Critical Components of the Complaint Management Lifecycle. Dick Roy Product Surveillance Director GE Healthcare
|
|
|
- Abner Haynes
- 6 years ago
- Views:
Transcription
1 Critical Components of the Complaint Management Lifecycle Dick Roy Product Surveillance Director GE Healthcare
2 About your speaker 25+ years in Medical Device Industry 18 years in Post Market Management dealing with Class II and Class III products Expertise in Complaint Handling, MDR and other reporting requirements, Quality Systems and Audit * The contents of this presentation are based on the experience and views of the presenter and do not necessarily reflect those of GE Healthcare. 2
3 Agenda Overview of the Complaint Handling Process Establishment of a Robust Complaint Handling System Additional Topics 3
4 Establishing a Robust Complaint Handling System Complaint Handling Findings Regulatory and Competitive Pressures Education and Training for the Team Scalable features of a High Volume Complaint System 4
5 Regulatory Requirements for Complaint Handling Complaint Handling requirements are documented in 21CFR , available at: fr/cfrsearch.cfm?fr= These include requirements for complaint files, procedures, investigations, communications with complaint Formally Designated Units, and evaluation for MDR reporting. 5
6 Where do Manufacturers encounter difficulties? FDA publishes 483 findings by fiscal year. FY2014 findings are available at: m The site* groups 4010 findings originating from 972 form 483s * There are 483 findings that are not listed on this site. 6

7 Where do Manufacturers encounter difficulties? Most frequent 483 findings by Quality System citation involving postmarket include: 7
8 Where do Manufacturers encounter difficulties? What should be in a complaint procedure? For example, your firm s complaint handling procedure document does not Adequately designate a formal unit to review and evaluate complaints Require complaints to be evaluated to determine whether a complaint represents an event that is required to be reported to FDA under 21 CFR 803 Require complaint files to be maintained for all received complaints. During the inspection, your firm s general manager, indicated that the complaints deemed insignificant or invalid upon initial review are not documented as complaints WL 10/10/12 8
9 Where do Manufacturers encounter difficulties? What should be in a complaint procedure? Specifically, your complaint handling procedure, does not: a. ensure that records of complaint investigations, when necessary, include the required information. For example, your firm failed to document the following elements in 10 complaints associated with your devices: i. adequate details or nature of the complaint including the setting in which the malfunction occurred; ii. iii. resultant patient adverse effects; and whether further medical intervention was require enquired. b. define timeliness of complaint investigations until after a subject device is returned to your firm. WL 2/9/15 9
10 Where do Manufacturers encounter difficulties? What should be in a complaint record? your firm's complaint form does not contain a field for an evaluation for MDR reportable events. WL 7//15 Information recorded on complaint records, was inaccurate, in that the complaint records concluded no corrective action was necessary when, in fact, a Corrective and Preventive Action (CAPA) had been initiated WL 1/29/14 For example, your Customer Complaint Handling and Vigilance procedure, fails to include the requirement for review and evaluation of complaints to determine the necessity of an investigation, or a record to include the rational for not conducting an investigation. WL 12/19/14 10
11 Where do Manufacturers encounter difficulties? Do complaint records comply with procedures or regulation? Specifically, 10 of the 11 non-complaint records reviewed by the FDA investigator meet the definition of a complaint. WL 2/21/12 Twenty-eight of twenty-eight Customer Service Reports were not adequately completed to include: name of the device, date the complaint was received, or customer address and phone number. WL 12/22/10 Your firm has not analyzed quality data from customer returns, including product return codes, complaint codes, and action codes in the Return Good Authorization (RGA) log to identify existing or potential causes of nonconforming product and other quality problems WL 8/23/12 11
12 Where do Manufacturers encounter difficulties? Do complaint records comply with procedures or regulation? Your firm failed to complete separate complaint records for complaints involving multiple lots or multiple patients. As a result, your complaint data is inaccurate and incomplete for analyzing trends.. WL 6/4/14 For example: 72% (18 of 25) complaint records including complaints, reviewed during our inspection attribute the root cause of the complaints to wear and tear during service calls, and no investigations were conducted. WL 8/8/14 12
13 Where do Manufacturers encounter difficulties? When must a manufacturer investigate? Any complaint involving the possible failure of a device, labeling, or packaging to meet any of its specifications shall be reviewed, evaluated, and investigated, unless such investigation has already been performed for a similar complaint and another investigation is not necessary c Any complaint that represents an event which must be reported to FDA under part 803 of this chapter shall be promptly reviewed, evaluated, and investigated by a designated individual(s) and shall be maintained in a separate portion of the complaint files or otherwise clearly identified d 13
14 Where do Manufacturers encounter difficulties? Are investigations adequate? a. Complaint Investigation Reports (CIR) for Customer Service Reports do not include investigations into the cause of non-conformities related to leaking vests and masques. b. CSR outlined a complaint that reported a ''patient had a very bad reaction to the product." There was no investigation. The CSR indicates that the action or investigation required was a ''refund.'' d. No investigation was conducted for the following complaints that reported missing device components: WL 12/22/102 14
15 Where do Manufacturers encounter difficulties? What can comprise an investigation? The procedure, Complaint Handling, does not require a review of Device History Records (DHR) for disposable items unless the event is reportable under the Medical Device Reporting regulations, even when the device is returned or the lot number is known. Specifically, complaints received for showed that lot numbers were provided, but DHRs were not reviewed prior to the complaints being closed. WL 1/29/14 The complaint record for complaint further identifies several individuals who were interviewed as part of the complaint investigation; however, there is no record documenting the details of these interviews. 3. Further, your firm subsequently received the suspect unit and conducted testing identifying the pressure relieve valve did not function as intended. There are no records of the dates the testing was conducted or any documentation of corrective actions taken as a result of the complaint and subsequent investigation. WL 12/8/14 15
16 Where do Manufacturers encounter difficulties? Consider having a disinterested person show where each clause in the Regulation exists in your procedures Be sure to use definitions as they exist in Regulation. If you have one procedure that combines requirements from many countries, be able to show clearly that each requirement is met. 16

17 Regulatory and Competitive Pressures FDA makes MDRs available on MAUDE 17

18 MAUDE Search Tools MAUDE data files may be downloaded from: RegulationandGuidance/PostmarketRequir ements/reportingadverseevents/ucm htm. These files include more data than is available in the online search tools 18
19 MAUDE Search Tools Who uses MAUDE? How do you use the data? Who else uses MAUDE? Is MAUDE ever incorrect? Remember, MAUDE data has a number of limitations delineated on cfdocs/cfmaude/textsearch.cfm 19
20 MAUDE Search Tools Searching MAUDE will identify User and Voluntary Reports that you may not have otherwise received. Be expansive User and Voluntary Reports may render your company s name in many different ways. Consider whether your company wants to echo User and Voluntary reports with a corresponding Manufacturer report. Consider reviewing MAUDE content for high-priority issues to confirm reports were captured accurately. 20
21 The Complaint Team: Education and Training Formally Designated Units should understand the product, the product s use, the therapy or diagnosis rendered, the patient and the clinician. Those preparing records should be able to explain the event, including impact to the patient before, during and after the event occurs. 21
22 The Complaint Team: Education and Training How do you do this? How long does it take to train a new person? What should be included in the training? How much of the training is desk training? How much is hands-on? 22
23 Overview of the Complaint Handling Process What are we trying to accomplish? Steps in the complaint process Reporting, trending and analysis of metrics 23
24 Overview of the Complaint Handling Process What are we trying to accomplish? Understand product performance throughout the product s lifecycle Accurately represent product information to internal audiences, regulators, and others 24
