Regulations governing the application of medical accelerators
|
|
|
- Norah Shields
- 6 years ago
- Views:
Transcription
1 Regulations governing the application of medical accelerators in 50 minutes.

2 2 1.The wonderland of STANDARDS AND REGULATIONS
 it all starts here. to ensure safety and effectiveness of devices on the market.")
 organizations.")
3 3 Laws and standards Medical devices (and systems) are subject to regulations (laws!) it all starts here. to ensure safety and effectiveness of devices on the market. Every country/region has its own set of laws/regulations. Standards are written by (international, private) organizations. In theory, standards are NOT MANDATORY, but In practice, officially recognized standards need to be followed.
 Technical File Harmonized Standards EN Dev. class I, II and III.")
4 4 Medical Devices Directive 93/42/EEC* (replaced by new MDR) Code of Fed. Regulations Title 21 Dev. class I, IIa, IIb and III Quality Syst. EN13485 CE Marking Notified Body (private) Technical File Harmonized Standards EN Dev. class I, II and III. QMS Title 21 Part (k) or PMA FDA clearance DHF, DMR and DHR Recognized consensus std. AAMI/ANSI
5 5 93/42/EEC Contents Some important contents: Definitions. Classification into I, IIa, IIb or III. List of essential requirements for safety and effectiveness for all devices. Processes to be followed by manufacturers, such as quality, risk management, post market, etc. Instructs manufacturers to create and maintain a Technical File for every device Foresees the adoption of harmonized standards. Legal mechanism for market approval and entitles notified bodies.
 6.")
6 6 Marking 1. Intended purpose and classification 2. Identify the essential requirements 3. Establish necessary processes in the organization 4. Develop the device accordingly 5. Verification and Validation Test against requirements (we ll talk about it latter) 6. Clinical evaluation: a) Find equivalent device and use available literature route and/or b) Clinical investigation route. 7. Finalize technical documentation 8. Contact notified body and perform certification 9. Monitor trough lifecycle
7 7 Harmonized Standards 1. Quality management system EN 13485:2016 Defines organization processes ++ Requires auditing the organization (not only the product!) 2. Risk management EN 14971: Electrical safety EN series (next slide) 4. Software lifecycle EN Usability EN Other standards for specific applications (labeling, packaging, transport, environmental, etc.)
 (EN/AAMI) IEC 60601-1 Section 4:")
 IEC 62304 Med.")

8 8 Simplified Standards Ecosystem FDA CFR Part 21 EC MDD 93/42/EEC ANSI/AAMI CEN/CENELEC QMS (820) (EN/AAMI) IEC Section 4: General Section 14: PEMS Section 16: ME Systems (EN) ISO (EN/AAMI) IEC Med. SW lifecycle Electron Linac EMC Risk Management Light Ion Beam Image guided X-ray Usability (EN/AAMI) IEC Application of Usability

9 9 Meet the family :2007 General Requirements Sections 1 to 17 Collateral Standards EMC X-Ray Usability Alarms Envir Home Emergency Particular Standards Electron ACC Surgical therapeutic X- ray Light Ion Beam Image guided radiotherapy
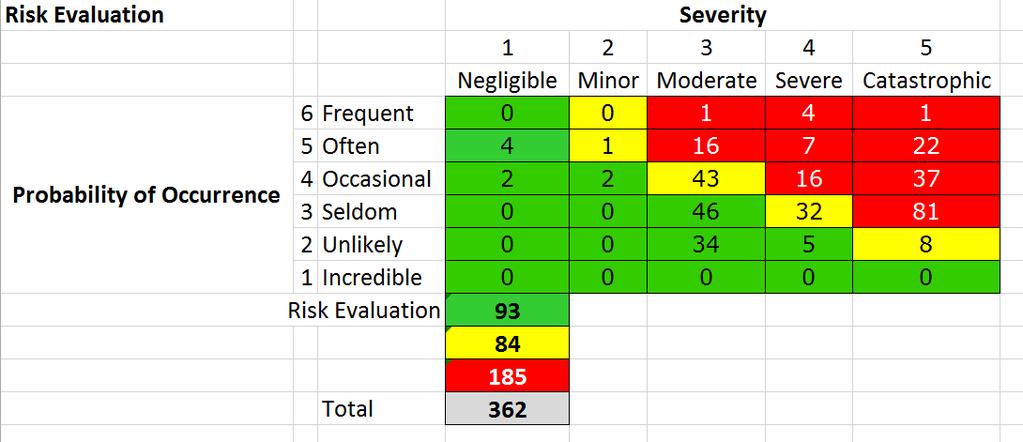
10 10 Risk management: ISO Most widely recognized RM standard for medical devices (and required by law). Not only design phase => whole device lifecycle. Based on the following concepts: HAZARD - potential source of harm HARM - physical injury or damage to the health of people, or damage to property or the environment RISK - combination of the probability of occurrence of harm and the severity of that harm. A damage-centric system => it aims to reduce the risk of harm (contrast with FMEA!) Not only failures to devices: also misuse is covered.
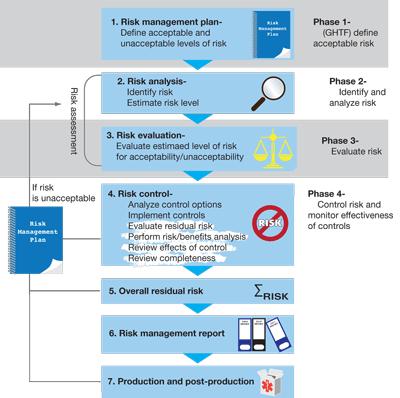
11 11 Risk Management Process
12 12 Some important aspects Risk = (Probability, Severity). Traceability is Key: Every risk must be identified Every hazardous situation must be evaluated Every risk must be linked to mitigation Every mitigation must be verified (e.g. tested ) Risk Management activities must continue after the product goes to the market (!!) RM team members must be defined in advance, with proper qualifications/training. After each RM cycle, a RM report must be released

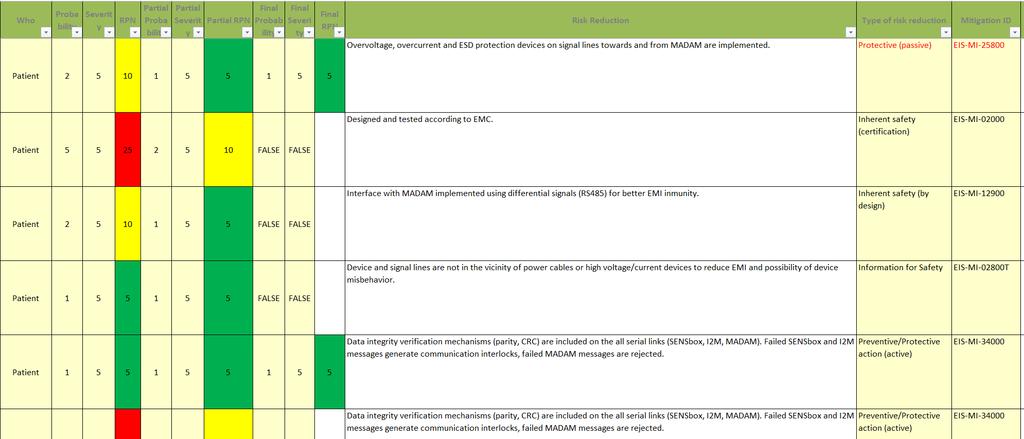
13 13 How does it look like?
