Europe s new approach to assurance of API quality and its implications for manufacturers and producing countries
|
|
|
- Cornelia French
- 5 years ago
- Views:
Transcription
1 Europe s new approach to assurance of API quality and its implications for manufacturers and producing countries IPA / EDQM / WHO Mumbai Conference 28 September 2012 Dr Florence Benoit-Guyod, EDQM Inspector, Certification of substances Division, EDQM 1
2 INTRODUCTION Overview Council of Europe and the European Union EDQM and the European Pharmacopeia The EU regulatory framework NEWS Submission of API Quality Data in EU The ICH Q8 Q9 Q10 guidelines The Falsified Medicines Directive 2

3 The Council of Europe Founded in Member States 800 million inhabitants Development of common and democratic principles: Democracy Rule of law Human rights Same flag as EU 2 different independent organisations 3
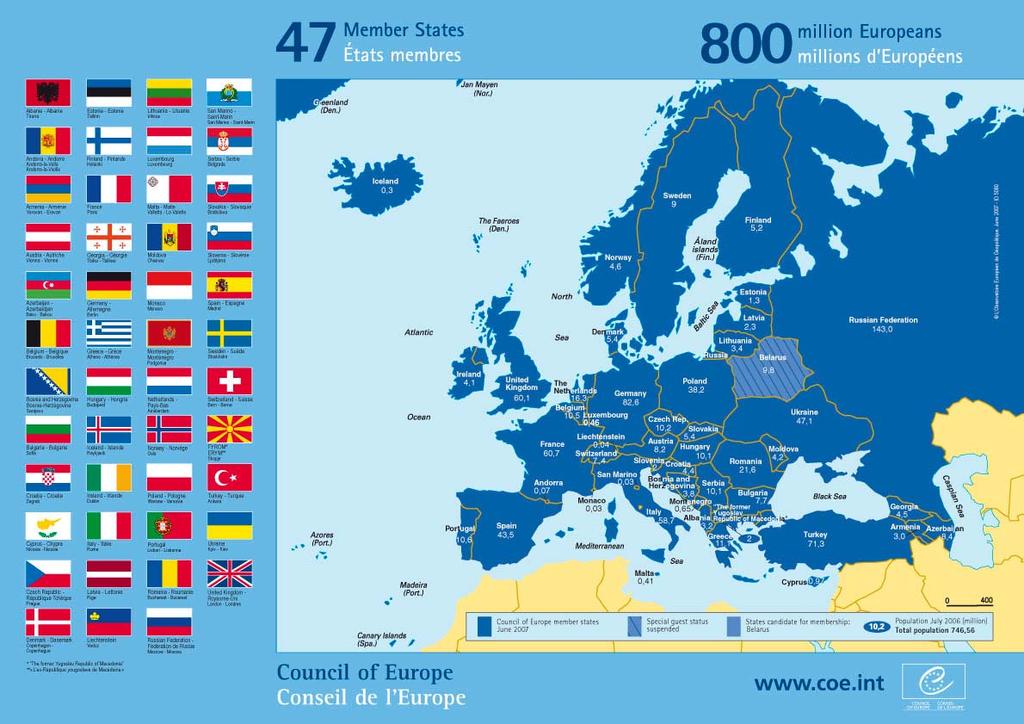
4 4
5 The European Union 27 Member States 500 million inhabitants Political and economic community Founded in the early 50s (European Coal and Steel Community formed among 6 countries in 1951 and Treaty of Rome in 1957) Current legal framework : Maastricht Treaty (1993) / Lisbon Treaty (2009) Comprises a single market created by a system of laws which apply in all Member States 5
6 EDQM and the European Pharmacopeia 6
7 The EDQM Not part of the European Union but a Council of Europe Directorate Not part of the European Union but collaboration 1964: Activities based on an International Convention from the Council of Europe to promote free movement of medicines in Europe Mandatory status reinforced in 1975 in the EU Regulations (Directives) (27 EU Member States) 1994: EU signed the EP Convention 7
 25 Observers (8")
 PDG Pharmacopeial Pharmacopeial discussion")
8 Ph. Eur. in the world 36 Members (+EU) 25 Observers (8 European countries, 16 non European countries +WHO) PDG Pharmacopeial Pharmacopeial discussion discussion group group (USP, (USP, JP, Ph.Eur.) 8
9 General information of the European regulatory framework 9
10 Current regulation of API in the EU Documentation of API quality in Marketing Authorisation Application (MAA) API manufactured according to GMP GMP inspections of API manufacturing sites triggered by national competent authorities Additional requirements after coming into force of EU Falsified Medicines Directive (Dir. 2011/62/EU) as of January 2, 2013 (July 2, 2013 for third country inspections) 10
11 Submission of API quality data in the EU 11
12 API quality data in the EU API quality has to be documented in the Marketing Authorisation Application (MAA) Three different options: Certificate of suitability (CEP) Active substance Master File (ASMF) Full details of manufacture in marketing authorisation application Whatever the route of submission, identical data requirements apply! 12
13 CTD-Q: Drug Substance 3.2.S S.1 General Information (Nomenclature, Structure, General Properties) S.2 Manufacture S.2.1 Manufacturer(s) S.2.2 Description of Manufacturing Process and Process Controls S.2.3 Control of Materials S.2.4 Controls for Critical Steps and Intermediates S.2.5 Process Validation and/or Evaluation S.2.6 Manufacturing Process Development S.3 Characterisation S.3.1 Elucidation of Structure and other Characteristics S.3.2 Impurities S.4 Control of Drug Substances S.5 Reference Standards or Materials S.6 Container Closure System S.7 Stability 13
14 S.2. Manufacture: S.2.1. Manufacturers/Sites Art. 46 f EU Directive 2001/83/ EC as amended Obligation of MA holders «( ) use as starting materials only active substances manufactured in accordance with ( ) GMP for starting materials» Currently not described how this is to be done QP declaration: responsibility of QP of the manufacturing authorisation holder to certify the above requirement. QP declaration cannot be replaced by an official GMP certificate Integral part of the Marketing Authorisation Application! 14
15 The ICH guidelines Q8 Q9 Q10 Encourage systematic approaches Science and risk-based Applicable over entire product lifecycle 3 guidelines intended to work together to enhance pharmaceutical product quality 15