25 Overview of the Complaint Handling Process Receive Complaint Customer Response Regulatory Reporting Review and Update Design / Risk Analysis Documents Complaint Investigation Coding Trend Analysis Corrective Action Returned Product Lab Analysis Verify Effectiveness Model for the post market process complaint handling is central to understanding performance 25
26 The Complaint Handling Process: Scalable Features Receive Complaint Customer Response Regulatory Reporting Review and Update Design / Risk Analysis Documents Complaint Investigation Coding Trend Analysis Corrective Action Returned Product Lab Analysis Verify Effectiveness What is the product? How many complaints come in each day? 26
27 The Complaint Handling Process: Scalable Features Can you build system tools to prepare records for evaluation? For edit checks prior to closure? To identify similar complaints or prior complaints for the same device or devices with the same production lot number? 27
28 The Complaint Handling Process: Scalable Features Which steps require lower- or higher-level skills? Are there many regulatory reports? Should report decisions and preparation take place in-line or with a separate group? Can some of these be outsourced or take place offshore? 28
29 The Complaint Handling Process: Possible Structures Partitioning complaint receipt can permit focus on preparing records for detailed review. Combining Investigation, Coding and Regulatory Reporting may be effective if a company files large numbers of MDRs. Close proximity to Lab Analysis will help with accurately tying analysis results to customer descriptions. 29
30 Complaint Capture and Processing Complaint Capture depends on accurate information from complaint sources. Manufacturers may receive information from field support staff, service staff and repair depots, clinicians, and other sources including: Marketing, Focus Groups Customer Service Newspapers, Technical Publications, Social Media Lawsuits Clinical Studies /Post Market Studies / Marketing Studies 30
31 Complaint Capture and Processing All employees must be trained on how to recognize a complaint and what to do with that complaint. Training can differ based on the employee s role and proximity to clinical use. Training to identify a customer complaint vs an inquiry, order or scheduled maintenance Consider using examples of what should or should not be forwarded. 31
32 Complaint Capture and Processing Tools can be set up to promote compliance: What information is required by the complaint group? Design smartforms or software tools to walk through a field event using workflow and language familiar to the field staff. Set decision points so that the field staff must enter only the required information Interfaces eliminate transcription errors, provide for data accuracy Consider systems to submit incomplete reports after a certain time interval. 32
33 Complaint Capture and Processing Raw information from the field may require merging with internal information, including: Prior complaints for that individual medical device Patient implant information Returned product analysis or field service for that device 33
34 Complaint Evaluation All complaints must be evaluated. Evaluation includes assessing whether the complaint is valid, an investigation is required or regulatory reporting is necessary. 34
35 Complaint Evaluation Investigations are necessary for: (c) Any complaint involving the possible failure of a device, labeling, or packaging to meet any of its specifications shall be reviewed, evaluated, and investigated, unless such investigation has already been performed for a similar complaint and another investigation is not necessary. 21CFR c (d) Any complaint that represents an event which must be reported to FDA under part 803 of this chapter shall be promptly reviewed, evaluated, and investigated by a designated individual(s) and shall be maintained in a separate portion of the complaint files or otherwise clearly identified. 21CFR d 35
36 Complaint Evaluation What comprises an investigation? 36
37 Complaint Evaluation / Root Cause Analysis How do I investigate a complaint if the product is not available or not returned? Refer to information that may be obtained from: Manufacturing records for that device Prior complaints for that device Similar devices that may be within your control, e.g. reserve samples 37
38 Complaint Evaluation / Root Cause Analysis A structured analysis process can help with the investigation. Quality tools such as Five Whys, Is/Is Not, and other elements can help. Investigation depth can be tailored to the type of field event. Defining the appropriate depth can help with application of resources. 38
39 Reporting, Trending and Metrics Metrics fall into two categories: Process Performance, including Incoming complaint volume, including volume by geography Time to milestones in the complaint process, including initial assessment, coding, regulatory reporting, customer response and closure Complaint WIP and Aging to include median age and age of oldest complaint Accuracy as shown by assessments of complaint records Product performance 39
40 Reporting, Trending and Metrics Metrics fall into two categories: Process Performance Product performance, including Product reliability Failure rates Run charts of as-reported and as-investigated Remember to use alert and action limits Signals should be traceable to some action Pareto charts of leading failure modes 40
41 Reporting, Trending and Metrics Trending has three objectives: Is it good enough? Is it getting better or getting worse? See design predictions, look for gradual rise or spikes Identify predominant causes of nonconformance What is causing this? Review Pareto charts and FMEA, determine if it is an early issue or a wearout problem Provide triggers for further action Do I need to do something? Run rules, Weibull predictions 41
42 Reporting, Trending and Metrics Have a Structured Process for Trending Define Who looks at the data Define how Frequently you will look at data Define your Criteria for initiating action Count of similar observations Rate of occurrence Risk analysis Define the next Activity you will undertake Define how you will Document the investigation Review progress at cross-functional meeting 42

43 Links to Risk Management ISO14971 provides guidance on applying risk to medical devices. Risk may be used for determining risk/benefit or setting triggers for taking action. New failure mode? Unanticipated changes in rates of occurrence or severity? ISO14971 requires manufacturers to collect and review information about medical devices in the production and post-production phase (ISO14971:2012) 43
44 CAPA All complaints require some assessment for Investigation; however, systemic or high severity issues may have to be elevated to a CAPA. CAPAs will have a rigorous process for determining root cause, action plans, dates, and effectiveness checks. CAPA review boards will monitor CAPAs as they work through the process and confirm the adequacy of actions. 44
45 Documentation Documentation should stand on its own, including procedures, complaint records and CAPA records. An SOP documenting the overall procedure can be valuable during inspections, when training new staff, and for other audiences outside the complaint department. Complaint records should be laid out to show a clear path from receipt to resolution. CAPAs / trend reports can frame the issue, bound the affected population, track investigation, identify actions to take and verify effectiveness. 45
46 Process Controls Variation can be minimized using tools: Decision Charts MDR decision trees MDR list of common situations System screen layout to promote a workflow System error checks, whether in-line or before record is closed Pivot tables: Compare specific codes to MDR decisions Compare as-reported codes to failure mode. Text Analytics to identify records with known keywords or to identify patterns 46

47 Expectations (a) Each manufacturer shall maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit. Such procedures shall ensure that: (1) All complaints are processed in a uniform and timely manner What is timely? I know it when I see it Justice Potter Stewart, Jacobellis v. Ohio 47
48 Additional Topics How do other companies do this? How do I know I have the right number of staff? When things go wrong Electronic MDRs The future 48
49 How do Other Companies do this? Compliance Alliance has surveyed Medical Device manufacturers on several occasions. These surveys focus on post-market activities. 103 responses were received from companies with <100 to >50,000 employees <5 to >25 FTEs directly involved with complaints <100 to >5,000 complaints per month Survey results are available at 49
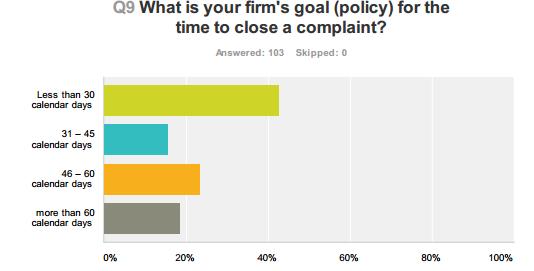
50 Compliance Alliance Survey Results 50

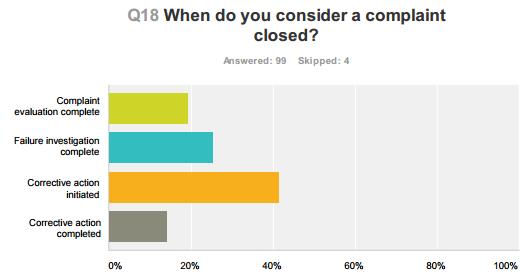
51 Compliance Alliance Survey Results 51

52 Compliance Alliance Survey Results 52

53 Compliance Alliance Survey Results 53
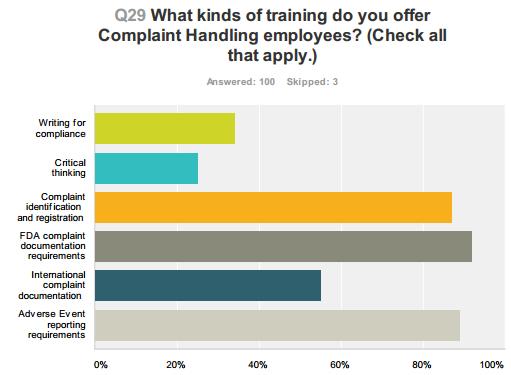
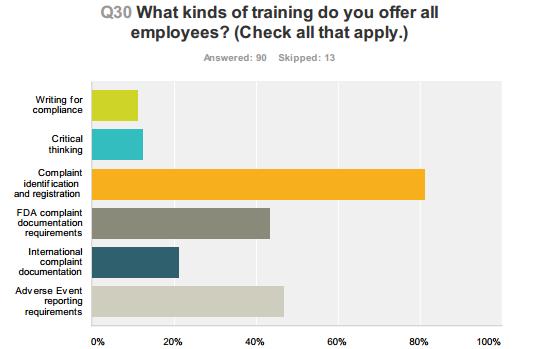
54 Compliance Alliance Survey Results 54
