The FDA Just Arrived... Are You Ready? Presented By Sandra Lueken Sr. Director, Quality AstraZeneca Biologics
|
|
|
- Abraham Hutchinson
- 5 years ago
- Views:
Transcription
1 The FDA Just Arrived... Are You Ready? Presented By Sandra Lueken Sr. Director, Quality AstraZeneca Biologics
2 Objectives Review Quality Systems Audit Approach and cgmp fundamentals Logistical Audit Preparation Develop your own FDA readiness plan Please Note: The views and opinions in today's presentation are those of the speaker and do not necessarily reflect the views of my employer.
3 FDA; who, what, and why The FDA is a consumer protection agency within the Department of Health & Human Services. They provide oversight for most food products (excluding meat & poultry), Drugs (human & animal), Therapeutic agents of biological origin (blood, vaccines, etc.), Medical devices, Radiation emitting products (x-ray, microwave), Cosmetics, as well as animal feed.
4 Quality Systems Auditing Quality System Inspection Technique: QSIT Still viable? Originally designed for Medical Device manufacturers, QSIT provides guidance to FDA field staff that is used to aid inspectors focus on key elements of a firm s quality system FDA: Guide to Inspections of Quality Systems, August 1999
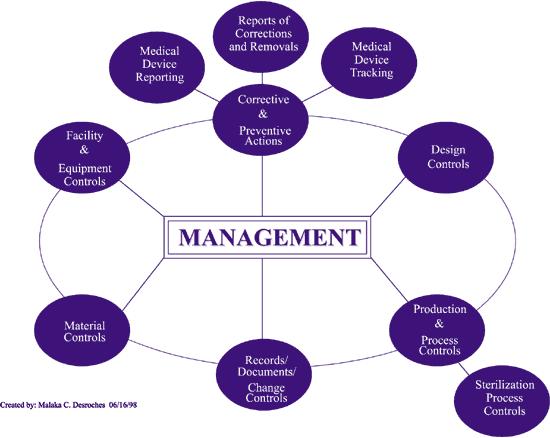
5 Accountable Management
6 Subsystem Top-Down Approach Focus placed on key elements of a firm s quality system The main subsystems covered: Management control Corrective and Preventive Actions (CAPA) Corrections and Removals (Recalls) Design Controls (Process Validation) Production and Process Controls
7 Subsystem Top-Down Approach The FDA is looking specifically to see if a firm has procedures in place that meet cgmp requirements and a detailed review of records to verify that said procedures are implemented in actual production.
8 cgmps Govern our Industry cgmps are the current minimum standards for methods to be used in, and facilities or controls to be used for the manufacture, processing, packing or holding of a drug to assure that it meets its required quality characteristics. Failure to comply shall render the drug to be considered adulterated and the drug and the persons responsible shall be subject to regulatory action.
9 The Purpose of cgmps To assure that all pharmaceutical, biologic, diagnostic and medical device products meet all the requirements of the Federal Food, Drug and Cosmetic Act as to safety and efficacy and have the identity and strength to meet the quality and purity characteristics which they purport to have - Safety - Efficacy - Identity - Strength - Quality - Purity TITLE 21 OF U.S. CODE OF FEDERAL REGULATIONS - "21 CFR"
10 cgmps cover: Organization Personnel Facility Equipment Components, Containers and Closures Production and Process Control Packaging and Labeling Product Holding and Distribution Laboratory Control Records
11 This is not all The FDA not only expects strict adherence to cgmps and CAPA effectiveness, they will audit to ensure management accountability; they are shifting towards innovation and continuous improvement. They are looking for sound data and a statistical approach in support of process validation with continuous monitoring of our processes.
12 Ready, Set, Go Determine who: CBER or CDER Center for Biologics Evaluation and Research (CBER) Center for Drug Evaluation and Research (CDER) Determine type of Audit expected; PAI General GMP For Cause
13 FDA Readiness Plan Pull the appropriate documents relating to the type of Audit expected and review for content Know your Landmines be professionally transparent and confident Prepare presenters and train support staff Perform Facility Walk-throughs know your site, clean inside and outside, paint/repair as needed Establish and know the tour route Dispose of ALL excess Hold and Reject material Review status labeling in ALL storage areas Always maintain proper perspective
14 Example Document Preparation List Manufacturing Quality Regulatory, CMC, Equipment Process validation protocols & reports Batch Production Records Raw material & component usage Cleaning validation Environmental Monitoring Complaints Investigations Failures Reworks/ reprocess Raw data Methods validation Stability Training Development Reports SOPs Change Control Investigations (OOS, Deviations) Field Alerts Recalls SMF DMF System validations (e.g., water, gas, steam computer) Critical Process Parameters (CPP) DQ/IQ/OQ/PQ Equipment Validation and Certifications (HEPA Filters) Preventive Maintenance
15 Logistics Determine the roles of individual members of the inspection team Pre-determine the Go To lead person within each area of the tour Have a contact list 2 levels deep Reserve conference rooms Review production schedules Schedule routine products and activities Best to avoid rework or other challenging activities
16 Example Checklist Audit logistics Plan (location) Documentation list Pre-staged documents Introductory presentation Verification of batch records Draft site communication templates (arrival, daily and final wrap-up) Opening and closing meeting attendees list Complete Hot Topics Identify SMEs, Compile documentation/presentations for each topic. Significant Deviation Presentations Area walk-throughs Remediation of facility issues (cosmetic)
17 After the inspection Prepare for the next inspection by: Maintain your level of cgmp compliance Start fixing problems the day you learn about them Start documenting everything you learn about your products and processes Implement a consistent CAPA system Perform regular, professional self-inspections Maintain senior management s involvement in quality issues Regularly hire outside experts to challenge your operation Follow current Industry trends & 483s, identify and address gaps
18 Being Audit Ready Audit readiness is more than reviewing records, training, cleaning, painting, and sprucing up your facility. Audit readiness is about the decisions you make daily. It s following cgmps and always having the objective evidence to support all components of product release decisions.
19 Questions? Thank you!
20 Acknowledgements/References Barry A. Friedman, Ph.D., Preparing for the Pre-Approval Inspection What to do Before the FDA Arrives. Guide to Inspections of Quality Systems, U.S. Department of Health and Human Services Food and Drug Administration, August 1999 Guidance for Industry Process Validation: General Principles and Practices, U.S. Department of Health and Human Services Food and Drug Administration, January 2011, Current Good Manufacturing Practices (CGMP) Revision 1