16 ICH Q9 Quality risk management Became annex 20 of EU GMP (applicable 2008) and WHO guideline Principles and general QRM process Methodology and tools (FMEA, FTA, HACCP ) Applicable to companies (development, distribution, manufacturing, labs, materials management...) Applicable to authorities (inspection & assessment) Scope: medicinal products, APIs, biological and biotech product 16
17 ICH Q9 Quality risk management Process development Control Strategy development Continuous improvement 17
18 ICH Q10 Pharmaceutical quality system Scope: - Pharmaceutical development - Technology transfer - Commercial manufacturing - Product discontinuation For API, formulation, IMP, delivery system, manufacturing process, analytical method Objectives: - Achieve appropriate quality attributes (product realisation) - Establish and maintain state of control - Facilitate continual improvement 18
19 ICH Q10 Pharmaceutical quality system The enablers (or facilitators): Knowledge Obtained at each step of the life cycle (from development through the commercial life up to and including product discontinuation) Quality risk management The enablers provide a proactive approach to identifying, scientifically evaluating and controlling potential risks to quality See ICH Q9 19
20 ICH Q10 Pharmaceutical quality system The management responsibility: ICH Q10 insists on Management commitment, Quality policy etc Concepts already presents in the GMP applicable to APIs and drug products but they have now a growing and crucial role to play 20
21 ICH Q 8 Pharmaceutical development Basic principles: Quality cannot be tested into a product; quality needs to be built in by design Pharmaceutical development is aimed to design a quality product and its manufacturing process to consistently deliver the intended performance of the product Information from pharmaceutical development studies can be a basis for QRM 21
22 ICH Q8 Pharmaceutical development Provides a tiered approach, from minimal approach to enhanced quality by design approach... 22
23 ICH Q8 Pharmaceutical development Minimal approach/baseline expectations: Set Quality Target Product Profile (QTPP) Determine potential Critical Quality Attributes (CQAs) Design and implement a control strategy 23
24 ICH Q8 Pharmaceutical development Enhanced approach: Link raw material attributes and process parameters to CQAs and perform risk assessment Develop a design space (optional) Manage product lifecycle, including continuous improvement Additional information provides an opportunity to demonstrate a higher degree of understanding of material attributes, manufacturing processes and their controls 24
25 Sample & Test Sample & Test Sample & Test API Excipient Excipient Blend Screen Blend Tablet Pass or Fail Fixed processes Quality criteria met if: Meets specifications (off-line QC tests) GMP guidelines followed 25
26 Characterise Adaptive processes API Excipient Blend Screen Blend Tablet 100% Pass Excipient PAT PAT Real time release Standards and acceptance criteria for a PAT/QbD approach are not the same as a Test to Document Quality approach 26
27 Quality by design as in Q8 A more systematic approach to development may include: Incorporation of prior knowledge Results of experimental studies using design of experiments Use of quality risk management Use of knowledge management (ICH Q10) throughout the lifecycle Demystification of QbD A systematic approach will facilitate quality achievement and generate more knowledge Not necessarily new requirements: Pharm. development has been a requirement in the EU for a long time QbD does not require establishment of design space or RTR testing The level of development will depend on the complexity of the process and product and on the opportunities chosen by the applicant 27
28 ICH Q8 Pharmaceutical Development argely composed of annexes with examples + glossary Design space for a drying operation (example 3 from ICH Q8) 28