14 14 Summary (I) Laws regulate the market of medical devices. Safety and effectiveness. Standards are used to demonstrate compliance with the laws. In EU, the process is called CE marking. It is about demonstrating fulfillment of Essential Requirements. Notified bodies. There are many standards; the most important ones are: Quality system Risk management The family Software lifecycle Design process must be documented.

15 15 2. Some practical aspects of a PT MACHINE AS A MEDICAL DEVICE
16 16 What device??? It is quite simple to think of an ECG machine, a thermometer or even an X-ray diagnostic. According to MDD, a PT system is a class IIb. medical device. But What is the medical device? Are the sub-components medical devices on their own? Also class IIb.? Do all the same standards apply for all subsystems and components? How to aggregate different components into a device with a single purpose?






17 A complex one Accelerator Beamline Machine Control System Machine Interlock Syst. Auxiliary systems Beam Diagnostics Patient Couch Gantry Positioning Lasers Nozzle Extender Collision Avoidance Control Electronics Imaging 2D X-Ray / CBCT Controls for Imaging Registration Software Patient protection system Fast interlocks Room locking, operator protection Scanning system Dose delivery Treatment Control System Medical Software, Interfaces to OIS

18 18 ME SYSTEMS The standard , section 16 offers an interesting view. ME system composed of medical and non medical equipment. 3 Conditions: The ME SYSTEM as a whole shall not have unacceptable RISK. Within the PATIENT ENVIRONMENT, the level of safety for Outside PATIENT ENVIRONMENT, the level of safety for applicable standards allows to exclude some parts of the system from the need to be developed according to medical standards.



19 19 System Architecture compliance is required PT System Might be excluded Beam Production and Transport Nozzle System Treatment and Positioning Control System Position Verification Accelerator Scanning Treatment Control X-Ray Beamline Dose Delivery Positioning Control X Ray tube Magnets SW Module 1 X Ray panel Beam Diag. SW Module 2 Registration SW Control System *but still needs to comply with all other applicable regulations for a device/system of its type Lasers
20 20 Use of Software Software plays a key role: system complexity is in fact driven by automation -> PEMS ( Section 14.) 1. EN PEMS clause applies, excepted: No BASIC SAFETY or ESSENTIAL PERFORMANCE* ISO14971 demonstrates no unacceptable risk Compliance requires that a development life-cycle be specified and followed 2. EN > Software life cycle processes. Verification and Validation activities need to be conducted.
21 21 Goals of System Architecture To define a wise system architecture, to group components with similar characteristics and functions in subsystems. Risk management is used to determine overall risk and to find what components are high risk and which are low risk. Also, risk aggregation. Some system components may not need to comply with (and related standards). Classify software according to into classes A, B and C. Translate User Requirements into Engineering Requirements/Specifications.
22 22 Two types of requirements Product Requirements System/ Subsystem Reqs Functional and performance requirements corresponding to the intended use of the product. Applicable regulatory requirements The sources for product requirements are: User requirements formulated by clients respect their medical and business goals. Requirements known from the market. Regulatory requirements imposed or recommended by authority. derived from the product definition requirements (functional, performance and regulatory), translating them onto technical specification. Technical requirements for the entire system, potentially with notes for the user manual and service manual. Safety relevant requirements The sources for translating regulatory requirements into technical requirements are the corresponding standards. Safety relevant requirements are derived from risk control measures defined in the risk management process, and applicable safety and essential performance standards for medical devices.

23 23 V&V at first sight The infamous V-model : A concept from the Software world Building the thing right vs. building the right thing
24 24 Test case traceability Every requirement should be covered by single or multiple test cases Requirement Unique ID single or multiple Arch. and design Unique ID single or multiple single or multiple RM Mitigation Unique ID Risk Identification Risk Evaluation Risk Control RM Hazard Unique ID Test case Unique ID Test report Unique ID Risk Control Verification
25 25 Summary (II) A PT System is one of the most complex medical devices. The regulations and standards are easier to apply to simpler devices; for complex systems sometimes it is necessary to do some magic. 3 useful standards: , sections 14 and 16 Risk Management EN IEC System level architecture plays a major role! Write good requirements; start EARLY in the process Traceability!


26 26 It s all about processeses 1. The laws and regulations request it 2. Technical standards require it 3. Good engineering practices recommend it 4. It gives you good tools and maneuvering space when things does not go as planned. 5. It s about finding the sweet spot between too much and too little :
27 THANK YOU! Marko Mehle COSYLAB
28 28 THOSE SLIDES AT THE END
29 29 Basic safety and essential performance Originally only safety, Essential Performance is new to the 3rd. Edition ESSENTIAL PERFORMANCE: performance necessary to achieve freedom from unacceptable RISK. [whether its absence or degradation causes unacceptable RISK.] BASIC SAFETY: freedom from unacceptable RISK directly caused by physical HAZARDS when ME EQUIPMENT is used under NORMAL CONDITION and SINGLE FAULT CONDITION. Only makes sense in the context of Risk Management! Effectiveness Essential performance Safety
30 30 V&V for FDA From 21. CFR Design controls Design verification: [ ] Design verification shall confirm that the design output meets the design input requirements. The results of the design verification, including identification of the design, method(s), the date, and the individual(s) performing the verification, shall be documented in the DHF. Design validation: [ ] Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation,
31 31 V&V for MDD MDD 93/42/EEC, 12.1a: For devices which incorporate software or which are medical software in themselves, the software must be validated according to the state of the art taking into account the principles of development lifecycle, risk management, validation and verification.