55 Compliance Alliance Survey Results What kind of efficiencies have you introduced in the last 12 months (partial list) Developed tools to assist with trending and escalation to CAPA; Established a single standardized procedure for complaints Trending is performed based upon normalized data for both disposable and repairable equipment Complaint file quality improvement Utilization of SAS for Pareto and trending Combining Complaints Handling with Customer Service - one single entry point f or all complaints into the system Standardized and Harmonize the complaint handling department across multiple business units. 55
56 Staffing Questions How many people do I need? Consider capacity studies for staffing Ask managers to document how many staff are required to do current tasks. Subdivide tasks to a meaningful level (probably <10 segments); understand that tasks will vary with the event. Know how many tasks will require reporting, follow up and other paramaters. 56
57 Staffing Questions What qualifications are required? Sufficient to perform the work product reporting specialist was making decisions about MDR reportability for the... The training record for this particular employee showed that this person only had a high school diploma with some additional in-house training. WL June 1, 2009 Qualifications can match the work to be performed Firms ARE NOT required to have every MDR report reviewed by a person qualified to make a medical judgment and/or a person with a medical degree or training. Compliance Program Guidance Manual
58 Staffing Questions How do I train the staff? Training must extent beyond procedure compliance; consider training on product, patient, disease process, therapy provided, and relationships with field, product analysis and engineering activities. Don t forget training on regulations. Testing may provide objective evidence that the specialist is qualified to perform the role. 58
59 When Things go Wrong Manufacturers may encounter situations in which a product issue expands widely. This may result in field actions with attendant increases in complaint volume sometimes two or three times baseline levels. Staffing may have to be increased quickly and work processes may be re-examined to ensure compliance needs are met for this higher volume. 59
60 When Things go Wrong Handle similar events consistently Develop standard language for events and MDRs within an advisory and events documenting field assessments Explore where automation can be used, but ensure a specialist will document investigation and reporting decisions and event closure. Assign data requests to a small number of staff. Retain query criteria and output files. 60
61 When Things go Wrong Re-evaluate your workflow Segment work to permit training for temporary staff Assign training and monitoring activities to technical leaders in the group. Implement stringent quality checks for work completed by temporary staff Assess metrics to ensure that work does not accumulate mid-process and perform reconciliations to ensure work does not get lost. 61
62 When Things go Wrong Assign trend or similar numbers to fields within an advisory. Run queries periodically to confirm similar coding, reporting decisions and language where appropriate. Generate and keep pdfs of all MDRs submitted. This will be easier if you are using emdr. You will receive many requests from regulators, legal and others for sets of MDRs. Similarly, generate and retain pdfs of all correspondence with FDA and other regulators. 62
63 When Things go Wrong Create queries with information that has been sent to FDA, including record number, MDR number and submission date. Review periodically for consistency. Review data on MAUDE periodically to ensure: Information in MAUDE records is accurate compared to submitted information You have captured all user reports. 63
64 When Things go Wrong Develop position paper on MAUDE analysis. Remember that MAUDE data cannot be used to compare data among manufacturers due to differences in Language used Reporting Criteria Reporting timelines FDA MAUDE update schedules Summary vs individual reports and loading supplemental reports in MAUDE 64
65 Electronic MDRs Electronic Manufacturer MDR reporting will be mandatory in August, Information is available at: ance/guidancedocuments/ucm htm ance/postmarketrequirements/reportingadverseevents/emd R%E2%80%93ElectronicMedicalDeviceReporting/ucm htm Consider applying for low volume solution as a backup even if you are using the HL-7 system 65
66 Observations on working with FDA Focus on the customer: What does FDA want? Enough detail so that they don t have to follow up with questions Enough information so that they can fulfill their mission to protect the public health Lessons from Inspections Review the Process as the inspection begins Make your Procedures simple and understandable Make your Investigation and Reporting Rationale clear Success means handing files to investigators for review and getting them back with no follow-up questions. 66

67 The Future /MedicalDevices/Safety/CDR HPostmarketSurveillance/UC M pdf FDA and others are working to improve the data and methods used to monitor product performance. This assessment proposes actions focused in four areas: Implementing Unique Device Identifiers (UDI) Registry development Modernizing Adverse Event Reporting and Analysis Develop new methods for evidence generation and synthesis 67

68 The Future Proposed Mission Statement: The National Medical Device Postmarket Surveillance System (MDS) supports optimal patient care by leveraging the experiences of patients to inform decisions about medical device safety, effectiveness, and quality in order to promote the public health. Goals: Coordinate medical device postmarket activities to ensure there is a harmonized national approach focused on improving /AboutFDA/CentersOffices/Off evidence and reducing burden. iceofmedicalproductsandtoba Build and facilitate access to data partners cco/cdrh/cdrhreports/uc using the emerging national electronic M pdf information infrastructure. 68

69 Any Questions? 69
Incorporating Risk Management into Complaint and Service Experience. Presented by: Richard J. DeRisio, M.S. Kinetic Concepts, Inc.
 Incorporating Risk Management into Complaint and Service Experience Presented by: Richard J. DeRisio, M.S. Kinetic Concepts, Inc. 1 Management of Complaint and Service Experience A Compliance Headache?
Incorporating Risk Management into Complaint and Service Experience Presented by: Richard J. DeRisio, M.S. Kinetic Concepts, Inc. 1 Management of Complaint and Service Experience A Compliance Headache?
AAMI Quality Systems White Paper: Comparison of 21 CFR Part 820 to ISO 13485:2016 1
 AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward, Owner, Ward Sciences
Best Practices: Integration of Risk Management and Corrective and Preventive Action
 Best Practices: Integration of Risk Management and Corrective and Preventive Action Presented by: Norman L. Collazo Worldwide Director of Strategic Quality Cordis Corporation, a Johnson & Johnson company
Best Practices: Integration of Risk Management and Corrective and Preventive Action Presented by: Norman L. Collazo Worldwide Director of Strategic Quality Cordis Corporation, a Johnson & Johnson company
Supplier Quality Agreements
 Supplier Quality Agreements Dan O Leary CBA, CQA, CQE, CRE, SSBB, CIRM President Ombu Enterprises, LLC Dan@OmbuEnterprises.com www.ombuenterprises.com 603-209-0600 OMBU ENTERPRISES, LLC 1 Outline QSR Requirements
Supplier Quality Agreements Dan O Leary CBA, CQA, CQE, CRE, SSBB, CIRM President Ombu Enterprises, LLC Dan@OmbuEnterprises.com www.ombuenterprises.com 603-209-0600 OMBU ENTERPRISES, LLC 1 Outline QSR Requirements
Post Market Surveillance
 Post Market Surveillance December 6 th, 2017 Michael Maier, Medidee Services Slide 1 Agenda Postmarket surveillance Postmarket clinical follow up (PMCF) Incident reporting What changes with MDR Slide 2
Post Market Surveillance December 6 th, 2017 Michael Maier, Medidee Services Slide 1 Agenda Postmarket surveillance Postmarket clinical follow up (PMCF) Incident reporting What changes with MDR Slide 2
Post market Surveillance ISO EU Medical Device Regulation
 Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
 ZOLL Document Number: 90E0021 Page 6 of 50 QUALITY SYSTEM MANUAL ZOLL Document Number: 90E0021 Page 7 of 50 1 EXECUTIVE MANAGEMENT APPROVAL... 10 2 INTRODUCTION... 11 3 QUALITY SYSTEM... 12 3.1 QUALITY
ZOLL Document Number: 90E0021 Page 6 of 50 QUALITY SYSTEM MANUAL ZOLL Document Number: 90E0021 Page 7 of 50 1 EXECUTIVE MANAGEMENT APPROVAL... 10 2 INTRODUCTION... 11 3 QUALITY SYSTEM... 12 3.1 QUALITY
Windchill Quality Management
 Windchill Quality Management Closed-Loop Quality for Medical Device Innovators Windchill Quality Management is purpose-built for medical device makers who seek to accelerate delivery of breakthrough medical
Windchill Quality Management Closed-Loop Quality for Medical Device Innovators Windchill Quality Management is purpose-built for medical device makers who seek to accelerate delivery of breakthrough medical
Overview of Good Food Laboratory Practices
 Overview of Good Food Laboratory Practices Dr. Anne Bridges Technical Director, AACC International International Workshop & Training Program On Good Food Laboratory Practices Jointly Organized By Food
Overview of Good Food Laboratory Practices Dr. Anne Bridges Technical Director, AACC International International Workshop & Training Program On Good Food Laboratory Practices Jointly Organized By Food
Quality Manual. This manual complies with the requirements of the ISO 9001:2015 International Standard. AW2 Logistics, Inc Ace Industrial Dr.
