Inefficient Translation Renders the Enterococcus faecalis fabk Enoyl-ACP Reductase Phenotypically Cryptic
|
|
|
- Gwenda Jones
- 6 years ago
- Views:
Transcription
1 JB Accepts, published online ahead of print on 25 October 2013 J. Bacteriol. doi: /jb Copyright 2013, American Society for Microbiology. All Rights Reserved. Inefficient Translation Renders the Enterococcus faecalis fabk Enoyl-ACP Reductase Phenotypically Cryptic Hongkai Bi 1, Lei Zhu 1,2, Haihong Wang 2* and John E. Cronan 1,3* Departments of 1 Microbiology and 3 Biochemistry, B103 Chemical and Life Sciences Laboratory, University of Illinois, 601 S. Goodwin Ave, Urbana, Illinois 61801, USA. 2 College of Life Science, South China Agricultural University, Guangzhou , China These authors contributed equally to this work. Running title: Cryptic E. faecalis fabk All correspondence should be addressed to: Haihong Wang, Ph.D. College of Life Science South China Agricultural University No.483 Wushan Road, Tianhe, Guangzhou , P. R. China Phone:(86) Fax:(86) wanghh36@scau.edu.cn OR John E. Cronan, Ph.D. Departments of Biochemistry and Microbiology
2 University of Illinois B103 CLSL, 601 S Goodwin Ave Urbana 61801, USA Phone: Fax: j-cronan@life.uiuc.edu.
3 Summary Enoyl-acyl carrier protein (ACP) reductase catalyzes the last step of the bacterial fatty acid elongation cycle. Enterococcus faecalis is unusual in that it encodes two unrelated enoyl-acp reductases, FabI and FabK. We recently reported that deletion of the gene encoding FabI results in an unsaturated fatty acid (UFA) auxotroph despite the presence of fabk, a gene encoding a second fully functional enoyl-acp reductase. By process of elimination the prior work argued that poor expression was the reason that fabk failed to functionally replace FabI. We now report that FabK is indeed poorly expressed and that the expression defect is at the level of translation rather than transcription. We isolated four spontaneous mutants that allowed growth of the E. faecalis fabi strain on fatty acid-free medium. Each mutational lesion (single base substitution or deletion) extended the fabk ribosome binding site. Inactivation of fabk blocked growth indicating that the mutations acted only on fabk rather than a downstream gene. The mutations activated fabk translation to levels that supported fatty acid synthesis and hence cell growth. Furthermore, site-directed and random mutagenesis experiments showed that point mutations that resulted in increased complementarity to the 3' end of the 16S ribosomal RNA increased FabK translation to levels sufficient to support growth whereas mutations that decreased complementarity block fabk translation.
4 Introduction The type II or dissociated fatty acid synthesis pathway is responsible for membrane fatty acid synthesis in most bacteria. Fatty acid synthesis is carried out by a series of discrete proteins encoded by separate genes (1, 2) and differs significantly from the type I mammalian and fungal systems in which multifunctional polypeptides catalyze fatty acid synthesis (3-5). This distinguishing characteristic makes the type II system a prime target for antibacterial agents. Enoyl-ACP reductases (ENRs) catalyze the last step of fatty acid elongation cycle, the reduction of trans-2-acyl-acps (an enoyl-acp) to the fully saturated acyl-acp species. Unlike the other FAS II enzymes, ENRs display an unusual diversity. In bacteria, four ENR isozymes have been identified; the products of the fabi (6, 7), fabl (8), fabv (9, 10), and fabk (11, 12) genes. FabI, FabL and FabV are diverse members of the short-chain dehydrogenase/reductase (SDR) superfamily whereas FabK is a TIM barrel flavoprotein (13). Genes encoding homologues of FabI are readily identified in most bacteria. These are about 40% identical to Escherichia coli FabI and contain a conserved Tyr-156 (Xaa) 6 Lys-163 (E. coli numbering) catalytic dyad (10, 14). For example, E. coli and Francisella tularensis contain a single ENR encoded by fabi, which is essential for bacterial viability (7, 15). In contrast two SDR superfamily ENRs, FabI and FabV have been characterized in Pseudomonas aeruginosa. Each of the encoding genes can support growth and functionally replace E. coli fabi (10) as also found for the two SDR superfamily ENRs of Bacillus subtilis (8). Thus, it seems that some bacteria have only a single ENR whereas others have two, either of which suffices for growth in the laboratory. However, this is not the case for Enterococcus faecalis, a Gram-positive commensal bacterium that normally inhabits the gastrointestinal tracts of humans and other mammals (16, 17). Recently we characterized two ENRs, FabI and FabK, that play different roles in E. faecalis. FabI is the major ENR of this bacterium in that deletion of fabi results in unsaturated fatty acid (UFA) auxotrophy whereas deletion of fabk has no effect on growth although the traces ENR activity remaining in the fabi strain do alter the cellular fatty acid composition (18). Both FabI and FabK are fully functional in vitro and functionally replace E. coli fabi. Moreover, when expressed from a plasmid construct, FabK restores growth of the fabi strain indicating that it is poorly expressed in its native chromosomal location.
5 In this report we demonstrate that the inability of fabk to support growth of E. faecalis is due to inefficient translation. We found that upon plating of E. faecalis fabi null mutants on fatty acid free medium, colonies arose that no longer required oleate supplementation. The mutations were found to extend the complementarity between the fabk ribosome binding region and the 3 end of the 16S rrna.