 at the isocentre")

32 32 Measurement of beam parameters The beam size shall be 3mm or less (1sigma) at the isocentre at the highest treatment energy at all gantry angles (Verification)


33 33 Measurement of beam parameters The maximum range of the beam in water shall be 37 g/cm^2 or greater for treatment, which is defined as 250MeV or greater in a water target. (Verification)

34 34 Mechanical tests and measurements The PPS shall be aligned with mechanical isocenter ±1mm. (Verification)
ISO 13485:2016 Medical Devices-Quality Management Systems- Requirements for Regulatory Purposes. Theresa McCarthy
 ISO 13485:2016 Medical Devices-Quality Management Systems- Requirements for Regulatory Purposes Theresa McCarthy Welcome and Introductions Please interview and collect: Name Primary Quality Management
ISO 13485:2016 Medical Devices-Quality Management Systems- Requirements for Regulatory Purposes Theresa McCarthy Welcome and Introductions Please interview and collect: Name Primary Quality Management
Medical Device Directive
 Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
International Standards and EU regulation of medical device software an update
 International Standards and EU regulation of medical device software an update Sherman Eagles Partner, SoftwareCPR seagles@softwarecpr.com 612 865 0107 1 Who am I? 18 years at Medtronic, retired 2008 Last
International Standards and EU regulation of medical device software an update Sherman Eagles Partner, SoftwareCPR seagles@softwarecpr.com 612 865 0107 1 Who am I? 18 years at Medtronic, retired 2008 Last
SOFTWARE APPS AS MEDICAL DEVICES. The Regulatory Landscape 2015 WHITE PAPER WHITE PAPER PRODUCED BY MAETRICS
 WHITE PAPER SOFTWARE APPS AS MEDICAL DEVICES The Regulatory Landscape 2015 WHITE PAPER PRODUCED BY MAETRICS For more information, please contact: USA Office: +1 317 706 1493 UK Office: +44 115 921 6200
WHITE PAPER SOFTWARE APPS AS MEDICAL DEVICES The Regulatory Landscape 2015 WHITE PAPER PRODUCED BY MAETRICS For more information, please contact: USA Office: +1 317 706 1493 UK Office: +44 115 921 6200
Medical Device Software: A new challenge
 Medical Device Software: A new challenge Daniela Luzi and Fabrizio Pecoraro National Research Council Institute for Research on Population and Social Policies, Rome Italy Main issues Q.1 Is a software
Medical Device Software: A new challenge Daniela Luzi and Fabrizio Pecoraro National Research Council Institute for Research on Population and Social Policies, Rome Italy Main issues Q.1 Is a software
Short intro to the MDR & IVDR (C)2018 QAdvis AB
2018 QAdvis AB") Short intro to the MDR & IVDR 2018-06-11 1 , MDR Major changes for medical device and IVD device industry EU regulations Non-EU regulations Standards Brexit Affecting all stakeholders: manufactuers, distributors,
Short intro to the MDR & IVDR 2018-06-11 1 , MDR Major changes for medical device and IVD device industry EU regulations Non-EU regulations Standards Brexit Affecting all stakeholders: manufactuers, distributors,
Design Quality. Indu Lakshman
 Design Quality Indu Lakshman Overview New product development (NPD) covers the complete process of bringing a new product to market. In commercial terms, new product development is described in the literature
Design Quality Indu Lakshman Overview New product development (NPD) covers the complete process of bringing a new product to market. In commercial terms, new product development is described in the literature
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS
 FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
(C) QAdvis. IEC and IEC how to make them work (and why so much attention on SW) QAdvis RMD, Prague, November 8th 2016
 QAdvis. IEC and IEC how to make them work (and why so much attention on SW) QAdvis RMD, Prague, November 8th 2016") IEC 62304 and IEC 82304-1 - how to make them work (and why so much attention on SW) QAdvis RMD, Prague, November 8th 2016 QMS in-the cloud Turn key QMS Digital signatures Efficient and lean Validated and
IEC 62304 and IEC 82304-1 - how to make them work (and why so much attention on SW) QAdvis RMD, Prague, November 8th 2016 QMS in-the cloud Turn key QMS Digital signatures Efficient and lean Validated and
1 Preface Introduction... 13
 1 Preface... 11 2 Introduction... 13 3 Legal Requirements... 15 3.1 Approach to determining the legal implications... 15 3.2 Software as a medical device... 17 3.2.1 Intended purpose... 18 3.2.2 Normal
1 Preface... 11 2 Introduction... 13 3 Legal Requirements... 15 3.1 Approach to determining the legal implications... 15 3.2 Software as a medical device... 17 3.2.1 Intended purpose... 18 3.2.2 Normal
Regulation of software a medical device
 Regulation of software a medical device IOF-TTO Event 17.10.2017 Steve Eglem Responsible of clinical investigation @ famhp Member of the medical device European Clinical and Evaluation (CIE) working group
Regulation of software a medical device IOF-TTO Event 17.10.2017 Steve Eglem Responsible of clinical investigation @ famhp Member of the medical device European Clinical and Evaluation (CIE) working group
The Complete Guide to FDA Design Controls
 The Complete Guide to FDA Design Controls Jon D. Speer Founder & VP QA/RA of greenlight.guru ABOUT THE PRESENTER Jon D. Speer is the founder and VP of QA/RA of greenlight.guru 20+ years in medical device
The Complete Guide to FDA Design Controls Jon D. Speer Founder & VP QA/RA of greenlight.guru ABOUT THE PRESENTER Jon D. Speer is the founder and VP of QA/RA of greenlight.guru 20+ years in medical device
Implications of the new MDR from a Product Testing and Certification Perspective
 Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
Automated System Validation By: Daniel P. Olivier & Curtis M. Egan
 Automated System Validation By: Daniel P. Olivier & Curtis M. Egan In today s technical environment validation practices are both a requirement and an important tool in the medical and pharmaceutical industry.
Automated System Validation By: Daniel P. Olivier & Curtis M. Egan In today s technical environment validation practices are both a requirement and an important tool in the medical and pharmaceutical industry.
The challenges of software medical device regulation.
 The challenges of software medical device regulation. david.grainger@mhra.gov.uk Introduction A brief history of software device regulation A look at the new device regulations 2 Current framework In Vitro
The challenges of software medical device regulation. david.grainger@mhra.gov.uk Introduction A brief history of software device regulation A look at the new device regulations 2 Current framework In Vitro
MEDICAL DEVICE. Technical file.
 MEDICAL DEVICE Technical file www.icaro-research.eu ICARO MDTF v1.0 1 Mar 2016 1. Do you plan to launch your medical device in Europe? If you re reading this, chances are good that you re considering introducing
MEDICAL DEVICE Technical file www.icaro-research.eu ICARO MDTF v1.0 1 Mar 2016 1. Do you plan to launch your medical device in Europe? If you re reading this, chances are good that you re considering introducing
Medical Device Software Standards
 Background Medical Device Software Standards By Peter Jordan, BA, C.Eng., MBCS Much medical device software is safety-related, and therefore needs to have high integrity (in other words its probability
Background Medical Device Software Standards By Peter Jordan, BA, C.Eng., MBCS Much medical device software is safety-related, and therefore needs to have high integrity (in other words its probability
Manufacturers. Factsheet for. of Medical Devices. Medical Devices Regulation (MDR) background. Act now to be ready on time!
 background. Act now to be ready on time!") Ref. Ares(2018)3873669-20/07/2018 Factsheet for Manufacturers of Medical Devices This Factsheet is aimed at manufacturers of medical devices. For a general overview of the impact of the In-Vitro Medical
Ref. Ares(2018)3873669-20/07/2018 Factsheet for Manufacturers of Medical Devices This Factsheet is aimed at manufacturers of medical devices. For a general overview of the impact of the In-Vitro Medical
Left to Our Own Devices Design Control & Risk Management Strategies for Combination Products
 Left to Our Own Devices Design Control & Risk Management Strategies for Combination Products DR. RICK WEDGE 12 MAR 2018 Overview Definitions Current Regulatory Landscape Design Controls Risk Management
Left to Our Own Devices Design Control & Risk Management Strategies for Combination Products DR. RICK WEDGE 12 MAR 2018 Overview Definitions Current Regulatory Landscape Design Controls Risk Management
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards. 26 th September / V. Fischer / Rev. 01
 ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
Manufacturers. Factsheet for. of Medical Devices. Medical Devices Regulation (MDR) background. Act now to be ready on time!