 Quality Manual This manual complies with the requirements of the ISO 9001:2015 International Standard. AW2 Logistics, Inc. 6001 Ace Industrial Dr. Cudahy, WI 53210 Quality Manual Rev 3 Page 1 of 30 Table
Quality Manual This manual complies with the requirements of the ISO 9001:2015 International Standard. AW2 Logistics, Inc. 6001 Ace Industrial Dr. Cudahy, WI 53210 Quality Manual Rev 3 Page 1 of 30 Table
MDSAP AUDIT PROCESS. A Manufacturer s Perspective. Connie Hoy EVP Regulatory Affairs Cynosure, Inc.
 MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
Adverse Event Reporting
 Adverse Event Reporting AE Case Receipt When we receive a case, we induct it through a well-oiled process that reduces the number of subsequent queries, classifies events appropriately, and increases the
Adverse Event Reporting AE Case Receipt When we receive a case, we induct it through a well-oiled process that reduces the number of subsequent queries, classifies events appropriately, and increases the
YOU RE CLOSER THAN YOU THINK
 YOU RE CLOSER THAN YOU THINK ISO 13485:2016 READINESS CHECKLIST We understand that some of our customers would like to be able to check how close they are to meeting the requirements of ISO 13485:2016.
YOU RE CLOSER THAN YOU THINK ISO 13485:2016 READINESS CHECKLIST We understand that some of our customers would like to be able to check how close they are to meeting the requirements of ISO 13485:2016.
Lifecycle Product Quality Risk Management
 Lifecycle Product Quality Risk Management Richard L. Friedman, M.S. Associate Director Office of Manufacturing and Product Quality Office of Compliance IFPAC Annual Meeting (Arlington, VA) January, 21-24,
Lifecycle Product Quality Risk Management Richard L. Friedman, M.S. Associate Director Office of Manufacturing and Product Quality Office of Compliance IFPAC Annual Meeting (Arlington, VA) January, 21-24,
Revision. Quality Manual. Multilayer Prototypes. Compliant to ISO / AS9100 Rev C
 1 of 29 Quality Manual Multilayer Prototypes Compliant to ISO 9001-2008 / AS9100 Rev C This Quality Manual sets forth the quality system policies and Defines compliance with the ISO 9001-2008 SAE AS 9100
1 of 29 Quality Manual Multilayer Prototypes Compliant to ISO 9001-2008 / AS9100 Rev C This Quality Manual sets forth the quality system policies and Defines compliance with the ISO 9001-2008 SAE AS 9100
The following is an example systems manual from a low volume (TE, but not an automotive supplier) company.
 company.") The following is an example systems manual from a low volume (TE, but not an automotive supplier) company. You will note that this is essentially a copy of ISO 9001:2000. I take this path because long
The following is an example systems manual from a low volume (TE, but not an automotive supplier) company. You will note that this is essentially a copy of ISO 9001:2000. I take this path because long
Association of American Railroads Quality Assurance System Evaluation (QASE) Checklist Rev. 1/12/2017
 Checklist Rev. 1/12/2017") Company: Prepared By: Date: Changes from previous version highlighted in yellow. Paragraph Element Objective Evidence 2.1 Objective of Quality Assurance Program 2.2 Applicability and Scope 2.3 QA Program
Company: Prepared By: Date: Changes from previous version highlighted in yellow. Paragraph Element Objective Evidence 2.1 Objective of Quality Assurance Program 2.2 Applicability and Scope 2.3 QA Program
Quality Management System MANUAL. SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702
 Quality Management System MANUAL SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702 Doc. No. G5500 Rev. 11 Status : APPROVED Effective: 09/07/2017 Page 2 of 32 Quality Manual Table of Contents
Quality Management System MANUAL SDIX, LLC Headquarters: 111 Pencader Drive Newark, Delaware 19702 Doc. No. G5500 Rev. 11 Status : APPROVED Effective: 09/07/2017 Page 2 of 32 Quality Manual Table of Contents
Audit of Weighing Services. Audit and Evaluation Services Final Report Canadian Grain Commission
 Audit and Evaluation Services Final Report Canadian Grain Commission November 2016 Table of Contents 1. EXECUTIVE SUMMARY... 2 Conclusion... 2 Statement of Assurance... 2 2. INTRODUCTION... 3 Authority
Audit and Evaluation Services Final Report Canadian Grain Commission November 2016 Table of Contents 1. EXECUTIVE SUMMARY... 2 Conclusion... 2 Statement of Assurance... 2 2. INTRODUCTION... 3 Authority
WHO Prequalification of In Vitro Diagnostics Programme
 P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Manufacturing Site(s) Inspection (Assessment of the Quality Management System) WHO Prequalification
P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Manufacturing Site(s) Inspection (Assessment of the Quality Management System) WHO Prequalification
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards. 26 th September / V. Fischer / Rev. 01
 ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
The power of the Converge platform lies in the ability to share data across all aspects of risk management over a secure workspace.
 Converge Platform The transition to value-based care is breaking down the barriers between the CNO, CMO, and Chief Legal Counsel in managing enterprise risk. It s time to take a proactive systems approach
Converge Platform The transition to value-based care is breaking down the barriers between the CNO, CMO, and Chief Legal Counsel in managing enterprise risk. It s time to take a proactive systems approach
QUALITY MANUAL Revision H
 QUALITY MANUAL Revision H JADE PRECISION MEDICAL COMPONENTS, LLC 3063 B Philmont Avenue Huntingdon Valley, PA 19006 Page: 1 of 15 Table of Contents 1. Purpose & Scope... 2 2. Applicable Standards... 2
QUALITY MANUAL Revision H JADE PRECISION MEDICAL COMPONENTS, LLC 3063 B Philmont Avenue Huntingdon Valley, PA 19006 Page: 1 of 15 Table of Contents 1. Purpose & Scope... 2 2. Applicable Standards... 2
RE: FDA-2011-N-0090: Food and Drug Administration; Unique Device Identification System
 November 7, 2012 Margaret Hamburg Commissioner Food and Drug Administration Department of Health and Human Services Room 2217 White Oak Building One 10903 New Hampshire Avenue Silver Spring, MD 20993 RE:
November 7, 2012 Margaret Hamburg Commissioner Food and Drug Administration Department of Health and Human Services Room 2217 White Oak Building One 10903 New Hampshire Avenue Silver Spring, MD 20993 RE:
Metrics in Microbiology Monitoring Practices
 Metrics in Microbiology Monitoring Practices Crystal Booth, M.M. Masters of Microbiology from North Carolina State University Former Associate Director of Microbiology at Novartis Metrics in Microbiology
Metrics in Microbiology Monitoring Practices Crystal Booth, M.M. Masters of Microbiology from North Carolina State University Former Associate Director of Microbiology at Novartis Metrics in Microbiology
9001:2015. Quality Manual 9001:2015. Engineering Value through Quality and Innovation.