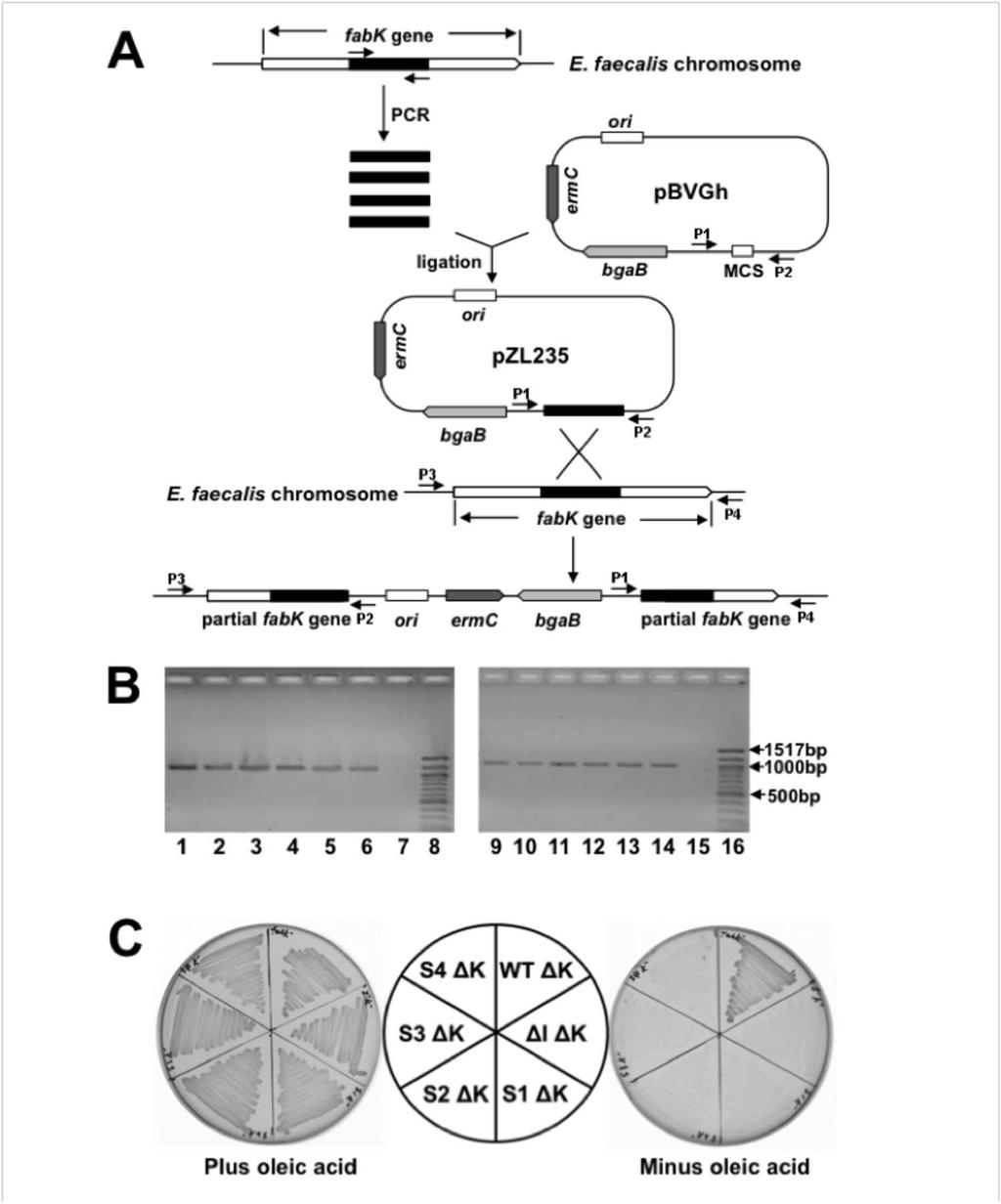
6 Materials and Methods Bacterial strain, plasmids and growth conditions The E. faecalis strains (Table 1) were all derived from FA2-2, a plasmid-free strain (30). Primers used in this study are given in Table 2. Generally, E. coli strains were grown at 37 C in Luria Bertani (LB) medium (tryptone, 10 g/l; yeast extract, 5 g/l; NaCl, 10 g/l, ph 7.0), whereas E. faecalis strains were maintained at 37 C or 42 C in GM17 medium (31). Antibiotics were added as required at the following concentrations (in mg/l): sodium ampicillin, 100 for E. coli; kanamycin sulfate, 25 for E. coli; erythromycin, 150 for E. coli and 5 for E. faecalis. Bacterial growth was determined by measuring optical density at 600 nm. Oligonucleotide primers were synthesized by Integrated DNA Technologies and cloned genes were verified by sequencing performed by ACGT, Inc. Gene sequences were amplified using Pfu Turbo DNA polymerase (Stratagene) according to the manufacturer s recommendations. The pcr2.1-topo vector (Invitrogen) was applied for PCR cloning, and E. coli strain DH5 is the corresponding recipient host (Table 1). Primers fabkup275 and fabkdo288 were used to amplify fabk fragments from E. faecalis FA2-2 and four spontaneous mutants and the PCR products were cloned to pcr2.2- TOPO vector to give plasmids pzl52, pzl53, pzl54, pzl55 and pzl56 for sequencing. These five plasmids were digested with EcoRV and BamHI and inserted into plasmid ptrkl2 to yield plasmids pzl157, pzl158, pzl159, pzl160 and pzl161, respectively, which were linearized and used to transform strain FAZL1. Qiagen provided plasmid isolation and PCR product purification kits. Construction of translational fusions The BamHI-digested DNA fragment containing a promoterless lacz from the plasmid pmc1871 (32)was ligated to the shuttle plasmid ptrkl2 cut with the same enzyme to give the plasmid pbhk322. The insertion direction was confirmed by DNA sequencing. Overlapping PCR was used to generate the fusion of the promoter P32 from Lactococcus lactis to fabk (-136 to +35 relative to the fabk ATG initiation codon). The promoter P32 from L. lactis and fabk (-136 to +35) from E. faecalis FA2-2 were first amplified from the genomic DNAs using primer sets P32- f/p32fabkfusion-r and P32fabKfusion-f/fabKfusion-r, respectively. The first step PCR fragments were purified from agarose gels and mixed to serve as templates to amplify chimera-p32fabk
7 with primer sets P32-f/fabKfusion-r. The PCR products after digestion with PstI and SalI were ligated into the plasmid pbhk322 to produce pbhk323. For the construction of other four translational fusion plasmids each containing a spontaneous mutation in the RBS region, fabk (- 136 to +35) was first amplified from the genomic DNAs of spontaneous mutants BHK260, BHK261, BHK262 or BHK263 and used as the first step PCR template. The following procedure was the same to the construction of the plasmid pbhk323 and finally the fusion plasmids pbhk324, pbhk325, pbhk326 and pbhk327 were obtained. Construction of E. faecalis strains having disrupted fabk genes E. faecalis strains having disruptions of fabk were constructed using the method described previously (33). A centrally located segment of fabk was obtained by amplification from the genomic DNA of E. faecalis FA2-2 strain with Pfu DNA polymerase and primers (Table 2) containing designed restriction sites (a SacI site in the forward primer and a PstI site in the reverse primer). This fragment was then used to construct plasmid pzl235 by insertion of the PstI/SacI digested PCR fragment into plasmid pbvgh cut with the same enzymes. The disruption plasmid was transformed into competent cells of E. faecalis wild-type strain FA2-2. After recovery in SGM17 medium at 30 C, the transformed cells were plated on SGM17 agar containing 5 µg/ml of erythromycin plus 100 µg/ml of X-Gal and incubated for 24 h at 30 C. The resulting blue colonies were cultured overnight in GM17 liquid medium containing erythromycin at 30 C and the cultures were plated on GM17 agar medium containing 5 µg/ml of erythromycin and 100 µg/ml of X-Gal and incubated for 24 h at 42 C. To construct fabk disruptions in strain carrying the fabi mutation 0.1% Tween 40, 0.1% Tween 80 and 0.01% oleate were added to the growth medium. The single-crossover homologous recombinant colonies were screened for disruption of the fabk gene by PCR analysis with pbvgh primers (P1 and P2) and fabk check primers (P3 and P4) and verified by sequencing. Transformation of E. faecalis Electro-competent cells were prepared as previously described (31) with minor changes. A single colony of E. faecalis was grown overnight in GM17 medium and then diluted 1:100 into 100 ml of SGM17 (GM17 with 0.5M sucrose and 8% glycine) medium in a 500 ml flask with vigorous