 background. Act now to be ready on time!") European Commission Factsheet for Manufacturers of Medical Devices This Factsheet is aimed at manufacturers of medical devices. For a general overview of the impact of the In Vitro Medical Devices Regulation
European Commission Factsheet for Manufacturers of Medical Devices This Factsheet is aimed at manufacturers of medical devices. For a general overview of the impact of the In Vitro Medical Devices Regulation
Software in Medical Devices
 Software in Medical Devices Module 1: Regulations, Guidance, Standards, and Terminology Planning Jeremy Jensen Fellow Software Quality Engineer Boston Scientific Irma Sandoval-Watt Senior Regulatory Strategist
Software in Medical Devices Module 1: Regulations, Guidance, Standards, and Terminology Planning Jeremy Jensen Fellow Software Quality Engineer Boston Scientific Irma Sandoval-Watt Senior Regulatory Strategist
V&V Best Practices. CASSS, CMC Strategy Forum Steven W. Badelt, PhD Managing Partner Suttons Creek, Inc. SUTTONSCREEK.COM
 V&V Best Practices CASSS, CMC Strategy Forum Steven W. Badelt, PhD Managing Partner Suttons Creek, Inc. Solving the problem of Complexity. 21CFR820.30 & FDA Guidance INCOSE System Engineering Handbook
V&V Best Practices CASSS, CMC Strategy Forum Steven W. Badelt, PhD Managing Partner Suttons Creek, Inc. Solving the problem of Complexity. 21CFR820.30 & FDA Guidance INCOSE System Engineering Handbook
Rocky Mountain Regulatory Affairs Society. Elements of and Maintenance/Remediation of the Design History File (DHF) March 15, 2017
 March 15, 2017") Rocky Mountain Regulatory Affairs Society Elements of and Maintenance/Remediation of the Design History File (DHF) March 15, 2017 Design History File Presenters: Lee Travis Design Solutions Jim Tartarini
Rocky Mountain Regulatory Affairs Society Elements of and Maintenance/Remediation of the Design History File (DHF) March 15, 2017 Design History File Presenters: Lee Travis Design Solutions Jim Tartarini
The Risk Management + Design Controls Connection: What Device Makers Need to Know
 !!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
!!! The Risk Management + Design Controls Connection: What Device Makers Need to Know Jon Speer Founder & VP of QA/RA greenlight.guru Table of Contents 1 Intended Use & User Needs 6 Verification, Validation,
BSI Audits for MDR Certification
 BSI Audits for MDR Certification Quality System Audits Unannounced Audits (ongoing per MDD / MDR) No changes as a result of MDR / IVDR New frequencies already applied Once per 3 years for Class III & Implants
BSI Audits for MDR Certification Quality System Audits Unannounced Audits (ongoing per MDD / MDR) No changes as a result of MDR / IVDR New frequencies already applied Once per 3 years for Class III & Implants
Medical Device Regulation
 Johner Institut Medical Device Regulation 7 Steps to MDR Compliance Astrid Schulze Graduate in economics 20 years entrepreneur in quality management and approval of medical devices and software Since 2017
Johner Institut Medical Device Regulation 7 Steps to MDR Compliance Astrid Schulze Graduate in economics 20 years entrepreneur in quality management and approval of medical devices and software Since 2017
Ready or Not: The New Medical Device Regulations Are Here!
 Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Auditdata Your Partner in Audiology Solutions
 Auditdata Your Partner in Audiology Solutions My PLM Road map 20 + years experience within medical device development, production, PLM and quality management (1988-) Agile PLM and QMS architect GN Otometrics
Auditdata Your Partner in Audiology Solutions My PLM Road map 20 + years experience within medical device development, production, PLM and quality management (1988-) Agile PLM and QMS architect GN Otometrics
Self-Care Medical Devices Framework. Dr. Simone Breitkopf Head HEOR, Governmental and Public Affairs
 Self-Care Medical Devices Framework Dr. Simone Breitkopf Head HEOR, Governmental and Public Affairs Disclaimer The view and opinions expressed in the following PowerPoint slides are those of the individual
Self-Care Medical Devices Framework Dr. Simone Breitkopf Head HEOR, Governmental and Public Affairs Disclaimer The view and opinions expressed in the following PowerPoint slides are those of the individual
Experience with Directive 93/42/EEC (MDD)
") (MDD) A medical device manufacturer s retrospective view on 19 years of experience with Directive 93/42/EEC Lübeck, 2012-09-05, Dr. Peter Gebhardt Preliminary Remarks Scope of this presentation is based
(MDD) A medical device manufacturer s retrospective view on 19 years of experience with Directive 93/42/EEC Lübeck, 2012-09-05, Dr. Peter Gebhardt Preliminary Remarks Scope of this presentation is based
AAMI Quality Systems White Paper
 AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017, Updated February 2018 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward,
AAMI s White Paper Comparison of 21 CFR Part 820 to ISO 13485:2016 February 2017, Updated February 2018 AUTHORS Seb Clerkin, GMP Advisory Services Nicola Martin, Owner, Nicola Martin Consulting Jack Ward,
Post market Surveillance ISO EU Medical Device Regulation
 Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
Standard and Directives on Medical Devices
 Standard and Directives on Medical Devices Definition A Medical Device is identified by means of its INTENDED PURPOSE Intended to treat, prevent or control physiological characteristics of a living being
Standard and Directives on Medical Devices Definition A Medical Device is identified by means of its INTENDED PURPOSE Intended to treat, prevent or control physiological characteristics of a living being
Regulatory Framework for Medical Device
 Regulatory Framework for Medical Device International Seminar Safety of Health Products French Ministry of Labour, Employment and Health Paris, 26 May 2011 Peter Bischoff-Everding European Commission EU
Regulatory Framework for Medical Device International Seminar Safety of Health Products French Ministry of Labour, Employment and Health Paris, 26 May 2011 Peter Bischoff-Everding European Commission EU
Contents. List of Acronyms Preface
 Contents List of Acronyms Preface xi xv PART I Introduction 1 1 Introduction 3 1.1 The evolution of medical purpose software 3 1.2 Product quality and software quality 4 1.3 On the need for quality in
