Prioritization: from vcf to finding the causative gene
|
|
|
- Stewart Reeves
- 5 years ago
- Views:
Transcription


1 Prioritization: from vcf to finding the causative gene vcf file making sense A vcf file from an exome sequencing project may easily contain thousand variants. In order to optimize the search for the causative variant it is essential to prioritize the list of variants and to start to consider those that are most likely to be relevant for the phenotype under analysis.
2 Anatomy of a vcf file A full description of vcf can be found at the following link: [HEADER LINES] #CHROM POS ID chr chr rs chr rs chr rs In the example shown above, chr1: chr1: chr1: chr1: is is is is a a a a novel known known known REF T A C T ALT G G T C QUAL FILTER PASS PASS PASS LowQual INFO [ANNOTATIONS] [ANNOTATIONS] [ANNOTATIONS] [ANNOTATIONS] FORMAT GT:AD:DP:GQ:PL GT:AD:DP:GQ:PL GT:AD:DP:GQ:PL GT:AD:DP:GQ:PL NA /1:173,141:282:99:255,0,255 1/1:0,105:94:99:255,255,0 0/1:1,3:4:25.92:103,0,26 0/1:14,4:14:60.91:61,0,255 here is how we interpret the line corresponding to each variant: T/G A/G C/T T/C variant, found with very high confidence (QUAL = ) SNP (named rs ), found with very high confidence (QUAL = ) SNP (named rs ), but has a relative low confidence (QUAL = 71.77) SNP but we have so little evidence that we cannot say anything about it Looking at that last column, here is what the tags mean: GT : The genotype of 0/0 - the sample 0/1 - the sample 1/1 - the sample this sample. For a diploid organism, GT will be either: is homozygous reference is heterozygous, carrying 1 copy of each of the REF and ALT alleles is homozygous alternate GQ: The Genotype Quality, or Phred-scaled confidence that the true genotype is the one provided in GT. AD and DP: These are complementary fields: AD (DepthPerAlleleBySample) and DP (Coverage). PL: This field provides the likelihoods of the given genotypes (here, 0/0, 0/1, and 1/1).
3 Needles in a haystack: finding relevant variants from exome sequencing To find the needle in the haystack (i.e. the relevant variants in the vcf file) we need some criteria of prioritization. For instance: Gene and disease. If the patient has a disease that is known in terms of possible candidate genes, then the simplest and easiest thing to do is to give a high priority to the analysis to the candidate genes. OMIM can be very useful for this approach, but often it is not enough. Genetics and zygosity. Sometimes, depending on the genetics of the disease, it is possible to restrict the analysis to homozygous variants or to the chromosome X. Chromosomal position. If we know genetic markers associated to the disease we can prioritize their chromosomal region. Variant frequency. An important point to consider is the frequency of the variant in the population. If the frequency is high, then it is unlikely that the variant is pathogenic. This is even more so if the phenotype shows a dominant inheritance pattern. Variant patogenicity. Some variants are already known to be associated to genetic diseases, therefore we can prioritize them. Variant effect on protein. If a mutation is an a protein-coding region, then we can estimate the effect of the variant on the protein. There are several programs that have been developed to do this task, such as PolyPhen and SIFT. Gene and Disease ontologies. It is also possible to prioritize genes associated to Gene Ontology terms, or to disease ontology or to other ontologies. Quality of the called variant. We can give low priority to the variants that were called with low quality, for instance because the coverage is low or because the reads are not good.
 Given a disease, you can find all the know genes related to the disease; 2) given a")
4 OMIM OMIM OMIM is an enciclopedia of genetic diseases and related genes. It can be useful in two main ways: 1) Given a disease, you can find all the know genes related to the disease; 2) given a gene you can find if it is associated to any disease.
5 Searching OMIM for diseases For instance, if you search for DENT DISEASE 1 you get this page: CLICK HERE TO SEE THE ENTIRE PAGE or HERE TO GO TO THE ACTUAL OMIM PAGE You can see that this disease is caused by mutations on the CLCN5 gene
6 Searching OMIM for genes You can also search OMIM for genes, for instance CLCN5: CLICK HERE TO SEE THE ENTIRE PAGE or HERE TO GO TO THE ACTUAL OMIM PAGE