 Quality Manual 9001:2015 9001:2015 Suburban Manufacturing, Inc. encourages comments from customers, suppliers, employees and friends in order to improve our business and its performance. Information contained
Quality Manual 9001:2015 9001:2015 Suburban Manufacturing, Inc. encourages comments from customers, suppliers, employees and friends in order to improve our business and its performance. Information contained
ISO 9001: 2000 (December 13, 2000) QUALITY MANAGEMENT SYSTEM DOCUMENTATION OVERVIEW MATRIX
 QUALITY MANAGEMENT SYSTEM DOCUMENTATION OVERVIEW MATRIX") In completing your Documented Quality Management System Review, it is important that the following matrix be completed and returned to us as soon as possible. This will save time during the review and
In completing your Documented Quality Management System Review, it is important that the following matrix be completed and returned to us as soon as possible. This will save time during the review and
SQF 2000 Guidance. Guidance for Food Sector Category 4 Fresh Produce Pack House Operations. 1st Edition
 SQF 2000 for Food Sector Category 4 Fresh Produce Pack House Operations 1st Edition SEPTEMBER 2010 Safe Quality Food Institute 2345 Crystal Drive, Suite 800 Arlington, VA 22202 USA 202-220-0635 www.sqfi.com
SQF 2000 for Food Sector Category 4 Fresh Produce Pack House Operations 1st Edition SEPTEMBER 2010 Safe Quality Food Institute 2345 Crystal Drive, Suite 800 Arlington, VA 22202 USA 202-220-0635 www.sqfi.com
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155
 MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
ISO General Requirements for Competence of Testing Laboratories Procedure
 Competence of Testing Laboratories Page: 1 of 24 ISO 17025 General Requirements for Competence of Testing Laboratories Procedure Competence of Testing Laboratories Page: 2 of 24 Table of Contents Para.
Competence of Testing Laboratories Page: 1 of 24 ISO 17025 General Requirements for Competence of Testing Laboratories Procedure Competence of Testing Laboratories Page: 2 of 24 Table of Contents Para.
15. Process improvement
 15. Process 15-1: Continual concept Role in quality management system Process, one of the 12 quality system essentials, establishes a programme for helping to ensure continual in laboratory quality over
15. Process 15-1: Continual concept Role in quality management system Process, one of the 12 quality system essentials, establishes a programme for helping to ensure continual in laboratory quality over
CSV Inspection Readiness through Effective Document Control. Eileen Cortes April 27, 2017
 CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
Joint WQHO/UNICEF/UNFPA suppliers meeting
 Joint WQHO/UNICEF/UNFPA suppliers meeting Pre-qualification of male latex condoms Tuesday 24 th November 2015 David Hill David Hill and Associates Pre-qualification of manufacturing sites UNFPA inspector
Joint WQHO/UNICEF/UNFPA suppliers meeting Pre-qualification of male latex condoms Tuesday 24 th November 2015 David Hill David Hill and Associates Pre-qualification of manufacturing sites UNFPA inspector
QUALITY SYSTEM MANUAL
 QUALITY SYSTEM MANUAL This Manual is a Proprietary Document and any Unauthorized Reproduction is prohibited. ISSUE DATE July 26, 2012 AUTHORIZED BY: Quality Management Representative Eric Hoff Managing
QUALITY SYSTEM MANUAL This Manual is a Proprietary Document and any Unauthorized Reproduction is prohibited. ISSUE DATE July 26, 2012 AUTHORIZED BY: Quality Management Representative Eric Hoff Managing
Systematic Risk Management: An Overview of ICH Q-9
 Systematic Risk Management: An Overview of ICH Q-9 March 12, 2014 12014 ParagonRx International LLC Today s Session Systematic Risk Management: An Overview of ICH Q-9 Speakers: Jeff Fetterman, President,
Systematic Risk Management: An Overview of ICH Q-9 March 12, 2014 12014 ParagonRx International LLC Today s Session Systematic Risk Management: An Overview of ICH Q-9 Speakers: Jeff Fetterman, President,
Post Market Surveillance (including PMCF): common non compliances
: common non compliances") Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
Vendor Qualification Survey
 1200 West 96 th St Minneapolis, MN 55431 Ph: 952-888-7900 Fax: 952-888-2719 Vendor Qualification Survey Vendor Information Company Name: Date: Address: City: Phone Number: email address: Product or Service
1200 West 96 th St Minneapolis, MN 55431 Ph: 952-888-7900 Fax: 952-888-2719 Vendor Qualification Survey Vendor Information Company Name: Date: Address: City: Phone Number: email address: Product or Service
Recommendations for Strengthening the Investigator Site Community
 Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
Immunization Information System (IIS) Trainer Sample Role Description
 Trainer Sample Role Description") Immunization Information System (IIS) Trainer Sample Role Description March 2016 0 Note: This role description is meant to offer sample language and a comprehensive list of potential desired responsibilities
Immunization Information System (IIS) Trainer Sample Role Description March 2016 0 Note: This role description is meant to offer sample language and a comprehensive list of potential desired responsibilities
Pass4sure.ITIL-F.347.QA
 Pass4sure.ITIL-F.347.QA Number: ITIL-F Passing Score: 800 Time Limit: 120 min File Version: 19.1 http://www.gratisexam.com/ ITIL-F.EN.dat ITIL Foundation Enjoy the real success with nicely written Questions
Pass4sure.ITIL-F.347.QA Number: ITIL-F Passing Score: 800 Time Limit: 120 min File Version: 19.1 http://www.gratisexam.com/ ITIL-F.EN.dat ITIL Foundation Enjoy the real success with nicely written Questions
Risk Based Approach To Complaint Handling
 Risk Based Approach To Complaint Handling Khaudeja Bano, M.D. Senior Medical Director, Medical Device Safety Head, AbbVie Inc. Life Sciences Product Complaints Congress Europe Dublin, Ireland December
Risk Based Approach To Complaint Handling Khaudeja Bano, M.D. Senior Medical Director, Medical Device Safety Head, AbbVie Inc. Life Sciences Product Complaints Congress Europe Dublin, Ireland December
A02 Assessment Rating Guide Revision 2.9 August 21, 2016
 Revision 2.9 August 21, 2016 Laboratory Name: Assessment Date: (Lead) Assessor: Signature: ! TABLE OF CONTENTS 1.0! INTRODUCTION... 1 2.0! ASSESSOR NOTES ON THE USE OF CALA A02 ASSESSMENT RATING GUIDE...
Revision 2.9 August 21, 2016 Laboratory Name: Assessment Date: (Lead) Assessor: Signature: ! TABLE OF CONTENTS 1.0! INTRODUCTION... 1 2.0! ASSESSOR NOTES ON THE USE OF CALA A02 ASSESSMENT RATING GUIDE...
- Rev 4 Date
 Product Safety Management Manual - Rev 4 Date 10.05.2013 - Revision Index Revision no Change description Date 0 Initial release 30.12.2008 1 Part identification guidelines for traceability 25.07.2009 included
Product Safety Management Manual - Rev 4 Date 10.05.2013 - Revision Index Revision no Change description Date 0 Initial release 30.12.2008 1 Part identification guidelines for traceability 25.07.2009 included
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order
 Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL. ISO 22000:2005 Quality Manual
 HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL ISO 22000:2005 Quality Manual QUALITY MANUAL ISO 22000:2005 Food Safety Management HACCPEuropa Publications 2012 Table of Contents Introduction...
HACCPEUROPA PUBLICATIONS ISO 22000:2005 FOOD SAFETY QUALITY MANUAL ISO 22000:2005 Quality Manual QUALITY MANUAL ISO 22000:2005 Food Safety Management HACCPEuropa Publications 2012 Table of Contents Introduction...
OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS
 D i a g n o s t i c s a n d L a b o r a t o r y T e c h n o l o g y OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS Prequalification of Male Circumcision Devices PQMC_007
D i a g n o s t i c s a n d L a b o r a t o r y T e c h n o l o g y OVERVIEW OF THE PREQUALIFICATION OF MALE CIRCUMCISION DEVICES ASSESSMENT PROCESS Prequalification of Male Circumcision Devices PQMC_007
4. RELATED DOCUMENTS AND FORMS 4.1. AS ISO Cambridge Valley Machining, Inc. QMS procedures, including, but not limited to:
 Approved By: Purchasing Manager Candice Lane (signature on file) Date: 5/26/17 Approved By: Process Owner James D. Moore (signature on file) Date: 5/26/17 1. PURPOSE 1.1. To define Cambridge Valley Machining,
Approved By: Purchasing Manager Candice Lane (signature on file) Date: 5/26/17 Approved By: Process Owner James D. Moore (signature on file) Date: 5/26/17 1. PURPOSE 1.1. To define Cambridge Valley Machining,
Medical Devices. Epicor for. Functionality. Meeting the Challenges for Medical Devices
 Epicor for Medical Devices Functionality XXGlobal operations XXTraceability and quality XXCost management XXRapid configuration XXProduct lifecycle management XXRegulatory compliance XXFDA, cgmp, ISO,
Epicor for Medical Devices Functionality XXGlobal operations XXTraceability and quality XXCost management XXRapid configuration XXProduct lifecycle management XXRegulatory compliance XXFDA, cgmp, ISO,
Chapter 2 GALP Implementation Assistance
 Chapter 2 GALP The GALP Implementation is based on established data management principles. 1. PRINCIPLES Control is the essential objective behind most data management principles. Effective management
Chapter 2 GALP The GALP Implementation is based on established data management principles. 1. PRINCIPLES Control is the essential objective behind most data management principles. Effective management
Prevent Quality System Deficiencies by Conducting Effective Internal Audits. Whitepaper
 Prevent Quality System Deficiencies by Conducting Effective Internal Audits Whitepaper An internal audit system is one of the most effective ways to monitor, analyze, control, and improve quality management
Prevent Quality System Deficiencies by Conducting Effective Internal Audits Whitepaper An internal audit system is one of the most effective ways to monitor, analyze, control, and improve quality management
The Impact of Quality Culture on Quality Risk Management. FDA Perspective on Quality Culture; how it Impacts Risk Management
 The Impact of Quality Culture on Quality Risk Management FDA Perspective on Quality Culture; how it Impacts Risk Management Teresa Gorecki Practice Lead Compliance Architects Agenda The WHAT Definitions
The Impact of Quality Culture on Quality Risk Management FDA Perspective on Quality Culture; how it Impacts Risk Management Teresa Gorecki Practice Lead Compliance Architects Agenda The WHAT Definitions
AUTOMOTIVE SPICE v3.1 POCKET GUIDE
 EXTENDED VDA SCOPE ASPICE v3.1 AUTOMOTIVE SPICE v3.1 POCKET GUIDE 4 5 6 7 8-9 10 11-13 14-15 16-19 20-43 44-49 50-51 52-69 70-93 94-103 104-105 106 Automotive SPICE at a glance Automotive SPICE application
EXTENDED VDA SCOPE ASPICE v3.1 AUTOMOTIVE SPICE v3.1 POCKET GUIDE 4 5 6 7 8-9 10 11-13 14-15 16-19 20-43 44-49 50-51 52-69 70-93 94-103 104-105 106 Automotive SPICE at a glance Automotive SPICE application
Pharmaceutical Quality for 21 st Century Initiative
 Quality Metrics Alicia Mozzachio, RPh, MPH Senior Advisor for International Activities Office of Policy for Pharmaceutical Quality (OPPQ) Center for Drug Evaluation and Research U.S. Food and Drug Administration
Quality Metrics Alicia Mozzachio, RPh, MPH Senior Advisor for International Activities Office of Policy for Pharmaceutical Quality (OPPQ) Center for Drug Evaluation and Research U.S. Food and Drug Administration
Moving from ISO/TS 16949:2009 to IATF 16949:2016. Transition Guide
 Moving from ISO/TS 16949:2009 to IATF 16949:2016 Transition Guide IATF 16949:2016 - Automotive Quality Management System - Transition Guide An effective Quality Management System is vital for organizations
Moving from ISO/TS 16949:2009 to IATF 16949:2016 Transition Guide IATF 16949:2016 - Automotive Quality Management System - Transition Guide An effective Quality Management System is vital for organizations
Project Management. Objectives 3/17/2015. David Borrill, MT, MBA, PMP
 Project Management David Borrill, MT, MBA, PMP Objectives Nobody falls asleep or walks out. Presentation of some of the main points in the management of projects in the healthcare environment. Everybody
Project Management David Borrill, MT, MBA, PMP Objectives Nobody falls asleep or walks out. Presentation of some of the main points in the management of projects in the healthcare environment. Everybody
Hong Kong Deposit Protection Board
 Hong Kong Deposit Protection Board Independent Assessment Program and Self-Declaration for Compliance with the Guideline on Information Required for Determining and Paying Compensation ( Program Guide
Hong Kong Deposit Protection Board Independent Assessment Program and Self-Declaration for Compliance with the Guideline on Information Required for Determining and Paying Compensation ( Program Guide
Medical Device Purchasing Controls Challenges of Compliance in a World Market June, 2013 OMTEC
 Medical Device Purchasing Controls Challenges of Compliance in a World Market June, 2013 OMTEC - 2013 Quality Business Acceptance Activities 1 ..FDA Across the Globe.. 2 ..Warning Letters and FDA 483 update..
Medical Device Purchasing Controls Challenges of Compliance in a World Market June, 2013 OMTEC - 2013 Quality Business Acceptance Activities 1 ..FDA Across the Globe.. 2 ..Warning Letters and FDA 483 update..
Business Management System Manual Conforms to ISO 9001:2015 Table of Contents
 Table of Contents 1.0 Welcome to Crystalfontz... 3 2.0 About the Crystalfontz Business Systems Manual... 4 3.0 Terms and Conditions... 5 4.0 Context of the Organization... 6 4.1. Understanding the Organization
Table of Contents 1.0 Welcome to Crystalfontz... 3 2.0 About the Crystalfontz Business Systems Manual... 4 3.0 Terms and Conditions... 5 4.0 Context of the Organization... 6 4.1. Understanding the Organization
Correspondence Between ISO 13485:2016 and 21 CFR Part 820 QMS Requirements
 Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
Laboratory Challenges with TNI Implementation and Laboratory Audits
 Laboratory Challenges with TNI Implementation and Laboratory Audits December 14, 2017 Dr. Donna Ferguson, Director, Monterey County Public Health Laboratory Dr. Martina McGarvey, Director, Pennsylvania
Laboratory Challenges with TNI Implementation and Laboratory Audits December 14, 2017 Dr. Donna Ferguson, Director, Monterey County Public Health Laboratory Dr. Martina McGarvey, Director, Pennsylvania
SUPPLIER GUIDELINES MANUAL
 1.0 INTRODUCTION 1.1 This guidelines manual defines the minimum requirements for Supreme Machined Products Co.; Inc. LTD (to be referenced as Supreme throughout the document) Supplier s and does not supersede
1.0 INTRODUCTION 1.1 This guidelines manual defines the minimum requirements for Supreme Machined Products Co.; Inc. LTD (to be referenced as Supreme throughout the document) Supplier s and does not supersede
FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru
 FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru FDA QSR (21 CFR Part 820) ISO 13485:2016 820.1 Scope 1 Scope 2 Normative References 820.3 Definitions 3 Terms and Definitions
FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru FDA QSR (21 CFR Part 820) ISO 13485:2016 820.1 Scope 1 Scope 2 Normative References 820.3 Definitions 3 Terms and Definitions
KPMG s Major Projects Advisory Project Leadership Series: Stakeholder Management and Communication
 KPMG Global Energy Institute KPMG International KPMG s Major Projects Advisory Project Leadership Series: Stakeholder Management and Communication Stakeholder management and communication is critical to
KPMG Global Energy Institute KPMG International KPMG s Major Projects Advisory Project Leadership Series: Stakeholder Management and Communication Stakeholder management and communication is critical to
Update on ISO/DIS 45001:2016 Migration from OHSAS 18001:2007. May 31, 2016 Our webinar will begin at 1:00 PM
 Update on ISO/DIS 45001:2016 Migration from OHSAS 18001:2007 May 31, 2016 Our webinar will begin at 1:00 PM Update on ISO/DIS 45001:2016 Migration from OHSAS 18001:2007 Carmine Liuzzi Industry Leader SAI
Update on ISO/DIS 45001:2016 Migration from OHSAS 18001:2007 May 31, 2016 Our webinar will begin at 1:00 PM Update on ISO/DIS 45001:2016 Migration from OHSAS 18001:2007 Carmine Liuzzi Industry Leader SAI
UPPLIER ANUAL. Issued: 01 Aug 13
 UPPLIER ANUAL Issued: 01 Aug 13 Table of Contents Our Company 3 Our Vision 3 Scope and Purpose 4 Responsibilities 4 General Expectations and Requirements 5 Supplier Quality Management System 6 Supplier
UPPLIER ANUAL Issued: 01 Aug 13 Table of Contents Our Company 3 Our Vision 3 Scope and Purpose 4 Responsibilities 4 General Expectations and Requirements 5 Supplier Quality Management System 6 Supplier
THE COMMANDER NAVY REGION, SOUTHWEST (CNRSW) HAZARDOUS MATERIALS (HAZMAT) OPERATIONS QUALITY ASSURANCE SURVEILLANCE PLAN (QASP) 20 June 2000
 HAZARDOUS MATERIALS (HAZMAT) OPERATIONS QUALITY ASSURANCE SURVEILLANCE PLAN (QASP) 20 June 2000") THE COMMANDER NAVY REGION, SOUTHWEST (CNRSW) HAZARDOUS MATERIALS (HAZMAT) OPERATIONS QUALITY ASSURANCE SURVEILLANCE PLAN (QASP) 20 June 2000 TABLE OF CONTENTS 1.0 INTRODUCTION 1 1.1 PURPOSE... 1 2.0 OVERVIEW.2
THE COMMANDER NAVY REGION, SOUTHWEST (CNRSW) HAZARDOUS MATERIALS (HAZMAT) OPERATIONS QUALITY ASSURANCE SURVEILLANCE PLAN (QASP) 20 June 2000 TABLE OF CONTENTS 1.0 INTRODUCTION 1 1.1 PURPOSE... 1 2.0 OVERVIEW.2
GUIDE TO INSPECTIONS OF QUALITY SYSTEMS
 FOOD AND DRUG ADMINISTRATION GUIDE TO INSPECTIONS OF QUALITY SYSTEMS 1 1 August 1999 2 Guide to Inspections of Quality Systems This document was developed by the Quality System Inspections Reengineering
FOOD AND DRUG ADMINISTRATION GUIDE TO INSPECTIONS OF QUALITY SYSTEMS 1 1 August 1999 2 Guide to Inspections of Quality Systems This document was developed by the Quality System Inspections Reengineering
CUSTOMER AND SUPPLIER ROLES AND RESPONSIBILITIES FOR 21 CFR 11 COMPLIANCE ASSESSMENT. 21 CFR Part 11 FAQ. (Frequently Asked Questions)
") 21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
9100 revision Changes presentation clause-by-clause. IAQG 9100 Team November 2016
 Changes presentation clause-by-clause IAQG 9100 Team November 2016 INTRODUCTION In September 2016, a revision of the 9100 standard has been published by the IAQG (International Aerospace Quality Group)
Changes presentation clause-by-clause IAQG 9100 Team November 2016 INTRODUCTION In September 2016, a revision of the 9100 standard has been published by the IAQG (International Aerospace Quality Group)
Supplier Quality Manual
 ALLIANCE ELECTRONICS DISTRIBUTOR Supplier Quality Manual 22412 Gilberto Rd. Rancho Santa Margarita, CA 92688 INTRODUCTION Welcome to Alliance Electronics Distributor (AED) Alliance Electronics Distributor
ALLIANCE ELECTRONICS DISTRIBUTOR Supplier Quality Manual 22412 Gilberto Rd. Rancho Santa Margarita, CA 92688 INTRODUCTION Welcome to Alliance Electronics Distributor (AED) Alliance Electronics Distributor
The Risk Management + Design Controls Connection: What Device Makers Need to Know
 !!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
!!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
ISO 14001: 2015 Environmental Gap Analysis
 Environmental Gap Analysis The revised ISO 14001 standard was published on 14 TH September 2015. How to use this document This document provides an overview of the changes between ISO 14001:2004 and ISO
Environmental Gap Analysis The revised ISO 14001 standard was published on 14 TH September 2015. How to use this document This document provides an overview of the changes between ISO 14001:2004 and ISO
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior
 Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Review of FMMIS and DSS Assessment Project Procurement Divisions of Operations and Medicaid
 Report No. 13-08 February 2013 Office of the Inspector General Bureau of Internal Audit Review of FMMIS and DSS Assessment Project Procurement Divisions of Operations and Medicaid Executive Summary At
Report No. 13-08 February 2013 Office of the Inspector General Bureau of Internal Audit Review of FMMIS and DSS Assessment Project Procurement Divisions of Operations and Medicaid Executive Summary At
AS9003A QUALITY MANUAL
 Your Logo AS9003A QUALITY MANUAL Origination Date: (month/year) Document Identifier: Date: Document Status: Document Link: AS9003A Quality Manual Latest Revision Date Draft, Redline, Released, Obsolete
Your Logo AS9003A QUALITY MANUAL Origination Date: (month/year) Document Identifier: Date: Document Status: Document Link: AS9003A Quality Manual Latest Revision Date Draft, Redline, Released, Obsolete
Quality Management System
 Quality Management System ZYQ Testing Laboratory 11111 Testing Avenue Testing Town, Alaska 22222 (555) 555-5555 Date Prepared: January 19, 2009 1 Please note that this Quality Management System (QM) was
Quality Management System ZYQ Testing Laboratory 11111 Testing Avenue Testing Town, Alaska 22222 (555) 555-5555 Date Prepared: January 19, 2009 1 Please note that this Quality Management System (QM) was
Guide for Distributors of Medical Devices
 Guide for Distributors of Medical Devices IA-G0004-1 8 FEBRUARY 2018 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes only. CONTENTS 1 SCOPE 3
Guide for Distributors of Medical Devices IA-G0004-1 8 FEBRUARY 2018 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes only. CONTENTS 1 SCOPE 3
INS QA Programme Requirements
 Specification Date: 20/3/17 INS QA Programme Requirements UNCONTROLLED WHEN PRINTED Author: J Cooch AUTHORISATION Date: 20/3/17 A Brown Owner: J Cooch (Signature) N.B. only required for hard copy If issued
Specification Date: 20/3/17 INS QA Programme Requirements UNCONTROLLED WHEN PRINTED Author: J Cooch AUTHORISATION Date: 20/3/17 A Brown Owner: J Cooch (Signature) N.B. only required for hard copy If issued
NEMA Standards Publication NEMA/MITA 1. Good Refurbishment Practices for Medical Imaging Equipment
 NEMA Standards Publication NEMA/MITA 1 Good Refurbishment Practices for Medical Imaging Equipment Published by: National Electrical Manufacturers Association 1300 North 17 th Street, Suite 900 Rosslyn,
NEMA Standards Publication NEMA/MITA 1 Good Refurbishment Practices for Medical Imaging Equipment Published by: National Electrical Manufacturers Association 1300 North 17 th Street, Suite 900 Rosslyn,
Quality Manual. This manual complies with the requirements of the ISO 9001:2015 International Standard.