Contents List of Acronyms Preface xi xv PART I Introduction 1 1 Introduction 3 1.1 The evolution of medical purpose software 3 1.2 Product quality and software quality 4 1.3 On the need for quality in
Manufacturers will not need EMC or conformity assessments but will need to illustrate the
 EMC Directive 2004/108/EC: Implications for Manufacturers Kenneth L. Boston, PE, NCE January 2011 As of July 2007, 2004/108/EEC replaced the previous standard, which required manufacturers to create a
EMC Directive 2004/108/EC: Implications for Manufacturers Kenneth L. Boston, PE, NCE January 2011 As of July 2007, 2004/108/EEC replaced the previous standard, which required manufacturers to create a
Bridging gaps: medical device directive vs regulation. Geert Corstens 1 November 2018
 Bridging gaps: medical device directive vs regulation Geert Corstens 1 November 2018 Agenda Current Medical Device regulatory landscape New Medical Device Regulation EU Interaction & impact Medical device
Bridging gaps: medical device directive vs regulation Geert Corstens 1 November 2018 Agenda Current Medical Device regulatory landscape New Medical Device Regulation EU Interaction & impact Medical device
QMS Aspects of the MDR (& IVDR)
") QMS Aspects of the MDR (& IVDR) Vicky Medley Global QMS Manager Medical Devices 27 February 2018 Copyright 2018 BSI. All rights reserved This Presentation 1. The clock is ticking! 2. Dates & priorities
QMS Aspects of the MDR (& IVDR) Vicky Medley Global QMS Manager Medical Devices 27 February 2018 Copyright 2018 BSI. All rights reserved This Presentation 1. The clock is ticking! 2. Dates & priorities
EU MDR 10 Things Packaging Engineers Should Know
 EU MDR 10 Things Packaging Engineers Should Know networkpartners.com NUMBER ONE THE TIME FOR ACTION IS NOW First, some background: The EU Medical Device Regulation (MDR) was approved by the European Parliament
EU MDR 10 Things Packaging Engineers Should Know networkpartners.com NUMBER ONE THE TIME FOR ACTION IS NOW First, some background: The EU Medical Device Regulation (MDR) was approved by the European Parliament
Development of Safety Related Systems
 July 2015 LatticeSemiconductor 7 th Floor,111SW5 th Avenue Portland,Oregon97204USA Telephone:(503)268I8000 www.latticesemi.com WP004 The increasing degree of automation brings a lot of comfort and flexibility
July 2015 LatticeSemiconductor 7 th Floor,111SW5 th Avenue Portland,Oregon97204USA Telephone:(503)268I8000 www.latticesemi.com WP004 The increasing degree of automation brings a lot of comfort and flexibility
Regulatory Affairs in Medical Technology
 Regulatory Affairs in Medical Technology Medical Device Regulation: Implementation on European Level, First Results Dr. Matthias Neumann Federal Ministry of Health Lübeck 2018 Summer Academy on Medical
Regulatory Affairs in Medical Technology Medical Device Regulation: Implementation on European Level, First Results Dr. Matthias Neumann Federal Ministry of Health Lübeck 2018 Summer Academy on Medical
Choices IEC rd Edition and Component Selection
 Choices IEC 60601-1 3rd Edition and Component Selection Choices IEC 60601-1 3rd Edition and Component Selection Abstract When the 3rd edition of IEC 60601-1 was published, it marked the beginning of a
Choices IEC 60601-1 3rd Edition and Component Selection Choices IEC 60601-1 3rd Edition and Component Selection Abstract When the 3rd edition of IEC 60601-1 was published, it marked the beginning of a
Copyright GCI, LLC Remediation of Legacy Medical Devices
 Copyright GCI, LLC. 2016 Remediation of Legacy Medical Devices Copyright GCI, LLC. 2016 What Drives Legacy Remediation? Proactive Recertification / CE Mark Audit Readiness M&A / Due Diligence Reactive
Copyright GCI, LLC. 2016 Remediation of Legacy Medical Devices Copyright GCI, LLC. 2016 What Drives Legacy Remediation? Proactive Recertification / CE Mark Audit Readiness M&A / Due Diligence Reactive
GS1 Ireland Healthcare User Group (HUG) Information Day
 Information Day") GS1 Ireland Healthcare User Group (HUG) Information Day Regulatory update Medical Devices and the impact for Irish Healthcare Sinead Duggan, HPRA 28 th March 2017 29/03/2017 2 Medical Devices Regulation
GS1 Ireland Healthcare User Group (HUG) Information Day Regulatory update Medical Devices and the impact for Irish Healthcare Sinead Duggan, HPRA 28 th March 2017 29/03/2017 2 Medical Devices Regulation
EU MDR Timeline. Dr. Christian B. Fulda Jones Day FDLI Annual Conference Access materials at fdli.org/annual2018
 EU MDR Timeline Dr. Christian B. Fulda Jones Day EU MDR - Timeline EU has entered into force, but will only apply for products as of May 26, 2020, with transitional period through 2024/2025? Look closer!
EU MDR Timeline Dr. Christian B. Fulda Jones Day EU MDR - Timeline EU has entered into force, but will only apply for products as of May 26, 2020, with transitional period through 2024/2025? Look closer!
Technical documentation in the new MDR. Gabriele Catignoli, Andreas Purde
 Technical documentation in the new MDR Annex I/II Gabriele Catignoli, Andreas Purde TÜV SÜD Product Service GmbH Slide 1 MDR Annex I, II 1 Core changes, Essential Requirements 2 Role of Common Specifications
Technical documentation in the new MDR Annex I/II Gabriele Catignoli, Andreas Purde TÜV SÜD Product Service GmbH Slide 1 MDR Annex I, II 1 Core changes, Essential Requirements 2 Role of Common Specifications
EURATOM Booklet. Ready for February 6th 2018?
 EURATOM Booklet Ready for February 6th 2018? February 6 th 2018 will see the introduction of the new EURATOM directive 201/59/. This is the deadline when you must have the necessary framework in place
EURATOM Booklet Ready for February 6th 2018? February 6 th 2018 will see the introduction of the new EURATOM directive 201/59/. This is the deadline when you must have the necessary framework in place
A Continuous Improvement Approach for Medical Device Software Development Companies. Dr. Özden Özcan-Top - Dr. Fergal Mc Caffery
 A Continuous Improvement Approach for Medical Device Software Development Companies Dr. Özden Özcan-Top - Dr. Fergal Mc Caffery 12/07/2017 Lero 2015 1 Dr. Özden Özcan - Top Dr. Fergal Mc Caffery Dundalk
A Continuous Improvement Approach for Medical Device Software Development Companies Dr. Özden Özcan-Top - Dr. Fergal Mc Caffery 12/07/2017 Lero 2015 1 Dr. Özden Özcan - Top Dr. Fergal Mc Caffery Dundalk
ISO 13485:2016. What s in it - and what does it mean?
