Standard Operating Procedure
|
|
|
- Sharlene Chapman
- 5 years ago
- Views:
Transcription
: Signature: On file Signature: On file Supersedes (if applicable): Date: 23/12/2015 Date: 23/12/2015 Version: 1 The signatures above")
1 Standard Operating Procedure Title: Clinical Site Monitoring Status: PRIVATE Author Name: Audrey Strader Approver Name: Christine Kubiak Document no.: CSM 02 Effective date: 11/01/2016 Review Date (if applicable): Signature: On file Signature: On file Supersedes (if applicable): Date: 23/12/2015 Date: 23/12/2015 Version: 1 The signatures above certify that this procedure has been reviewed and accepted, and demonstrates that the signatories are aware of all the requirements contained herein and are committed to ensuring their provision. Amendment Record This procedure is reviewed to ensure its continuing relevance to the systems and process that it describes. A record of contextual additions or omissions is given below: Version no. Date Reason for change Main modifications SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 1/14
2 Table of Contents 1.0 PURPOSE SCOPE DEFINITIONS AND ABBREVIATIONS PROCEDURE Extent of monitoring CRA Responsibilities Monitoring Report REFERENCES APPENDICES Monitoring Visit Report Form Template Contact Form Template NOTE TO FILE Template SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 2/14
3 1.0 PURPOSE The purpose of this SOP is to describe monitoring procedures for clinical trials monitored by CRAs in order that Clinical Trials conducted within different institutions comply with European Law (European Union Directive 2001/20/CE and its transpositions to each member country) Monitoring is defined as the act of overseeing the progress of a clinical trial, and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, SOP s, GCP, and the applicable regulatory requirement(s). The purpose of monitoring is to verify: The rights and well-being of the human subjects are protected The reported trial data are accurate, complete and verifiable from source documents The conduct of the trial is in compliance with the currently approved protocol/amendment (s), GCP and the applicable regulatory requirements. Monitoring has an integral role in the quality control (QC) of a clinical trial and is designed to verify the ongoing quality of the study. 2.0 SCOPE All clinical trials sponsored by one or more academic trial unit or other institution will be monitored as described in this SOP. Trials sponsored by other organizations other than those above may be also monitored according to this SOP. Monitoring will be conducted by ECRIN scientific partners. 3.0 DEFINITIONS AND ABBREVIATIONS Adverse Event (AE): Any untoward medical occurrence in a subject to whom a medicinal product (or medical device) including occurrences which are not necessarily caused by or related to that product. Case Report Form (CRF): a printed, optical or electronic document designed to record all of the protocol required information to be reported to the sponsor on each trial subject Clinical Trial: any investigation in human subjects, other than a non-interventional trial, to discover or verify the clinical, pharmacological or other pharmacodynamics effects of one or more medicinal product or to identify any adverse reactions to one of more such products and to study absorption, distribution metabolism and excretion in one of more such products with the object of ascertaining the safety or efficacy of those products. SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 3/14
4 Clinical Research Associates (CRA s) / Monitor: An individual by training responsible for ensuring compliance with the Regulations, GCP and SOPs, by monitoring clinical trials. Contact Form (CF): A form used to record communication with a trial site which requires documentation. Ethics Committee (EC) The EC that undertakes the review of the research protocol, including the content of the patient information sheet and consent form rather than just site specific approval for each center. Good Clinical Practice (GCP): defined in the ICH-GCP regulations Informed Consent Form (ICF): A document which is signed by the participant/legal representative as well as the person who conducted the informed consent discussion confirming the willingness of the volunteer s to participate in the particular trial after having been informed of all aspects of the trial that are relevant to their decision. Investigational Medicinal Products (IMP): A pharmaceutical form of an active substance or placebo being tested, or used a reference in a clinical trial. This includes a medicinal product which has a marketing authorization but is, for the purposes of the trial- A. Used or assembled (formulated or packaged) in a way different from the form of the product authorized under the authorization, B. Used for an indication not included in the summary of product characteristics (SmPC) under the authorization of that product, or C. Used to gain further information about the form of that product as authorized under the authorization. Investigator Site File (ISF): A standard filing system which allows the effective storage and location of essential administrative and regulatory documents related to an individual trial site. Monitoring Plan: A document, detailing how all the monitoring activities for the trial will be carried out based upon the trial risk assessment and as defined in the trial protocol, if applicable. SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 4/14