8 shaking at 37 C. When the OD600 reached , the cultures were chilled on ice for 30 min and then collected by centrifugation at 3600 g for 12 min at 4 C. The harvested pellets in each tube were suspended in 100 ml of ice-cold electroporation buffer (0.5 M sucrose and 10% glycerol) and then centrifuged at 3800 g for 16 min at 4 C. Following three rounds of washing the cells with electroporation buffer, the bacterial pellets were suspended in 1 ml of ice-cold electroporation buffer, divided into small aliquots which were frozen on dry ice and kept at - 80 C until use. For the preparation of strain FAZL1 ( fabi of E. faecalis FA2-2) electrocompetent cells, 0.1% Tween 40, 0.1% Tween 80, and 0.01% oleate were added to the growth medium. Prior to electroporation the frozen cell suspensions were thawed on ice, mixed with plasmid DNA ( ng for delivery of plasmids) and then placed on ice for no more than 30 min prior to transfer into a chilled 2 cm gap cuvette (Bio-Rad). Electroporation parameters for the Gene Pulser apparatus (Bio-Rad) were 25 µf capacitance, 200 resistance and 2.5 kv electrical pulse to give a field strength of 12.5 kv cm-1 with an exponential decay constant of approximately 4 5 ms. After immediate placement on ice for 5 min, the pulsed cells were quickly diluted with 1 ml of SGM17MC medium (31) grown at 37 C for 2 h and plated on SR agar plates containing the appropriate antibiotics. The plates were incubated at 37 C for h. Transformants containing plasmids were verified by plasmid isolation. Acyl-ACP preparations The ACP thioester of trans-2-decenoic acid was synthesized as described previously (34). Briefly, a typical reaction mixture consisted of 25 M holo-acp, 200 M fatty acid and 170 nm V. harveyi AasS in a buffer containing 100 mm Tris-HCl (ph 7.8), 10 mm MgCl 2, 1 mm tris(2- carboxyethyl) phosphine HCl, and 10 mm ATP in a reaction volume of 1 ml. The reaction mixtures were incubated at 37 o C for 4 h and stopped by addition of two volumes of acetone and the proteins allowed to precipitate at -20 o C overnight. The precipitates were pelleted at 20,000xg for 30 min at 4 o C and then washed twice with 3 volumes of acetone. The pellets were air dried and resuspended in 20 mm Tris-HCl (ph 7.4) containing 1 mm tris(2-carboxyethyl) phosphine HCl. The final samples were concentrated with an Amicon ultracentrifugation filter device from Millipore (5,000 MWCO). Acyl-ACP synthesis was verified by electrophoresis on 18% gels
9 containing 2.5 M urea (2, 35) and by electrospray mass spectrometry. Note that the ACP preparations retained the N-terminal methionine NADH oxidation assay ENR activity was monitored spectrophotometrically by the decrease in the absorbance at 340 nm using an NADH extinction coefficient of 6,220 M -1 (9). Cultures grown to mid-log phase in 100 ml of GM17 medium were collected by centrifugation, washed twice with 0.9% KCl, repelleted, and treated for 30 min with lysozyme (15 mg/g, wet weight) in an osmotically protected medium consisting of 0.4 M NaCl, 0.05 M phosphate buffer, (ph 7.2), and 0.5 M sucrose. The cyclopropane fatty acid, cis-9,10-methylenehexadecanic acid (0.005%) was added to GM17 medium to support the growth of the fabi strain FAZL1. After lysozyme treatment, the cells were resuspended in 1 ml of 100 mm Tris-HCl (ph 8.0) containing 400 mm NaCl and lysed by passage three times through a French pressure cell press at 11,000 psi. The crude extract was collected on ice and cleared by centrifugation, and the protein concentration was determined by the Bio-Rad Bradford assay using a standard curve of bovine serum albumen (36). Crude extract protein preparations (1 to 10 µg) were added directly to UV-transparent microcuvettes containing the assay reaction mixture (volume, 100 µl), consisting of 0.1 M sodium phosphate buffer (ph 7.0), 100 µm trans-2-decenoyl-acp, 150 µm NADH, and 0.1 M LiCl. Kinetic constants were determined using GraphPad Prism software, version 4. Radioactive labeling, phospholipid extraction and fatty acid analysis Fatty acid biosynthesis was analyzed by [1-14 C]acetate incorporation. E. faecalis strains were cultured in GM17 medium (containing 0.005% cis-9,10-methylenehexadecanic acid for growth for fabi mutant FAZL1) at 37 o C overnight. Cells were washed twice with water, resuspended in the same volume of GM17 medium and 0.1 ml this suspension was added to 5 ml of GM17 with containing 5 Ci of sodium [1-14 C]acetate. These cultures were grown at 37 o C to mid-log phase and the cells were collected by centrifugation. The phospholipids were extracted by the method of Bligh and Dyer (37) and labeled fatty acid methyl esters were prepared and analyzed by thin layer chromatography and autoradiography as described previously (38). -Galactosidase Assays