 ISO 13485:2016 What s in it - and what does it mean? QAdvis key competence areas Turn key quality systems Sharepoint based Digital signatures Efficient and lean Validated and compliant QMS in-the cloud
ISO 13485:2016 What s in it - and what does it mean? QAdvis key competence areas Turn key quality systems Sharepoint based Digital signatures Efficient and lean Validated and compliant QMS in-the cloud
Applying ISO14971 / IEC62304 / IEC A Practical Guide On How To Implement Risk Management
 Applying ISO14971 / IEC62304 / IEC62366-1 - A Practical Guide On How To Implement Risk Management *** LIMITED TIME OFFER: FREE $100 AMAZON GIFT CARD! *** REGISTER TODAY! Risk management is a mandatory
Applying ISO14971 / IEC62304 / IEC62366-1 - A Practical Guide On How To Implement Risk Management *** LIMITED TIME OFFER: FREE $100 AMAZON GIFT CARD! *** REGISTER TODAY! Risk management is a mandatory
Safety cannot rely on testing
 Standards 1 Computer-based systems (generically referred to as programmable electronic systems) are being used in all application sectors to perform non-safety functions and, increasingly, to perform safety
Standards 1 Computer-based systems (generically referred to as programmable electronic systems) are being used in all application sectors to perform non-safety functions and, increasingly, to perform safety
Medical Device Software Development:
 Intertek Cleeve Road, Leatherhead, Surrey KT22 7SB UK info.uk@intertek.com 01372 370900 www.intertek.com Medical software Many electrical medical devices are like household appliances in some ways and
Intertek Cleeve Road, Leatherhead, Surrey KT22 7SB UK info.uk@intertek.com 01372 370900 www.intertek.com Medical software Many electrical medical devices are like household appliances in some ways and
SPECIFICATION STATEMENT OF WORK. Performance Specification for a radiotherapy linear accelerator
 Specifications / Statement of Work / Terms of Reference SPECIFICATION STATEMENT OF WORK Performance Specification for a radiotherapy linear accelerator 1. Scope These specifications describe the requirements
Specifications / Statement of Work / Terms of Reference SPECIFICATION STATEMENT OF WORK Performance Specification for a radiotherapy linear accelerator 1. Scope These specifications describe the requirements
ehealth Suisse Checklists Addendum to the guideline for app developers, manufacturers and distributors
 ehealth Suisse Checklists Addendum to the guideline for app developers, manufacturers and distributors Bern, 2 March 2018 Page 1 Legal notice ehealth Suisse, Swiss Competence and Coordination Centre of
ehealth Suisse Checklists Addendum to the guideline for app developers, manufacturers and distributors Bern, 2 March 2018 Page 1 Legal notice ehealth Suisse, Swiss Competence and Coordination Centre of
Accelerating Software Development
 Accelerating Software Development in Digital Therapeutics Patrick Alff Patrick.alff@voluntis.com voelter@acm.org www.voelter.de @markusvoelter Markus Völter 1 Software Medical Devices and Digital Therapeutics
Accelerating Software Development in Digital Therapeutics Patrick Alff Patrick.alff@voluntis.com voelter@acm.org www.voelter.de @markusvoelter Markus Völter 1 Software Medical Devices and Digital Therapeutics
Technical Documentation
 Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
Medidée Services SA. Nano-Tera.ch. 05 February 2015 part 3. Regulatory fundamentals. Michael Maier
 Nano-Tera.ch 05 February 2015 part 3 Regulatory fundamentals Michael Maier michael.maier@medidee.com www.medidee.com Regulatory Fundamentals Medical Devices EU Regulatory System Conformity Assessment US
Nano-Tera.ch 05 February 2015 part 3 Regulatory fundamentals Michael Maier michael.maier@medidee.com www.medidee.com Regulatory Fundamentals Medical Devices EU Regulatory System Conformity Assessment US
Results of the IEC Functional Safety Assessment
 Results of the IEC 61508 Functional Safety Assessment Project: 3051S Electronic Remote Sensors (ERS ) System Customer: Emerson Automation Solutions (Rosemount, Inc.) Shakopee, MN USA Contract No.: Q16/12-041
Results of the IEC 61508 Functional Safety Assessment Project: 3051S Electronic Remote Sensors (ERS ) System Customer: Emerson Automation Solutions (Rosemount, Inc.) Shakopee, MN USA Contract No.: Q16/12-041
IVDR Breakout. Copyright 2017 BSI. All rights reserved.
 IVDR Breakout 1 IVDR Annex I (SPRs) & Annex II (Tech documentation) 2 IVDR Annex I General Safety & performance requirements 3 Overview Context of the General Safety & Performance Requirements (SPRs) [our
IVDR Breakout 1 IVDR Annex I (SPRs) & Annex II (Tech documentation) 2 IVDR Annex I General Safety & performance requirements 3 Overview Context of the General Safety & Performance Requirements (SPRs) [our
Mapping Your Success Staying Current on Standards Under the EU Approach
 Mapping Your Success Staying Current on Standards Under the EU Approach Overview What is a Standard? Why use Standards? EU Harmonized Standards what does it mean? EU Standards Harmonization Process Annex
Mapping Your Success Staying Current on Standards Under the EU Approach Overview What is a Standard? Why use Standards? EU Harmonized Standards what does it mean? EU Standards Harmonization Process Annex
Post Market Surveillance
 Post Market Surveillance December 6 th, 2017 Michael Maier, Medidee Services Slide 1 Agenda Postmarket surveillance Postmarket clinical follow up (PMCF) Incident reporting What changes with MDR Slide 2
Post Market Surveillance December 6 th, 2017 Michael Maier, Medidee Services Slide 1 Agenda Postmarket surveillance Postmarket clinical follow up (PMCF) Incident reporting What changes with MDR Slide 2
Overview of the 2nd Edition of ISO 26262: Functional Safety Road Vehicles
 Overview of the 2nd Edition of ISO 26262: Functional Safety Road Vehicles Rami Debouk GM Research and Development rami.debouk@gm.com August 16 th, 2018 2010 ISSC Functional Minneapolis, Safety Road Vehicles
Overview of the 2nd Edition of ISO 26262: Functional Safety Road Vehicles Rami Debouk GM Research and Development rami.debouk@gm.com August 16 th, 2018 2010 ISSC Functional Minneapolis, Safety Road Vehicles
Understanding IEC 62304
 Understanding IEC 62304 Co-authored by MethodSense, Inc. and Medical Equipment Compliance Associates, LLC www.methodsense.com 919.313.3960 Understanding IEC 62304 By: Rita King, Chief Executive Officer,
Understanding IEC 62304 Co-authored by MethodSense, Inc. and Medical Equipment Compliance Associates, LLC www.methodsense.com 919.313.3960 Understanding IEC 62304 By: Rita King, Chief Executive Officer,
Food and Drug Administration Modernization Act of 1997: Modifications to the List of
 This document is scheduled to be published in the Federal Register on 01/14/2014 and available online at http://federalregister.gov/a/2014-00477, and on FDsys.gov 4160-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 01/14/2014 and available online at http://federalregister.gov/a/2014-00477, and on FDsys.gov 4160-01-P DEPARTMENT OF HEALTH AND HUMAN
Certificating a safety related part of a control system
 Certificating a safety related part of a control system Marita Hietikko, Mika Riihimaa VTT Expert Services Ltd, P.O. Box 345, FI-33101 Tampere, Finland Tel: +358 20 722 111, E-mail: marita.hietikko@vtt.fi,
Certificating a safety related part of a control system Marita Hietikko, Mika Riihimaa VTT Expert Services Ltd, P.O. Box 345, FI-33101 Tampere, Finland Tel: +358 20 722 111, E-mail: marita.hietikko@vtt.fi,
Laboratorio di Tecnologie Biomediche
 Laboratorio di Tecnologie Biomediche Introduction to medical devices Carmelo De Maria carmelo.demaria@unipi.it Medical Device A Medical Device is identified by means of its INTENDED PURPOSE Intended to
Laboratorio di Tecnologie Biomediche Introduction to medical devices Carmelo De Maria carmelo.demaria@unipi.it Medical Device A Medical Device is identified by means of its INTENDED PURPOSE Intended to
Model-Based Design for ISO Applications. April 2010
 Model-Based Design for ISO 26262 Applications April 2010 Agenda Introduction Certification, Standards, and Compliance Demonstration ISO 26262 & Qualification of Software Tools Verification & Validation
Model-Based Design for ISO 26262 Applications April 2010 Agenda Introduction Certification, Standards, and Compliance Demonstration ISO 26262 & Qualification of Software Tools Verification & Validation
Contents. Regulatory Bodies... 13
 Contents UNITED STATES... 3 Regulatory Bodies... 3 Product Classification and Regulatory Control... 3 Application Documents... 3 Quality System Requirements... 4 Product and Manufacturer s License... 4