29 Opportunities in pharmaceutical development Depending on the level of development/scientific understanding and a robust quality system in place, opportunities exist to consider more flexible regulatory approaches, for example, to facilitate: risk-based regulatory decisions (assessment and inspection) manufacturing process improvements, within the approved design space described in the dossier, without further regulatory review reduction of post-approval submissions real-time release testing, leading to a reduction of endproduct release testing 29
30 The ICH guidelines Q8 Q9 Q10: impact for certification division So far 2 QbD dossiers received and assessed at EDQM: Real time release, and Rapid microbiological method One of them was performed in conjunction with an inspection Risk assessment is a steering concept during assessment and inspection Risk assessment approach is used when preparing the inspection programme 30

31 The Falsified Medicines Directive 31
32 The Falsified Medicines Directive ( FMD ) EU Falsified Medicines Directive 2011/62/EU amends Directive 2001/83/EC...The harmonised principles and guidelines for inspections of manufacturers and wholesale distributors of medicinal products as well as active substances should be strengthened 32
33 FMD at a glance - Summary Introduction of safety features on drug packaging Stronger rules on importing APIs Verification by national licensees that manufacture of APIs is GMP & GDP compliant Stronger requirements for control and inspections of APIs manufacturers Stronger record-keeping for wholesale distributors Obligation for manufacturers and distributors to report any suspicion of falsified medicines 33
34 DEFINITION of falsified medicine Any medicinal product with a false representation of Its identity, including its packaging and labelling, its name or its composition as regards any of the ingredients including excipients and strength of those ingredients; Its source, including its manufacturer, its country of manufacturing, its country of origin or its marketing authorisation holder; Its history, including the records and documents relating to the distribution channels used 34
35 DEFINITION of falsified active substance Falsified active substance is an active substance where the labelling on the packaging does not show the real content or where the accompanying documentation does either not reflect all involved manufacturers or not reflect the factual distribution channel 35
36 Obligations of manufacturers in EU New principles of GDP for API to be enforced New registration obligation for importers, manufacturers and distributors of APIs Obligation for the MAH to audit manufacturers and distributors of APIs Imported APIs should be accompanied by a written confirmation from the competent authorities 36
37 Obligations of authorities European Commission adopts guidelines of GMP and GDP New activities to inspect: importers, distributors, excipients New inspection trigger: risk based principles New obligation to inspect in 3 rd countries 37
38 Importation preconditioned to: Written confirmation from local authorities accompanying the API Or Exporting country included in list of equivalence of GMP requirements / supervision Or Plant inspected by an EU member state (as an exception and where necessary to ensure the availability of medicinal products) 38
39 Written confirmation (art. 46b) Should be issued by the 3 rd country authority at either central or regional or local level Should be issued per manufacturing plant and the API(s) manufactured on this site A copy of it should accompany each consignment Does not apply to: veterinary API, API which are already introduced in finished products, API for IMP, API for R&D trials 39
40 Written confirmation (art. 46b) To be issued by the competent authority of the exporting 3 rd country; confirms that: GMP standards applicable to the plant are at least equivalent to EU requirements Plant is subject to regular, strict and transparent controls and strict enforcement of GMP, including repeated and unannounced inspections In case of non-compliance, information is supplied to EMA by the exporting country 40
41 Written confirmation (art. 46b) More details available in the QUESTIONS AND ANSWERS document issued by the EC on 10 th July
42 Thank you for your attention Website: 42