10 The cultures were grown overnight and then diluted 1:100 into fresh medium having the same composition and grown to the mid-log phase before assays were performed. Mid-log phase cultures were collected by centrifugation, washed twice with Z Buffer and assayed for - galactosidase activity after lysis with sodium dodecyl sulfate-chloroform (39). The data were recorded in triplicate with more than three independent experiments. RNA Isolation and Reverse Transcription-Polymerase Chain Reaction Total RNA preparations were isolated from the mid-log phase cells of E. faecalis FA2-2 and its derivatives grown in GM17 medium using the RNeasy bacterial RNA isolation kit (Qiagen). The contaminating DNA was digested using RNase-Free DNase Set (Qiagen) according to the manufacturer s protocol. The isolated RNA samples were analyzed by agarose gel electrophoresis to assess the quality of the rrna preparations. To further rule out the possibility of trace DNA contamination of the RNA, a general PCR-based detection was also conducted using total RNA as template with primers Ef16S-f plus Ef16S-r (Table 2). The concentration and purity of RNA were determined using a NanoDrop 2000C spectrophotometer (Thermo Scientific). RNA preparations were stored at -80 C until use. Synthesis of cdna was performed in 20 µl of a mixture using the Omniscript Reverse Transcription Kit (Qiagen) according to the manufacturer s protocol. The resulting cdna (0.5 µl) served as template for PCR amplification of the fatty acid synthesis genes (fab genes) using specific primers (Table 2) on an Eppendorf thermal cycler. Real-time quantitative RT-PCR To test if the mrna levels of fabk were altered in the spontaneous mutants, real-time quantitative PCR (qpcr) assays were conducted by the SYBR Green dye method as previously reported (40). The qpcr reaction system (20 µl) contained the following components: 10 µl of iq SYBR Green Supermix, 1 µl each of the primers, 1 µl of 50-fold diluted cdna sample, and 8 µl of sterile water. Data analyses were done in triplicate on a Mastercycler ep realplex (Eppendorf), instrument using a program of denaturing cycle at 95 C for 2 min, 45 cycles comprising 94 C for 20 s, 53 C for 20 s, and 72 C for 20s and a final step in which temperature was elevated in gradient from 60 C to 90 C for dissociating double stranded DNA products. The mrna levels relative to those of the wild type were calculated using the method of Livak and
11 Schmittgen (41) with the 16S rrna as an internal control gene and water functioned as blank control to monitor cross-contamination of the cdna samples. Site-Directed Mutagenesis of fabk Plasmids pbhk368, pbhk369, pbhk370, pbhk371, pbhk372 and pbhk373 each carrying a single mutation within the RBS sequence were obtained using the QuickChange site-directed mutagenesis kit (Stratagene) with pzl157 as the PCR template following the manufacturer s instructions (Stratagene). The primers used in PCR and in mutagenesis are listed in Table 2. The constructed plasmids were transformed into E. coli DH5 by CaCl 2 treatment. Plasmids pbhk334, pbhk335, pbhk336, pbhk337, pbhk338 and pbhk339 each carrying a single mutation within the RBS sequence were obtained using the QuickChange site-directed mutagenesis kit (Stratagene) with pbhk323 as PCR template following the manufacturer s instructions (Stratagene). All constructs were verified by DNA sequencing. Random mutagenesis of fabk and isolation of mutant plasmids for complementation Random mutagenesis of the fabk gene plus 275 bp upstream was performed with the GeneMorph II random mutagenesis kit using primers fabk-random-f and fabk-random-r (Table 2) and plasmid pzl157 as template at a concentration of 500 ng in a total reaction volume of 50 µl. The mutagenesis condition designed to give low mutation frequency was used because we expected single mutations would restore growth of E. faecalis fabi mutant in the absence of oleate. The PCR products of the mutagenesis reactions were then digested with BamHI and PstI and inserted into vector ptrkl2 digested with the same enzymes. The ligation products were then transformed into E. coli DH5 with selection on BHI plates for resistance to erythromycin. The transformants of about 15,000 clones were collected and the mixture of mutant plasmids was purified using the Qiagen Spin Miniprep kit. The plasmids were electroporated to the E. faecalis fabi mutant strain FAZL1 as described above. The 10,000 clones obtained were screened for growth on GM17 medium without fatty acid supplementation. The plasmids of the growthcomplemented strains were then isolated and again transformed to the strain FAZL1 to confirm
12 the growth complementation. Finally the confirmed complementing plasmids were sequenced to identify the mutations.