Contents UNITED STATES... 3 Regulatory Bodies... 3 Product Classification and Regulatory Control... 3 Application Documents... 3 Quality System Requirements... 4 Product and Manufacturer s License... 4
Pre-Clinical Testing for Devices and Diagnostics
 Pre-Clinical Testing for Devices and Diagnostics Larry O. Blankenship Evergreen Research, Inc. April, 2016 1 Many Levels, Many Approaches The requirements differ dramatically by class and category and
Pre-Clinical Testing for Devices and Diagnostics Larry O. Blankenship Evergreen Research, Inc. April, 2016 1 Many Levels, Many Approaches The requirements differ dramatically by class and category and
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior
 Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Course Title ID Duration Basic Premium. Biological Evaluation of Medical Devices: A Risk-Based Approach N mins
 Pre-Clinical Basic Level : CMDA Certified Medical Device Associate Biological Evaluation of Medical Devices: A Risk-Based Approach N134 63 mins 186.00 144.00 Introduction to Process Validation N135 75
Pre-Clinical Basic Level : CMDA Certified Medical Device Associate Biological Evaluation of Medical Devices: A Risk-Based Approach N134 63 mins 186.00 144.00 Introduction to Process Validation N135 75
Changes to the International Regulatory Environment
 Changes to the International Regulatory Environment Martin McHugh, Fergal McCaffery, Valentine Casey Regulated Software Research Group & Lero, Department of Mathematics and Computing, Dundalk Institute
Changes to the International Regulatory Environment Martin McHugh, Fergal McCaffery, Valentine Casey Regulated Software Research Group & Lero, Department of Mathematics and Computing, Dundalk Institute
Concept of Risk Management in Medical Equipment Application of ISO in IEC rd Edition
 THE FLAGSHIP OF THE IEC SYSTEM OF CONFORMITY ASSESSMENT IN THE FILED OF THE ELECTROTECHNICAL SECTOR Concept of Risk Management in Medical Equipment Application of ISO 14971 in IEC 6060-1 3rd Edition By
THE FLAGSHIP OF THE IEC SYSTEM OF CONFORMITY ASSESSMENT IN THE FILED OF THE ELECTROTECHNICAL SECTOR Concept of Risk Management in Medical Equipment Application of ISO 14971 in IEC 6060-1 3rd Edition By
Technical documentation. Capita Selecta proposed MDR
 Technical documentation Capita Selecta proposed MDR 25-03-2013 1 Technical Documentation MDD / AIMDD (In daily practice) technical documentation = design dossier = technical (construction) file = design
Technical documentation Capita Selecta proposed MDR 25-03-2013 1 Technical Documentation MDD / AIMDD (In daily practice) technical documentation = design dossier = technical (construction) file = design
A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO COMPLIANCE
 BYLINED ARTICLE ON: A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO 14971 COMPLIANCE By Mark Leimbeck PE Underwriters Laboratories and Larry Kessler Sc.D.
BYLINED ARTICLE ON: A REVIEW OF MARKET ENTRY REQUIREMENTS FOR RISK MANAGEMENT, WITH SPECIAL EMPHASIS ON FDA AND ISO 14971 COMPLIANCE By Mark Leimbeck PE Underwriters Laboratories and Larry Kessler Sc.D.
Essential Requirements Straumann Custom-made Dental Restorative Devices According to 93/42/EEC annex I as amended by 2007/47/EC
 Straumann's CAD (Computer Aided Design) software CARES Visual has built-in parameter checks to assure that a dental restorative device basically meets the generally applicable requirements. However, Institut
Straumann's CAD (Computer Aided Design) software CARES Visual has built-in parameter checks to assure that a dental restorative device basically meets the generally applicable requirements. However, Institut
MDSAP AUDIT PROCESS. A Manufacturer s Perspective. Connie Hoy EVP Regulatory Affairs Cynosure, Inc.
 MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
MDSAP AUDIT PROCESS A Manufacturer s Perspective Connie Hoy EVP Regulatory Affairs Cynosure, Inc. Cynosure Located in Westford, MA Largest manufacturer of Medical Lasers Second location in Hicksville,
RE: Docket No. FDA 2017 N 0041: Agency Information Collection Activities; Proposed Collection; Comment Request; Safety Assurance Case
 701 Pennsylvania Avenue, NW Suite 800 Washington, D.C. 20004 2654 Tel: 202 783 8700 Fax: 202 783 8750 www.advamed.org Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers
701 Pennsylvania Avenue, NW Suite 800 Washington, D.C. 20004 2654 Tel: 202 783 8700 Fax: 202 783 8750 www.advamed.org Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers
IEC Functional Safety Assessment
 IEC 61508 Functional Safety Assessment Project: Rosemount 5300 Series 4-20mA HART Guided Wave Radar Level and Interface Transmitter Device Label SW 2.A1 2.J0 Customer: Rosemount Tank Radar (an Emerson
IEC 61508 Functional Safety Assessment Project: Rosemount 5300 Series 4-20mA HART Guided Wave Radar Level and Interface Transmitter Device Label SW 2.A1 2.J0 Customer: Rosemount Tank Radar (an Emerson
FDA Medical Device HFE Guidance
 W H I T E P A P E R www.makrocare.com U S FDA has established a new Draft Guidance; Applying Human Factors and Usability Engineering to Medical Devices to Optimize Safety and Effectiveness in Design. Manufacturers
W H I T E P A P E R www.makrocare.com U S FDA has established a new Draft Guidance; Applying Human Factors and Usability Engineering to Medical Devices to Optimize Safety and Effectiveness in Design. Manufacturers
11th International Workshop on the Application of FPGAs in Nuclear Power Plants
 11th International Workshop on the Application of FPGAs in Nuclear Power Plants Case Study for Tailoring and Adapting IEEE Std 1012 Software Verification and Validation Requirements for FPGA Technology
11th International Workshop on the Application of FPGAs in Nuclear Power Plants Case Study for Tailoring and Adapting IEEE Std 1012 Software Verification and Validation Requirements for FPGA Technology
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS
 Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Checklist for the assessment based on the standards
 ISO & MDD & Checklist for the assessment based on the standards ISO :2016 ISO :2016 associate with EC Directive 93/42 EEC Where applicable EC Directive 93/42/EEC Annex II/V/VI Company: Audit date Auditor:
ISO & MDD & Checklist for the assessment based on the standards ISO :2016 ISO :2016 associate with EC Directive 93/42 EEC Where applicable EC Directive 93/42/EEC Annex II/V/VI Company: Audit date Auditor:
The New EU Medical Device Regulation (MDR) An Active Device Lifecycle Approach Implementation and Remediation Activities
 An Active Device Lifecycle Approach Implementation and Remediation Activities") visit usdm.com The New EU Medical Device Regulation (MDR) An Active Device Lifecycle Approach Implementation and Remediation Activities Jay Crowley VP and Practice Lead UDI Services and Solutions jcrowley@usdm.com
visit usdm.com The New EU Medical Device Regulation (MDR) An Active Device Lifecycle Approach Implementation and Remediation Activities Jay Crowley VP and Practice Lead UDI Services and Solutions jcrowley@usdm.com
The Theranos 483s. What can we learn? David Amor, MS, CQA Principal, Medgineering
 The Theranos 483s. What can we learn? David Amor, MS, CQA Principal, Medgineering www.medgineering.com david@medgineering.com @Medgineering The good. The bad and the ugly. What can we learn? 2006. The
The Theranos 483s. What can we learn? David Amor, MS, CQA Principal, Medgineering www.medgineering.com david@medgineering.com @Medgineering The good. The bad and the ugly. What can we learn? 2006. The
Conformity Assessment of Medical Devices Under The New MDR
 Conformity Assessment of Medical Devices Under The New MDR (Dec. 06, 2017) Tina Lochner, Medcert Slide 1 Agenda Scope: MDR Article 1 and 2 MDR Conformity Assessment: MHRA* (UK Competent Authority) MDR
Conformity Assessment of Medical Devices Under The New MDR (Dec. 06, 2017) Tina Lochner, Medcert Slide 1 Agenda Scope: MDR Article 1 and 2 MDR Conformity Assessment: MHRA* (UK Competent Authority) MDR
The Quality System Regulation
 Integrating Medical Device Product Development, Design for Six Sigma and Quality Systems Regulation Part II By Dr Vinny Sastri This article was originally published in Medical Device & Diagnostic Industry,
Integrating Medical Device Product Development, Design for Six Sigma and Quality Systems Regulation Part II By Dr Vinny Sastri This article was originally published in Medical Device & Diagnostic Industry,
CE marking is regarded as a passport for products to enter and circulate freely within the 30 counties forming part of the EEA.