 Quality Manual This manual complies with the requirements of the ISO 9001:2015 International Standard. Northeast Power Systems, Inc. 66 Carey Road Queensbury, New York 12804 Quality Manual Rev 0 Printed
Quality Manual This manual complies with the requirements of the ISO 9001:2015 International Standard. Northeast Power Systems, Inc. 66 Carey Road Queensbury, New York 12804 Quality Manual Rev 0 Printed
Intland s Medical IEC & ISO Template
 Intland s Medical IEC 62304 & ISO 14971 Template Intland s Medical IEC 62304 & ISO 14971 Template codebeamer ALM for Medical Device Development Intland s Medical IEC 62304 & ISO 14971 Template Medical
Intland s Medical IEC 62304 & ISO 14971 Template Intland s Medical IEC 62304 & ISO 14971 Template codebeamer ALM for Medical Device Development Intland s Medical IEC 62304 & ISO 14971 Template Medical
Quality Management System Guidance. ISO 9001:2015 Clause-by-clause Interpretation
 Quality Management System Guidance ISO 9001:2015 Clause-by-clause Interpretation Table of Contents 1 INTRODUCTION... 4 1.1 IMPLEMENTATION & DEVELOPMENT... 5 1.2 MANAGING THE CHANGE... 5 1.3 TOP MANAGEMENT
Quality Management System Guidance ISO 9001:2015 Clause-by-clause Interpretation Table of Contents 1 INTRODUCTION... 4 1.1 IMPLEMENTATION & DEVELOPMENT... 5 1.2 MANAGING THE CHANGE... 5 1.3 TOP MANAGEMENT
General Guidance for Developing, Documenting, Implementing, Maintaining, and Auditing an SQF Quality System. Quality Code. SQF Quality Code, Edition 8
 General Guidance for Developing, Documenting, Implementing, Maintaining, and Auditing an SQF Quality System Quality Code SQF Quality Code, Edition 8 October 2017 2014 Safe Quality Food Institute 2345 Crystal
General Guidance for Developing, Documenting, Implementing, Maintaining, and Auditing an SQF Quality System Quality Code SQF Quality Code, Edition 8 October 2017 2014 Safe Quality Food Institute 2345 Crystal
J. McCann & Co (Nottm) Ltd McCann House 110 Nottingham Road Chilwell Nottingham NG9 6DQ. Quality Manual
 Ltd McCann House 110 Nottingham Road Chilwell Nottingham NG9 6DQ. Quality Manual") McCann House 110 Nottingham Road Chilwell Nottingham NG9 6DQ Quality Manual Quality Manual Issue I Dated 8 th October 2012 Contents Page Section 1 - Introduction 2 Section 2 - Company Details 2 Section
McCann House 110 Nottingham Road Chilwell Nottingham NG9 6DQ Quality Manual Quality Manual Issue I Dated 8 th October 2012 Contents Page Section 1 - Introduction 2 Section 2 - Company Details 2 Section
SUPPLIER QUALITY ASSESSMENT
 Supplier Organization Name: Supplier Number: Street Address: Date of This Audit: City, State, Zip Code: Date of Last Audit: Country: of Employees: Main Phone Number: of Buildings/Size: Fax Number: Principal
Supplier Organization Name: Supplier Number: Street Address: Date of This Audit: City, State, Zip Code: Date of Last Audit: Country: of Employees: Main Phone Number: of Buildings/Size: Fax Number: Principal
PRODUCT COMPLAINTS MANAGEMENT. Infosys Handbook For Life Sciences
 PRODUCT COMPLAINTS MANAGEMENT Infosys Handbook For Life Sciences Table of Contents Introduction 3 Infosys Point of View 4 Success Story - Complaint management for one of the world s top 5 bio-pharmaceutical
PRODUCT COMPLAINTS MANAGEMENT Infosys Handbook For Life Sciences Table of Contents Introduction 3 Infosys Point of View 4 Success Story - Complaint management for one of the world s top 5 bio-pharmaceutical
FINAL DOCUMENT. International Medical Device Regulators Forum. Medical Device Regulatory Audit Reports
 FINAL DOCUMENT International Medical Device Regulators Forum Title: Authoring Group: Medical Device Regulatory Audit Reports IMDRF MDSAP Working Group Date: 2 October 2015 Toshiyoshi Tominaga, IMDRF Chair
FINAL DOCUMENT International Medical Device Regulators Forum Title: Authoring Group: Medical Device Regulatory Audit Reports IMDRF MDSAP Working Group Date: 2 October 2015 Toshiyoshi Tominaga, IMDRF Chair
Larry Strouse Railroad Safety Specialist (HM) Federal Railroad Administration
 Federal Railroad Administration") Transportation Technology Center, Inc., a subsidiary of the Association of American Railroads Larry Strouse Railroad Safety Specialist (HM) Federal Railroad Administration Root Cause Analysis 3:00 to 3:30
Transportation Technology Center, Inc., a subsidiary of the Association of American Railroads Larry Strouse Railroad Safety Specialist (HM) Federal Railroad Administration Root Cause Analysis 3:00 to 3:30
On the Path to ISO Accreditation
 On the Path to ISO 17025 Accreditation What We Wish We d Known Before We Started And Some Definitions: Language of ISO 17025 Version: 2013-08-29 1 Susan Humphries, QA Officer Bureau of Food Laboratories,
On the Path to ISO 17025 Accreditation What We Wish We d Known Before We Started And Some Definitions: Language of ISO 17025 Version: 2013-08-29 1 Susan Humphries, QA Officer Bureau of Food Laboratories,
Current Trends at FDA: Implications for Data Requirements
 Introduction The environment surrounding medical device regulation in the United States has always been rigorous, but recent events including well-publicized quality issues associated with implantable
Introduction The environment surrounding medical device regulation in the United States has always been rigorous, but recent events including well-publicized quality issues associated with implantable
 www.ulehssustainability.com YOUR PARTNER IN EHS, SUSTAINABILITY AND SUCCESS UL EHS Sustainability is the leading environmental, health, safety and sustainability software provider for enterprise clients
www.ulehssustainability.com YOUR PARTNER IN EHS, SUSTAINABILITY AND SUCCESS UL EHS Sustainability is the leading environmental, health, safety and sustainability software provider for enterprise clients
Process validation in medical devices
 Process validation in medical devices Fulfil requirements with expert regulatory guidance 1TÜV SÜD Contents INTRODUCTION 4 VALIDATION PLANNING 5 INSTALLATION QUALIFICATION 7 OPERATIONAL QUALIFICATION 9
Process validation in medical devices Fulfil requirements with expert regulatory guidance 1TÜV SÜD Contents INTRODUCTION 4 VALIDATION PLANNING 5 INSTALLATION QUALIFICATION 7 OPERATIONAL QUALIFICATION 9
NR CHECKLIST Rev. 1. QAM IMP References NBIC Part 3, 1.8 Y N Y N a. Organization. Company Name/Certificate Number: Page 1 of 26
 Company Name/Certificate Number: Page 1 of 26 a. Organization a.1. Has the Organizational Structure of the program identified the levels of management responsible for the Quality System Program, including
Company Name/Certificate Number: Page 1 of 26 a. Organization a.1. Has the Organizational Structure of the program identified the levels of management responsible for the Quality System Program, including
EPICOR, INCORPORATED QUALITY ASSURANCE MANUAL
 EPICOR, INCORPORATED QUALITY ASSURANCE MANUAL Revision: 6 Date 05/18/09 EPICOR, INCORPORATED 1414 E. Linden Avenue P.O. Box 1608 Linden, NJ. 07036-0006 Tel. 1-908-925-0800 Fax 1-908-925-7795 Table of Contents:
EPICOR, INCORPORATED QUALITY ASSURANCE MANUAL Revision: 6 Date 05/18/09 EPICOR, INCORPORATED 1414 E. Linden Avenue P.O. Box 1608 Linden, NJ. 07036-0006 Tel. 1-908-925-0800 Fax 1-908-925-7795 Table of Contents:
A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO COMPLIANCE
 BYLINED ARTICLE ON: A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO 14971 COMPLIANCE By Mark Leimbeck PE Underwriters Laboratories and Larry Kessler Sc.D.
BYLINED ARTICLE ON: A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO 14971 COMPLIANCE By Mark Leimbeck PE Underwriters Laboratories and Larry Kessler Sc.D.
21 CFR Ch. I ( Edition) Personnel Design controls Identification Traceability.
 Personnel Design controls Identification Traceability.") Pt. 820 device together with an explanation of the basis for the estimate; (iv) Information describing the applicant s clinical experience with the device since the HDE was initially approved. This information
Pt. 820 device together with an explanation of the basis for the estimate; (iv) Information describing the applicant s clinical experience with the device since the HDE was initially approved. This information
Quality Manual Revision: C Effective: 03/01/10
 TABLE OF CONTENTS DESCRIPTION SECTION PAGE INTRODUCTION 1.0 1 APPROVAL SIGNATURE PAGE 1.1 1 AMENDMENT RECORD 1.2 2 SCOPE 2.0 3 EXCLUSIONS 2.1 3 CORPORATE POLICY 3.0 3 QUALITY MANAGEMENT SYSTEM 4.0 4 GENERAL
TABLE OF CONTENTS DESCRIPTION SECTION PAGE INTRODUCTION 1.0 1 APPROVAL SIGNATURE PAGE 1.1 1 AMENDMENT RECORD 1.2 2 SCOPE 2.0 3 EXCLUSIONS 2.1 3 CORPORATE POLICY 3.0 3 QUALITY MANAGEMENT SYSTEM 4.0 4 GENERAL
25 D.L. Martin Drive Mercersburg, PA (717)
") QUALITY MANUAL D. L. MARTIN CO. 25 D.L. Martin Drive Mercersburg, PA 17236 (717) 328-2141 Revision 14 August 2012 Michael A. White Manager, QA & Engineering D.L. Martin Co. Quality Manual UNCONTROLLED
QUALITY MANUAL D. L. MARTIN CO. 25 D.L. Martin Drive Mercersburg, PA 17236 (717) 328-2141 Revision 14 August 2012 Michael A. White Manager, QA & Engineering D.L. Martin Co. Quality Manual UNCONTROLLED