13 Results Spontaneously arising mutants that restore fatty acid synthesis to the fabi strain contain point mutations within the fabk ribosome binding site region The two E. faecalis proteins catalyzing the ENR reaction (Fig. 1A) are located in different genome neighborhoods. The fabk gene is found within a putative operon responsible for saturated fatty acid synthesis whereas fabi is located in a smaller cluster with two genes involved in unsaturated fatty acid synthesis (19) (Fig. 1B). The fabi strain FAZL1 is severely defective in fatty acid synthesis and is unable to grow on GM17 medium unless the medium is supplemented with an unsaturated fatty acid such as oleic acid (18). A culture of strain FAZL1 was plated on GM17 medium lacking oleate supplementation at 37 C. Colonies appeared at a frequency of approximately 10-9 and purified isolates were shown to retain the fabi mutation. Four isolates, strains BHK260, BHK261, BHK262 and BHK263 were chosen for further study. Since overproduction of FabK alleviates the growth defect of a fabi null mutant strain (18), the most likely explanation for the spontaneous mutations was increased FabK expression. We first asked how fabk is transcribed. The cluster of fatty acid synthesis genes in which fabk is located appears to be an operon although there is an unusually large intergenic region (136 bp) between the acpp and fabk genes. To test if acpp and fabk are cotranscribed together with the upstream genes we performed RT-PCR analyses using primers bracketing each intergenic region (Fig. 1C). The same patterns were seen in both RT-PCR and PCR analyses indicating that at least these four genes are cotranscribed. ACP is generally produced at much higher levels than the Fab proteins because it acts as a coenzyme rather than an enzyme. Hence, acpp would be expected to be transcribed at a higher level than the other genes which we did not observe. However, E. faecalis has a second acpp gene (Fig. 1) which could be expressed at high levels. We are currently studying the relative contributions of the two acpp genes. We also asked if the acpp-fabk intergenic region contained a promoter by using the intergenic sequence to drive expression of a fabk-lacz translational fusion carried on a plasmid (Fig. 1D). The intergenic sequence gave no increase in ß-galactosidase activity over that seen with the reporter construct lacking a promoter whereas a construct driven by the Lactococcus lactis P32 promoter gave high levels of ß-galactosidase activity.
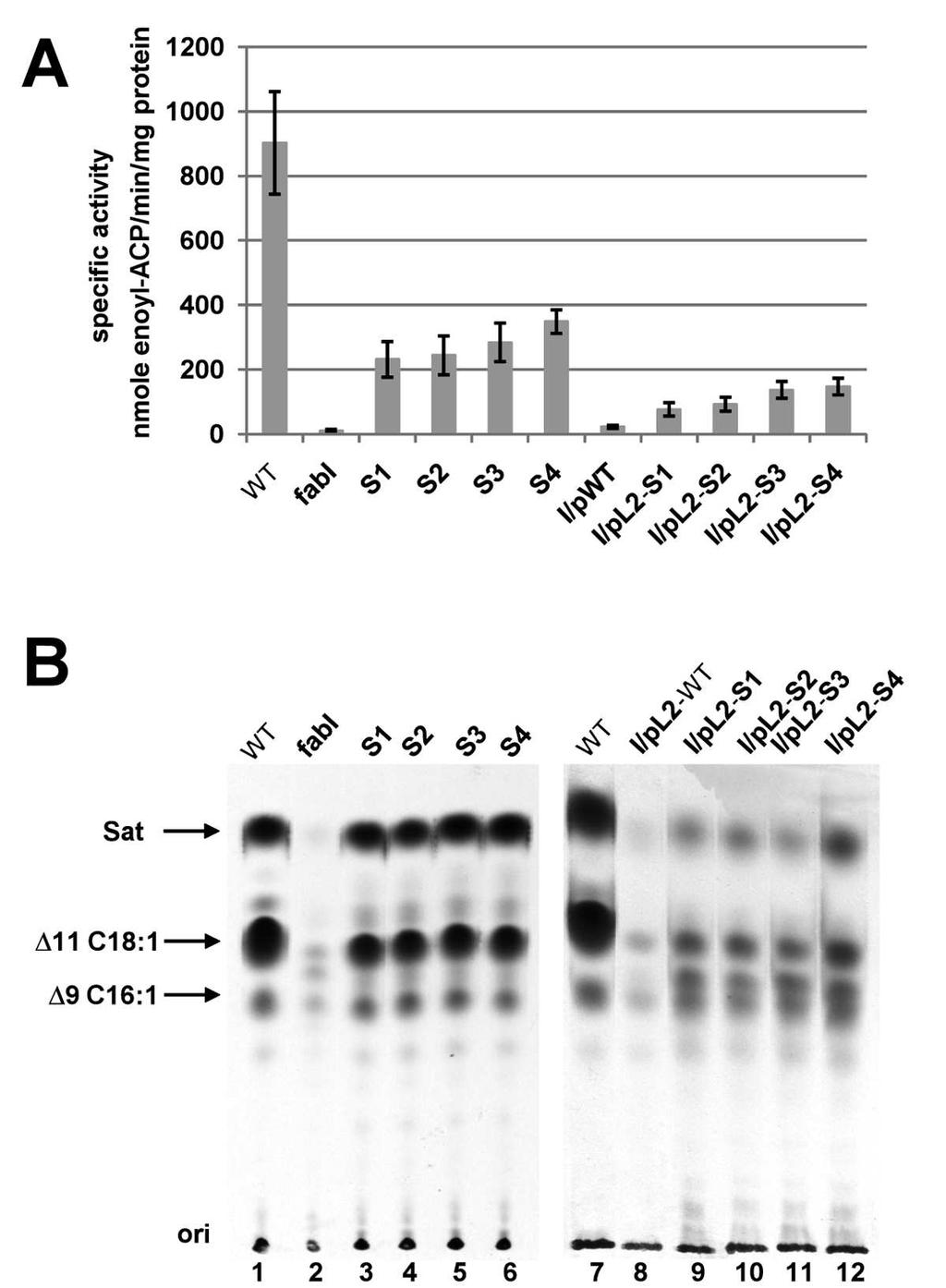
14 Given these background experiments we asked if growth of these four spontaneous mutants (Fig. 2A) was mediated by FabK. This was first tested by insertion of the fabk gene (plus about 300 bp of upstream and downstream sequence) from each of the spontaneous mutants into the low-copy-number shuttle vector ptrkl2. All four plasmids allowed the fabi mutant strain to grow on GM17 medium whereas the plasmid carrying the wild-type genomic segment failed to support growth (Fig. 2B). We then sequenced the fabk regions of each of the spontaneous mutants. All four mutant strains were found to carry a unique point mutation within the fabk ribosome binding region, each of which would extend base pairing with the 3 end of the 16S rrna (Fig. 2C). The mutations were C-12G (strain BHK260), a C12 single base deletion (strain BHK261), A-9G (strain BHK262) and T-19G (strain BHK263) (Fig. 2C). These data raised the question of whether or not our wild type strain FA2-2 is representative of E. faecalis. It seemed possible that our strain had acquired mutations in this region that resulted in decreased fabk translation and these were overcome or reversed by the spontaneous mutations. However, the sequence of the strain FA2-2 genomic segment located between the end of the acpp coding sequence and the beginning of the fabk sequence is either completely or almost completely conserved in the 295 complete and draft E. faecalis genome sequences currently available. Those few sequences that do not completely match the strain FA2-2 sequence have only a single base change located 63 bp upstream of the fabk initiation codon and hence well removed from the ribosome binding region. Moreover, the strain FA2-2 fabk coding sequence is at least 99% conserved at both the nucleotide and protein levels in the 295 genome sequences. Inactivation of fabk eliminates growth of the mutant strains. Although it seemed likely that growth of the spontaneously arising mutants of the fabi strain was due to increased fabk expression per se, it was possible that the effects of the mutations were indirect. If the entire cluster of genes in which fabk is located is indeed an operon, then binding of ribosomes to the fabk sequence of the polycistronic mrna might protect the mrna resulting in increased expression of the downstream genes. We tested this possibility by nonpolar disruption of the fabk coding sequence. If increased FabK ENR activity was responsible for growth, then fabk disruption should block growth in the absence of oleate whereas if increased downstream expression was the important factor, growth should proceed. A suicide plasmid carrying an internal fabk fragment was inserted into fabk by single crossover
