Medical Device Reporting for Manufacturers Issued on November 8, 2016 Final Guidance Supersedes 1997 MDR Guidance when final Available at 34
35 1997 MDR Guidance Previous MDR Guidance had not been revised since New final guidance follows issuance of updated draft guidance released in 2013 for public comment. New guidance reiterates certain longstanding interpretations and changes others. 35
36 Key Points Presumption of reportability Once a malfunction causes/contributes to a death/serious injury, future occurrences are presumed to be likely to cause/contribute to a death/serious injury (and therefore are reportable). Two-Year Rule Under the 1997 Guidance, if a malfunction does not cause/contribute to any deaths/serious injuries for two years, the presumption is automatically lifted. Elimination of the Two-Year Rule Two-Year Rule is eliminated, but Firms can submit evidence documenting that there have been no additional deaths/serious injuries and request an exemption from further MDR reporting for the malfunction. 36
37 Assigning MDR Responsibility Between Two Entities Firms can request exemptions so that only one entity reports MDRs for which both are responsible. Specification developers and manufacturers who distribute or market for the specification developer. Importers and foreign manufacturers. Entities should submit a joint request for the exemption. If an exemption is not obtained, then both parties must submit MDRs. Assigning responsibility by written agreement alone is not sufficient. Absent an exemption, failure to file an MDR for a reportable event is considered a violation for both firms. 37
38 Likely FDA s interpretation of when a malfunction is likely to cause/contribute to a death/serious injury is more conservative than many firms realize. New guidance reiterates longstanding agency view. The definition of likely includes: Chance of death/serious injury is not remote. Catastrophic malfunction that may lead to death/serious injury. Failure to perform essential function which could cause/contribute to death/serious injury. 38
39 Delays in Surgery According to FDA, some firms routinely report any delay in surgery as a MDR. Draft Guidance states that an event is not reportable solely on the basis of a delay in surgery. But delays in surgery are reportable if: Delay may have caused/contributed to a death/serious injury. Delay due to a malfunction would be likely to cause/contribute to death/serious injury upon recurrence. Question remains: what types of delays may cause/contribute to a death/serious injury? How long of a delay? Prolongation of anesthesia? 39
40 Scientific Literature Articles Firms must investigate events reported in literature articles to determine if they represent an MDR reportable event. If an event described in an article has already been reported, another report does not need to be submitted. Where a literature article describes multiple events in general terms, but does not provide specific patient/device information and a firm s investigation fails to yield specific information it may be acceptable to submit one MDR report for multiple reportable events. 40
41 Similar Devices Draft Guidance defines similar devices in the context of reporting malfunctions. Malfunctions are reportable if the device or a similar device marketed by the manufacturer would be likely to cause/contribute to a death/serious injury upon recurrence. Devices are generally considered similar if they have the same: Basic design and performance characteristics related to device safety and effectiveness, Intended use and function, and Device classification and product code. Other factors: brand name, common name, same 510(k) or same PMA. Need to consider similar devices sold OUS 41
42 Investigation of Events Draft Guidance says good faith effort is expected for investigation of reported events. Not determined solely by number of attempts to obtain information; common industry practice has been at least 3 attempts. Should include at least one written attempt. FDA makes the ultimate determination of whether a firm made a good faith effort. But FDA does not provide clear guidance about what will be considered a good faith effort. Level of effort... depends largely on the nature and severity of the event reported. The event must be analyzed even if the device isn t returned. You should undertake activities such as review of other similar events, device history review, review of appropriate manufacturing processes, etc., where they will help you analyze the event. 42
43 Other Areas Warranty period does not determine the expected life of a device. Expected life is the time a device is expected to remain functional after it is placed into use, including any and all calibration and maintenance cycles. Labeled complications and risks are reportable as MDRs. Events that were anticipated or intrinsically caused by a device are not exempt from reporting. Off Label Use and User Error continue to be reportable events when reportable criteria are met. 43
44 Update on MDSAP and ISO 13485:
45 MDSAP Consortium The International Medical Devices Regulators Forum (IMDRF), identified a Work Group consisting of an international coalition of countries dedicated to pooling technology, resources, to develop a harmonized single audit program (MDSAP). MDSAP would be piloted for 3 years starting in Health Canada indicated early on that would formally adopt the MDSAP approach upon completion of the pilot, therefore CMDCAS auditors were first to be authorized to conduct audits. As of January 2019, to do business in Canada, you must participate in MDSAP and operate in compliance with ISO 13485:



 Pharmaceuticals")
 U.S.")
46 MDSAP International Consortium The international consortium of countries for MDSAP as of June 2015 : Therapeutic Goods Administration (TGA) Agência Nacional de Vigilância Sanitária (ANVISA) Health Canada (HC) Pharmaceuticals and Medical Devices Agency (PMDA) U.S. Food and Drug Administration (FDA) 8 46
47 MDSAP International Consortium The mission of the MDSAP International Consortium is to jointly leverage regulatory resources to manage an efficient, effective, and sustainable single audit program focused on the oversight of medical device manufacturers Government resources can then focus on high risk or problematic medical devices, manufacturers that are not in compliance with the regulations, and oversight of the third party auditing organizations 47 47
48 Participating Manufacturers (Feb. 2016) As of Feb over 200 Corporations: 41 Individual sites:
49 4 9 Background ISO Revision International Organization Standardization (ISO) standard published first in 1996, revised in 2003, and now in 2016 Sector specific standard based off ISO 9001 Represents the requirements for a comprehensive quality management system (QMS) for the design and manufacture of medical devices and in vitro diagnostic devices, as well as their related processes and services
50 Objectives of ISO Revision Improve clarity of the requirements Increase confidence that the requirements are consistent with cgmps or current Quality System regulatory requirements/objectives Increase harmonization of QMS regulatory requirements Increase medical device manufacturers ability to meet customer requirements 50
51 ISO 13485:2016 and MDSAP Timeline ISO 13485:2003 or 2016 Certifications 13485:2003 New Certifications Discouraged No 13485:2003 Certifications or Re-Certifications Allowed ISO 13485:2016 Mar. 1, 2016 Mar. 1, 2017 Mar. 1, 2018 Mar. 1, 2019 MDSAP Jan. 1, 2016 Jan. 1, 2017 Jan. 1, 2018 January 1, 2019 MDSAP Certificate to ISO 13485:2016 Mandatory in Health Canada
52 ISO 13485:2016 Harmonization ISO 13485:2016 is closely aligned with 21CFR Part 820, Quality System regulation for Medical Devices Some preliminary thought is being given by FDA to acknowledge ISO 13485:2016, and perhaps accept it as a replacement to 21CFR Part 820 Whether to adopt it as a recognized standard, or adopt it as the Quality System required by the Food, Drug and Cosmetic Act are decisions that will need to be discussed as thought continues 52
53 In closing There are many and diverse activities ongoing affecting FDA regulated industries It is important that you stay abreast of them being cognizant that you will held accountable not only by FDA, but perhaps foreign authorities as we continue to grow in a globalized and harmonized environment Never lose sight that we all are involved in the life sciences industries, and our patients and physicians depend on the safety and effectiveness of the products we sell It is our responsibility to be responsive to their complaints, address them timely, identify their root cause and take the appropriate steps to reduce or eliminate them The public both expects and deserves it! 53
54 Questions? Thank you! 54