 CE Marking Services What is CE Marking? Many products that are placed on the single market in the European Economic Area (EEA) (28 EU countries, together with Iceland, Liechtenstein and Norway) contain
CE Marking Services What is CE Marking? Many products that are placed on the single market in the European Economic Area (EEA) (28 EU countries, together with Iceland, Liechtenstein and Norway) contain
Fill the gap! New product standard: IEC (C) 2016 QAdvis AB
 2016 QAdvis AB") Fill the gap! New product standard: IEC 82304-1 Maria Rickardsson QAdvis AB Stockholm maria.rickardsson@qadvis.com www.qadvis.com QAdvis team QAdvis key competence areas QMS in-the cloud Turn key quality
Fill the gap! New product standard: IEC 82304-1 Maria Rickardsson QAdvis AB Stockholm maria.rickardsson@qadvis.com www.qadvis.com QAdvis team QAdvis key competence areas QMS in-the cloud Turn key quality
CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT
 TF N 548 Rev1 2012-03-29 CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT How to draft clauses on Assessment and Verification of the Constancy of Performance (AVCP) in harmonized standards for construction
TF N 548 Rev1 2012-03-29 CONSTRUCTION SECTOR STANDARDIZATION GUIDANCE DOCUMENT How to draft clauses on Assessment and Verification of the Constancy of Performance (AVCP) in harmonized standards for construction
FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru
 FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru FDA QSR (21 CFR Part 820) ISO 13485:2016 820.1 Scope 1 Scope 2 Normative References 820.3 Definitions 3 Terms and Definitions
FDA 21 CFR Part 820 vs. ISO 13485:2016 Comparison Table created by greenlight.guru FDA QSR (21 CFR Part 820) ISO 13485:2016 820.1 Scope 1 Scope 2 Normative References 820.3 Definitions 3 Terms and Definitions
GENEESS Guideline. Best Practices for Electronics in Medical Devices. V1.3 October 2016
 V1.3 October 2016 Contact Filip Ponsaerts Phone: +32 16 283412 Mobile: +32 474 921351 Filip.Ponsaerts@imec.be IMEC Kapeldreef 75 B3001 Heverlee Copyright imec 2016 All rights reserved. Only an authorized
V1.3 October 2016 Contact Filip Ponsaerts Phone: +32 16 283412 Mobile: +32 474 921351 Filip.Ponsaerts@imec.be IMEC Kapeldreef 75 B3001 Heverlee Copyright imec 2016 All rights reserved. Only an authorized
Co-ordination of Notified Bodies Medical Devices (NB-MED) on Council Directives 90/385/EEC, 93/42/EEC and 98/79/EC
 on Council Directives 90/385/EEC, 93/42/EEC and 98/79/EC") Chapter: 2.7 Clinical investigations, clinical evaluation Text:... Key words: Clinical data, Evaluation 1. Introduction and purpose It is the primary purpose of this document to provide guidance to Notified
Chapter: 2.7 Clinical investigations, clinical evaluation Text:... Key words: Clinical data, Evaluation 1. Introduction and purpose It is the primary purpose of this document to provide guidance to Notified
REQUIREMENTS FOR ACCREDITATION OF INSPECTION BODIES
 NATIONAL CENTRE FOR ACCREDITATION REQUIREMENTS FOR ACCREDITATION OF INSPECTION BODIES according to SM SR EN ISO/IEC 17020:2013 Code DR-OI-07 Edition 2 Page 1/15 Approved by Technical Certification Committee
NATIONAL CENTRE FOR ACCREDITATION REQUIREMENTS FOR ACCREDITATION OF INSPECTION BODIES according to SM SR EN ISO/IEC 17020:2013 Code DR-OI-07 Edition 2 Page 1/15 Approved by Technical Certification Committee
A roadmap to ISO implementation
 A roadmap to ISO 14971 implementation Derek Flood *,, Fergal Mc Caffery, Valentine Casey, Ruth McKeever and Peter Rust Dundalk Institute of Technology Dundalk, Ireland ABSTRACT Medical device standards
A roadmap to ISO 14971 implementation Derek Flood *,, Fergal Mc Caffery, Valentine Casey, Ruth McKeever and Peter Rust Dundalk Institute of Technology Dundalk, Ireland ABSTRACT Medical device standards
U.S. Food and Drug Administration (FDA) Regulatory Pathways for Medical Devices
 Regulatory Pathways for Medical Devices") Lisa L. Michels General Counsel & Regulatory Expert Susan J. Schniepp, Distinguished Fellow Regulatory Compliance Associates Inc. U.S. Food and Drug Administration (FDA) Regulatory Pathways for Medical
Lisa L. Michels General Counsel & Regulatory Expert Susan J. Schniepp, Distinguished Fellow Regulatory Compliance Associates Inc. U.S. Food and Drug Administration (FDA) Regulatory Pathways for Medical
Commissioning of the Heidelberg Ion Beam Therapy Centre
 Commissioning of the Heidelberg Ion Beam Therapy Centre Jacksonville, May 2008 O. Jäkel 1,2, M. Ellerbrock 1, P. Heeg 1, B. Ackermann 1, M. Winter 1, K. Parodi 1, S. Brons 1, A. Mairani 1,2, T. Haberer
Commissioning of the Heidelberg Ion Beam Therapy Centre Jacksonville, May 2008 O. Jäkel 1,2, M. Ellerbrock 1, P. Heeg 1, B. Ackermann 1, M. Winter 1, K. Parodi 1, S. Brons 1, A. Mairani 1,2, T. Haberer
Medical Devices for Clinical Use
 Medical Devices for Clinical Use Andrew Sims Head of Department Northern Medical Physics and Clinical Engineering Newcastle upon Tyne Hospitals NHS Foundation Trust & Institute of Cellular Medicine Newcastle
Medical Devices for Clinical Use Andrew Sims Head of Department Northern Medical Physics and Clinical Engineering Newcastle upon Tyne Hospitals NHS Foundation Trust & Institute of Cellular Medicine Newcastle
ELECtro-Medical devices. in trusted hands. sgs is the world s leading
 ELECTRICAL & ELECTRONICS electro MEDICAL DEVICES ELECtro-Medical devices in trusted hands sgs is the world s leading testing and certification company 1 sgs medical network: THE As a medical devices manufacturer
ELECTRICAL & ELECTRONICS electro MEDICAL DEVICES ELECtro-Medical devices in trusted hands sgs is the world s leading testing and certification company 1 sgs medical network: THE As a medical devices manufacturer