15 homologous recombination events resulting in two disrupted copies of fabk bracketing the inserted plasmid (Fig. 3A and B). Upon insertion of the disruption plasmid all four spontaneous mutants lost the ability to grow without oleate (Fig. 3C) whereas the wild type strain grew well (due to the presence of FabI) thereby demonstrating the lack of a polar effect on expression of the downstream fatty acid synthetic genes. Strains carrying a chromosomal or plasmid borne mutant allele contain increased ENR activity Our previous results showed that E. faecalis FabI catalyzes an essential step in fatty acid biosynthesis and deletion of fabi results in almost complete loss of ENR activity (18). To directly test if growth of the spontaneous mutant strains was due to restoration of ENR activity, the ENR activities in cells from cultures grown on GM17 medium were assayed. The four spontaneous mutant strains had ENR activities that were 26-38% that of the FabI containing wild-type strain FA2-2 whereas the ENR activities of the derivatives of the fabi strain carrying the plasmid borne mutant segments were restored to 9%-16% that of wild-type strain (Fig. 4A). We also used [1-14 C]acetate labeling and argentation thin layer chromatographic analysis of the de novo synthesized fatty acids to demonstrate ENR activity in vivo (Fig. 4B) and found that the spontaneously arising mutants synthesized levels of saturated and unsaturated fatty acids comparable to that of the wild-type strain. However, consistent with their lower ENR activities and poorer growth, the strains carrying the plasmid borne mutant segments incorporated less 14 C- acetate into fatty acids than the wild-type strain (Fig. 4B) and no increase in ENR activity or 14 C- acetate incorporation was seen when the plasmid carried the wild-type genome segment. Hence the increased ENR activity due to mutations within the fabk RBS sequence was sufficient to restore growth and fatty acid synthesis to the fabi strain. The spontaneous mutations increase fabk translation Although the location of the nucleotide changes in the mutant strains argued in favor of increased translation rather than increased transcription, we measured the mrna levels of fabk in the mutant and wild-type strains. No significant change in mrna levels was observed (Fig. 5A) and thus the primary effect of the mutations was indeed at the translational level. We constructed fabk-lacz translational fusions that contained the acpp-fabk intergenic 136 bp plus
16 the first 35 bp of the fabk coding sequence under control of Lactococcus lactis promoter P32 and measured fabk translation by -galactosidase activity. The -galactosidase levels in cultures expressing fusions with the spontaneous fabk mutant sequences were 4-7-fold higher than that of the fusion plasmid carrying the wild-type genomic segment (Fig. 5B) whereas the lacz mrna levels were essentially the same for the wild type and mutant constructs (Fig. 5A). Hence, the spontaneous mutations overcome the normally weak translational initiation of fabk. Note that the 136 bp intergenic DNA fragment chosen for construction of the translational fusions was based on two considerations. The observed three intergenic amplicons from RT-PCR analysis (Fig. 1C) showed that at least the first four genes of the fab cluster comprise a transcription unit and hence constitute an operon. We also performed RLM-RACE, an improved version of 5 - RACE for amplification of the 5' end of the cdna and failed to amplify a PCR product (data not shown). Therefore, as in Fig. 1D, we were unable to detect a promoter between acpp and fabk. Effect of site-directed and random mutagenesis of the RBS sequence on fabk expression and cell growth The increased complementarity of the fabk RBS to 3' end of the 16S ribosomal RNA (Fig. 2C) seemed very likely to be responsible for the increased ENR activity of the spontaneous mutants. If so, then substitution of the mutant base with another base should result in loss of growth of the fabi null strain in the absence of oleic acid. Therefore, the mutant base was replaced with other nucleotides and as expected this resulted in loss of the abilities of plasmids carrying these constructs to complement growth of the fabi strain (Fig. 6A) and to increase fabk translation (Fig. 6B). We also randomly mutagenized the wild type fabk coding sequence plus the upstream 275 bp by error-prone PCR and isolated plasmids that suppressed the fabi phenotype. A total of 13 colonies were isolated and their plasmids sequenced. Two isolates had the A-9G mutation found in the spontaneous mutant BHK262. Another 11 isolates had the A-9G or C-12G mutations found in the spontaneously isolated strains and also contained phenotypically silent coding sequence mutations that were generally conservative residue substitutions (Table 3). The re-isolation of the mutations found in the spontaneous arising strains confirmed their abilities to increase the efficiency of mrna-ribosome pairing.