5 Monitoring visit report (MVR): A report written by the CRA to the sponsor (or representative) after each site visit, or if applicable, a remote visit. Patient Information Sheet (PIS): This explains all relevant study information to assist the trial participant in understanding the expectations and requirements of participation in a clinical trial. The sheet accompanies the Patient Informed Consent (PIC). Trial Master File (TMF): a standard filing system which allows the effective storage and location of essential documents, that is the large volume of regulatory documents and approvals needed for clinical research. The filing system can be in the form of a single project file or a number of files / filing cabinets, depending on what is deemed most appropriate for a particular clinical trial given its size and complexity. The regulatory documents and approval within the TMF will be maintained alongside case report forms and source documentation. 4.0 PROCEDURE The CRA s are the main line of communication between the trial site investigator and the sponsor. The CRA ensures that the Investigator conducts the clinical study in compliance with the final protocol and subsequent protocol amendments if any, as well as GCP and applicable safety reporting and regulatory requirements. 4.1 Extent of monitoring The Sponsor is responsible for determining the appropriate level and nature of monitoring required for the clinical trial. Monitoring will be proportional to the objective, purpose, design, size, complexity, blinding, endpoints and risks of the clinical study. It will be the Sponsor s responsibility to determine the appropriate level and nature of monitoring required for their clinical trial by risk assessment. Further to the risk assessment, detailed monitoring requirements will be documented in the trial specific monitoring plan. Before each visit, and as detailed in the monitoring plan, the CRA will confirm the either by letter or specifying who is expected to be present. After each visit, the CRA will send a follow up letter confirming what was discussed, outstanding action items, or deficiencies noted during the visit. If contact has been made with the site, a contact form will be used to document what was discussed. SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 5/14
6 4.2 CRA Responsibilities 1. The CRA will act as the main line of communication between the investigator and trial site and the sponsor. 2. The CRA will ensure that the Investigator provides all the required reports, notifications, applications, and submissions and that these documents are accurate, complete, timely, and legible, dated and identify the trial. 3. The CRA will ensure that all documents and trial supplies needed to conduct the trial properly, and to comply with the applicable regulatory requirements, are available and filed in the ISF. 4. The CRA will verify that any new Investigator has adequate qualifications, and that the resources and facilities, including laboratories, equipment and staff, to safely and properly conduct the trial and that these remain adequate throughout the study period. 5. The CRA will verify that trial functions are performed as designated and not delegated to unauthorized individuals. 6. The CRA will verify that informed consent was obtained and documented prior to subject participation in the trial and that only eligible subjects are enrolled as detailed in the trial monitoring plan. 7. The CRA ensures that the Investigator follow the approved protocol and any approved amendment(s), relevant regulatory requirements and is adequately informed about the study. Deviations will be communicated to the Investigator and appropriate action designed to prevent recurrence of the detected deviations taken. 8. The CRA will verify that source documents and other trial records are accurate, complete and up-to-date, and check the accuracy and completeness of the CRF entries. The CRA will examine a proportion of CRFs as specified in the monitoring plan. Where required by the monitoring plan with respect to the CRFs, the CRA will verify: a. The data required by the protocol are reported accurately in the CRFs and are consistent with the source documents according to the monitoring plan b. Any dose and/or therapy modifications are well documented for each of the trial subjects c. Adverse events, concomitant medications and concurrent illnesses are reported in accordance with the protocol in the CRFs. d. Visits that the subjects fail to make, tests that are not conducted, and examinations that are not performed are clearly reported as such in the CRFs. SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 6/14
7 e. All withdrawals and dropouts of enrolled subjects from the trial are reported and explained on the CRFs 9. The CRA will inform the Investigator of any CRF entry error, omission or illegibility and ensure that appropriate corrections, additions or deletions are made, dated, explained (if necessary) and initialed by the Investigator or an authorized individual. The CRA is not permitted to make such changes. 10. Verification and collection of subject data should be performed according to data protection laws and requirements. 11. The CRA will determine whether all SAEs are appropriately reported within the time periods required by GCP, the protocol, the EC, the sponsor, and the applicable regulatory requirements. 12. Where required with respect to the IMP, the CRA will ensure: a. Storage times and conditions are acceptable and that supplies are sufficient b. IMP is supplied only to subjects who are eligible, at the protocol specified doe(s). c. Subjects are provided with necessary instruction on properly using, handling, storing and returning IMP(s) d. The receipt, use and return of any IMP(s) at the trial sites are controlled and documented adequately. e. Disposal of unused IMP(s) complies with applicable regulatory requirements(s) and is in accordance with the sponsors SOP. 13. The CRA will ensure that any unblinding is properly documented according to the protocol. The CRA team will immediately notify the sponsor in the event of any suspicion of scientific misconduct, fraud or breach of GCP. 4.3 Monitoring Report Following a monitoring visit, the CRA will submit a written report to the sponsor within 14 days. This will be done using the Monitoring Visit Report Form (see section 5.1). Any other communication with the study site which requires documentation will be recorded on a Contact Form or (see section 5.2) A Note-to-File (see section 5.3) will be used to document any deviations from the protocol or other issues and these should be filed in the ISF and/or TMF with copies submitted to the sponsor. SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 7/14
8 According to the monitoring plan, the report or form will be reviewed promptly after the visit or communication and signed by an authorized individual. The investigator will be informed in writing of any problem(s) that were identified during the monitoring visit. If there is evidence of systematic failure to comply with GCP retraining will be given and trial management/sponsor informed. 5.0 REFERENCES 1. ICH E6 Good Clinical Practices 2. Directive 2001/20/EC 6.0 APPENDICES 6.1 Monitoring Visit Report Form Template 6.2 Contact Form Template 6.3 Note to File Template SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 8/14