7 ClinVar is a public resource to link variants to clinically relevant diseaes

8 ClinVar web site: the book : local copy of the book
9 Needles in a haystack: finding relevant variants from exome sequencing To find the needle in the haystack (i.e. the relevant variants in the vcf file) we need some criteria of prioritization. For instance: Gene and disease. If the patient has a disease that is known in terms of possible candidate genes, then the simplest and easiest thing to do is to give a high priority to the analysis to the candidate genes. OMIM can be very useful for this approach, but often it is not enough. Genetics and zygosity. Sometimes, depending on the genetics of the disease, it is possible to restrict the analysis to homozygous variants or to the chromosome X. Chromosomal position. If we know genetic markers associated to the disease we can prioritize their chromosomal region. Variant frequency. An important point to consider is the frequency of the variant in the population. If the frequency is high, then it is unlikely that the variant is pathogenic. This is even more so if the phenotype shows a dominant inheritance pattern. Variant patogenicity. Some variants are already known to be associated to genetic diseases, therefore we can prioritize them. Variant effect on protein. If a mutation is an a protein-coding region, then we can estimate the effect of the variant on the protein. There are several programs that have been developed to do this task, such as PolyPhen and SIFT. Gene and Disease ontologies. It is also possible to prioritize genes associated to Gene Ontology terms, or to disease ontology or to other ontologies. Quality of the called variant. We can give low priority to the variants that were called with low quality, for instance because the coverage is low or because the reads are not good.
10 Several resources are becoming available for assessing the frequency of variants in the population. NCBI is curating several such resources, for instance: dbsnp 1000 genomes

11
12 Needles in a haystack: finding relevant variants from exome sequencing To find the needle in the haystack (i.e. the relevant variants in the vcf file) we need some criteria of prioritization. For instance: Gene and disease. If the patient has a disease that is known in terms of possible candidate genes, then the simplest and easiest thing to do is to give a high priority to the analysis to the candidate genes. OMIM can be very useful for this approach, but often it is not enough. Genetics and zygosity. Sometimes, depending on the genetics of the disease, it is possible to restrict the analysis to homozygous variants or to the chromosome X. Chromosomal position. If we know genetic markers associated to the disease we can prioritize their chromosomal region. Variant frequency. An important point to consider is the frequency of the variant in the population. If the frequency is high, then it is unlikely that the variant is pathogenic. This is even more so if the phenotype shows a dominant inheritance pattern. Variant patogenicity. Some variants are already known to be associated to genetic diseases, therefore we can prioritize them. Variant effect on protein. If a mutation is an a protein-coding region, then we can estimate the effect of the variant on the protein. There are several programs that have been developed to do this task, such as PolyPhen and SIFT. Gene and Disease ontologies. It is also possible to prioritize genes associated to Gene Ontology terms, or to disease ontology or to other ontologies. Quality of the called variant. We can give low priority to the variants that were called with low quality, for instance because the coverage is low or because the reads are not good.
13 SIFT Sorting Tolerant From Intolerant (SIFT) algorithm flowchart for scoring individual amino acid substitutions. The process flow isdescribed for the amino acid sequence IRRLRPMD. The general rationale of SIFT is that homologous proteins have similar structure and conserved amino acids in some positions. Thus, the impact of an amino acid substitution can be evaluated, taking into consideration how conserved is that amino acid position in homologous proteins. From: Kumar P, Henikoff S, Ng PC. Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):
 of homologous proteins, but it considers also protein structure predictions.")
14 PolyPhen-2 PolyPhen is also a program for predicting damaging effects of missense mutations. Also this program uses information from Multiple Sequence Alignment (MSA) of homologous proteins, but it considers also protein structure predictions. Then the program uses an advanced statistical analysis to predict the general impact of the variant on the function of the mutated protein. From: Adzhubei et al. Nat Methods 7(4): (2010).
15 Needles in a haystack: finding relevant variants from exome sequencing To find the needle in the haystack (i.e. the relevant variants in the vcf file) we need some criteria of prioritization. For instance: Gene and disease. If the patient has a disease that is known in terms of possible candidate genes, then the simplest and easiest thing to do is to give a high priority to the analysis to the candidate genes. OMIM can be very useful for this approach, but often it is not enough. Genetics and zygosity. Sometimes, depending on the genetics of the disease, it is possible to restrict the analysis to homozygous variants or to the chromosome X. Chromosomal position. If we know genetic markers associated to the disease we can prioritize their chromosomal region. Variant frequency. An important point to consider is the frequency of the variant in the population. If the frequency is high, then it is unlikely that the variant is pathogenic. This is even more so if the phenotype shows a dominant inheritance pattern. Variant patogenicity. Some variants are already known to be associated to genetic diseases, therefore we can prioritize them. Variant effect on protein. If a mutation is an a protein-coding region, then we can estimate the effect of the variant on the protein. There are several programs that have been developed to do this task, such as PolyPhen and SIFT. Gene and Disease ontologies. It is also possible to prioritize genes associated to Gene Ontology terms, or to disease ontology or to other ontologies. Quality of the called variant. We can give low priority to the variants that were called with low quality, for instance because the coverage is low or because the reads are not good.