17 Discussion Selection for growth of the E. faecalis fabi strain in the absence of fatty acid supplementation gave four spontaneous mutant strains. The mutations in these strains were four different single base changes (three single-base substitutions and one single-base deletion) in the ribosome binding region of fabk which encodes a fully functional, but physiologically cryptic ENR. The discrete nature of our mutations is strikingly different from previously reported spontaneous translation activating mutants in which large DNA fragments carrying RBS sequences were inserted or deleted (20-22). Translation begins with the association between a 30S ribosomal subunit and the mrna. Translational efficiency is in part determined by the RBS interaction, i.e. the base pairing of the 3 end of 16S rrna to a stretch of complementary nucleotides in the messenger, located upstream of the initiation codon (23, 24). Indeed, as a result of the C -12 single base change of mutant strain BHK260, the substitution with G extended the pairing potential with the 3 end of the 16S rrna by 3 bp (Fig. 2C) and increased fabk expression (Fig. 5B). Moreover, site directed mutagenesis in which A or T was substituted for C -12 resulted in decreased fabk translation (Fig. 6B). The other three single mutations appeared to have similar effects on base pairing and translation. Moreover, mutagenesis by error prone PCR produced two of the same mutations isolated spontaneously (Table 3). Previous studies suggest that Gram-positive ribosomes require a stronger interaction between 16S rrna and mrna than Gram-negative ribosomes in order to initiate translation (21, 25). Our findings are also in agreement with a proposed scheme for the movement of mrna at different stages of translation, in which a lengthening of the RBS duplex takes place in order to contact ribosomal protein S2 following translation initiation (26). The increased extent of complementarity between the RBS sequence and the 16S rrna 3 end (Fig. 2C) resulting from the mutations aids mrna-16s rrna interaction and perhaps also aids the lengthening process. The broad question is why all E. faecalis genomes encode a fully functional FabK when the enzyme activity is both redundant and is not expressed at physiologically useful levels (at least under laboratory conditions). There seems no selection for retention of this silent gene because FabI can fully support E. faecalis fatty acid synthesis (18). Given this situation the fabk sequence would be expected to first degenerate to encode inactive proteins and finally be lost (as
18 may have been the case in the related bacterium, E. faecium). Instead, the fabk sequence has been retained intact and still encodes an active ENR. The only straightforward explanation for this puzzling situation is that FabK expression must be activated by physiological conditions that are not duplicated in the laboratory. However, our data argue that the RBS-16S rrna interaction is hardwired and, if that is the case, how can FabK expression be activated? Increased transcription from a cryptic promoter is a possible means, although since fabk is in an operon, increased expression of downstream genes would be expected and in other bacteria overproduction of some of the encoded proteins is toxic. A common means of translational control is sequestration of the RBS by base pairing to an mrna sequence located upstream of the RBS. The sequestration is released upon synthesis of a small RNA that base pairs with the inhibitory sequence and thereby releases the RBS for rrna interaction and translational initiation. Another possibility is activation of a translational enhancer sequence such as those studied in E. coli (26-29). E. coli enhancers are pyrimidine rich sequences located upstream of the RBS which interact with ribosomal protein S1 and their effects can be augmented by tracts of adenosine bases between the RBS and enhancer (28). To our knowledge no translational enhancer sequences have been reported in Firmicutes bacteria but this is not surprising considering the dearth of data on translational initiation in these bacteria. However, even in the E. coli system the details of enhancer function remain unclear because S1 protein is not present in the extant mrna/ribosome crystal structures. If an E. faecalis translational enhancer is present upstream of fabk, then it could be sequestered by base pairing with a complementary sequence in the mrna (RNA folding programs predict a high degree of base pairing within the acpp-fabk intergenic region transcript). The putative sequestration by base pairing could be disrupted by a small RNA (or perhaps a protein) that binds the complement of the enhancer sequence and thereby free the enhancer to bind ribosomal protein S1. However, it should be noted that our mutational data argue against the above scenarios because no mutations, either spontaneous or those obtained by error prone PCR, mapped in the region upstream of the RBS. The PCR mutagenesis produced 15,000 clones and those that survived the selection for both increased fabk translation and preservation of FabK enzymatic activity, all contained two of the RBS-extending mutations we had isolated spontaneously. Moreover, the fact that several of the error-prone PCR mutants have multiple base changes within the coding sequence despite the fact that FabK function was required for their isolation
19 indicates that the mutagenesis was robust.
20 ACKNOWLEDGMENTS This work was supported by National Institutes of Health (NIH) Grant AI15650 from National Institute of Allergy and Infectious Diseases (to J.E.C.) and a grant from National Natural Science Foundation of China ( and ) and the Specialized Research Fund for the Doctoral Program of Higher Education of China ( ) (to H.W.). We thank Dr. Carin Vanderpool for valuable comments on the manuscript.