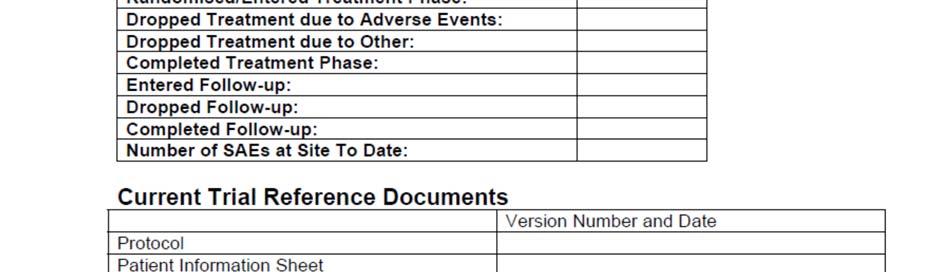
9 6.1 Monitoring Visit Report Form Template SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 9/14


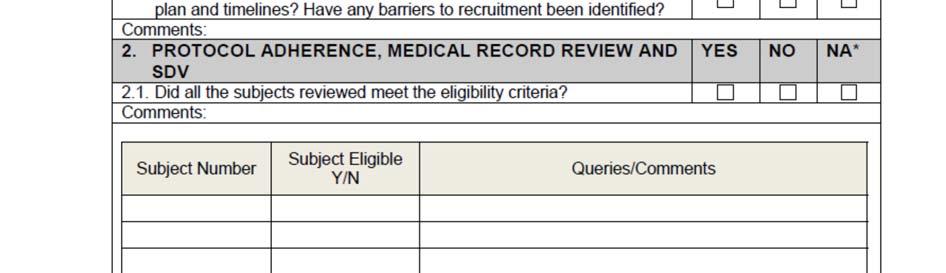
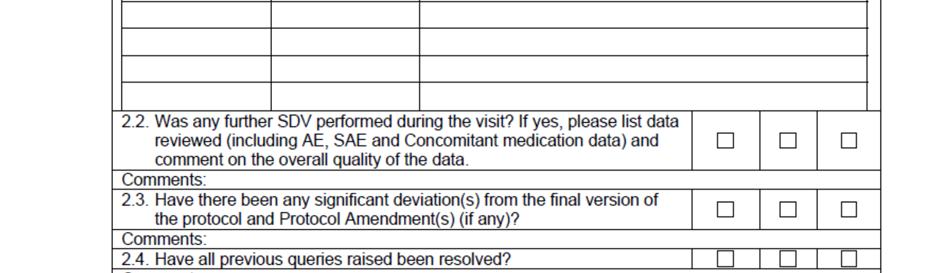
10 SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 10/14

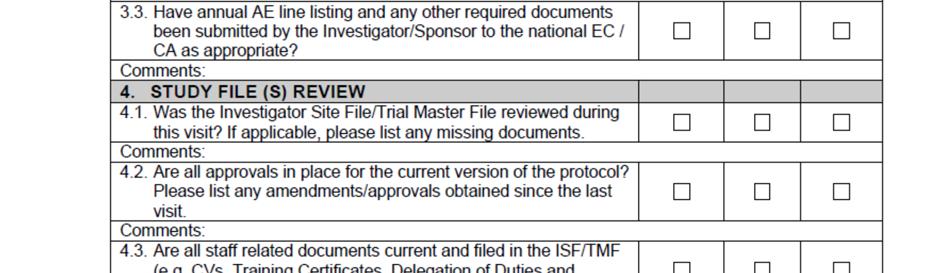
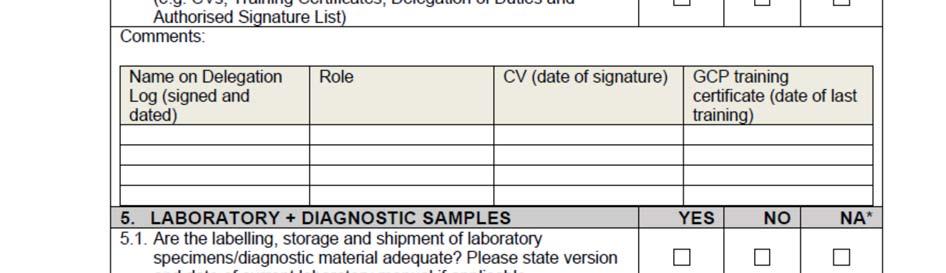

11 SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 11/14



12 SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 12/14


13 6.2 Contact Form Template SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 13/14
14 6.3 NOTE TO FILE Template SOP_02_Clinical_Site_Monitoring_V1_2015_12_23 Page 14/14