16 The Disease Ontology may be useful to classify diseases in a computer friendly manner
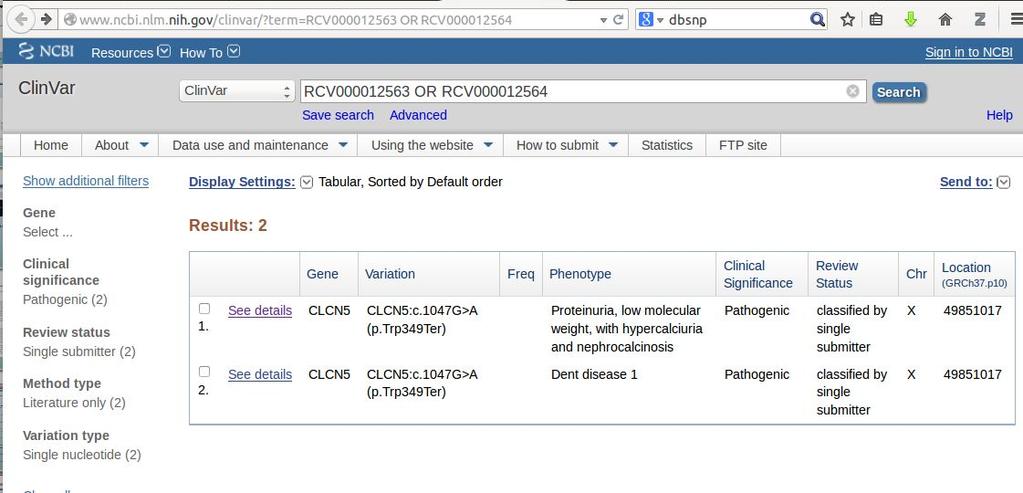
17
18 The prioritization process requires many analyses and the final integration of the results that in many cases cannot be done just by the computer because it needs a direct manual supervison to progressively refine the search. For this purpose we have developed QueryOR Other similar programs are becoming available.

19
20 QueryOR
21 Data integration The full explanation of QueryOR is available at the web site

22 You can select amongst many filters and queryor will return the list of genes that best satisfy your selection. You can also give dfferent weights to each filter.

23 You can visualize different individual to check whether they share the same variants (for instance a trio mather, father and child). You should also consider that each gene may produce different isoforms of a transcript, therefore the effect of a variant may depend on the alternative isoform. You can select the isoform and go to the next page (Transcript report page) that will show you the specific effect of the variant on that isoform.

24

25 Please visit our Queryor home page
26 When the short list of candidate genes is reduced to very few possibilities, then it is recommendable to check manually the quality of the reads and the coverage.

27 Visualizing vcf and bam files with the IGV genome browser VCF BAM SEQ A vcf and the original bam files were loaded onto the IGV genome browser. The vcf track, at the top, shows the variants that have been called, while the bam track shows all the reads and the original mismatches ith the reference genome.
28 Prioritization sequencing Illumina Proton Solid fastq file alignment BWA many others sam file variant call vcf file GATK IonProtonCaller make sense QueryOR some other These analyses require a comprehensive knowledge of different field for this reason they are still quite difficult to carry out.