21 References 1. Lu YJ, Zhang YM, Rock CO Product diversity and regulation of type II fatty acid synthases. Biochem Cell Biol 82: Cronan JE, Thomas J Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol 459: Cronan JE, Jr The structure of mammalian fatty acid synthase turned back to front. Chem Biol 11: Cronan JE Remarkable structural variation within fatty acid megasynthases. Nat Chem Biol 2: Schweizer E, Hofmann J Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev 68: , table of contents. 6. Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Hogenauer G, Turnowsky F Protein EnvM is the NADH-dependent enoyl- ACP reductase (FabI) of Escherichia coli. J Biol Chem 269: Turnowsky F, Fuchs K, Jeschek C, Hogenauer G envm genes of Salmonella typhimurium and Escherichia coli. J Bacteriol 171: Heath RJ, Su N, Murphy CK, Rock CO The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J Biol Chem 275: Massengo-Tiasse RP, Cronan JE Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J Biol Chem 283: Zhu L, Lin J, Ma J, Cronan JE, Wang H Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob Agents Chemother 54: Heath RJ, Rock CO A triclosan-resistant bacterial enzyme. Nature 406: Marrakchi H, Dewolf WE, Jr., Quinn C, West J, Polizzi BJ, So CY, Holmes DJ, Reed SL, Heath RJ, Payne DJ, Rock CO, Wallis NG Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem J 370: Massengo-Tiasse RP, Cronan JE Diversity in enoyl-acyl carrier protein reductases. Cell Mol Life Sci 66: Heath RJ, White SW, Rock CO Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res 40: Kingry LC, Cummings JE, Brookman KW, Bommineni GR, Tonge PJ, Slayden RA The Francisella tularensis FabI enoyl-acp-reductase gene is essential to bacterial viability and is expressed during infection. Journal of Bacteriology. 16. Jett BD, Huycke MM, Gilmore MS Virulence of enterococci. Clin Microbiol Rev 7: Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:
22 18. Zhu L, Bi H, Ma J, Hu Z, Zhang W, Cronan J, Wang H The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. mbio in press. 19. Wang H, Cronan JE Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J Biol Chem 279: Anderson C, Potter AA, Gerlach GF Isolation and molecular characterization of spontaneously occurring cytolysin-negative mutants of Actinobacillus pleuropneumoniae serotype 7. Infect Immun 59: Lin CK, Goldfarb DS, Doi RH, Rodriguez RL Mutations that affect the translation efficiency of Tn9-derived cat gene in Bacillus subtilis. Proc Natl Acad Sci U S A 82: Moniruzzaman M, Lai X, York SW, Ingram LO Isolation and molecular characterization of high-performance cellobiose-fermenting spontaneous mutants of ethanologenic Escherichia coli KO11 containing the Klebsiella oxytoca casab operon. Appl Environ Microbiol 63: Hui A, de Boer HA Specialized ribosome system: preferential translation of a single mrna species by a subpopulation of mutated ribosomes in Escherichia coli. Proc Natl Acad Sci U S A 84: Shine J, Dalgarno L The 3'-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A 71: Stallcup MR, Sharrock WJ, Rabinowitz JC Specificity of bacterial ribosomes and messenger ribonucleic acids in protein synthesis reactions in vitro. J Biol Chem 251: Yusupova G, Jenner L, Rees B, Moras D, Yusupov M Structural basis for messenger RNA movement on the ribosome. Nature 444: Komarova AV, Tchufistova LS, Dreyfus M, Boni IV AU-rich sequences within 5' untranslated leaders enhance translation and stabilize mrna in Escherichia coli. J Bacteriol 187: Takahashi S, Furusawa H, Ueda T, Okahata Y Translation enhancer improves the ribosome liberation from translation initiation. J Am Chem Soc 135: Vimberg V, Tats A, Remm M, Tenson T Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol 8: Clewell DB, Tomich PK, Gawron-Burke MC, Franke AE, Yagi Y, An FY Mapping of Streptococcus faecalis plasmids pad1 and pad2 and studies relating to transposition of Tn917. J Bacteriol 152: Shepard BD, Gilmore MS Electroporation and efficient transformation of Enterococcus faecalis grown in high concentrations of glycine. Methods Mol Biol 47: Casadaban MJ, Martinez-Arias A, Shapira SK, Chou J Beta-galactosidase gene fusions for analyzing gene expression in Escherichia coli and yeast. Methods Enzymol 100: Blancato VS, Magni C A chimeric vector for efficient chromosomal modification in Enterococcus faecalis and other lactic acid bacteria. Lett Appl Microbiol 50:
23 34. Bi H, Christensen QH, Feng Y, Wang H, Cronan JE The Burkholderia cenocepacia BDSF quorum sensing fatty acid is synthesized by a bifunctional crotonase homologue having both dehydratase and thioesterase activities. Mol Microbiol 83: Jiang Y, Chan CH, Cronan JE The soluble acyl-acyl carrier protein synthetase of Vibrio harveyi B392 is a member of the medium chain acyl-coa synthetase family. Biochemistry 45: Bradford MM A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: Bligh EG, Dyer WJ A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: Feng Y, Cronan JE Escherichia coli unsaturated fatty acid synthesis: complex transcription of the faba gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem 284: Miller JH Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 40. Feng Y, Cronan JE A new member of the Escherichia coli fad regulon: transcriptional regulation of fadm (ybaw). J Bacteriol 191: Livak KJ, Schmittgen TD Analysis of relative gene expression data using realtime quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25: O'Sullivan DJ, Klaenhammer TR High- and low-copy-number Lactococcus shuttle cloning vectors with features for clone screening. Gene 137: Jerga A, Rock CO Acyl-Acyl carrier protein regulates transcription of fatty acid biosynthetic genes via the FabT repressor in Streptococcus pneumoniae. J Biol Chem 284: Lu YJ, Rock CO Transcriptional regulation of fatty acid biosynthesis in Streptococcus pneumoniae. Mol Microbiol 59:
24 Table 1. Bacterial strains and plasmids used in this study Strains Genotype Reference or E. coli source DH5 EC1000 (argf lac)169 80dlacZ58(M15) phoa8 glnv44 deor481 gyra96, reca1 enda1, hsdr17 Em R ; MC1000 derivative carrying a single copy of the pwv01repagene in glgb Lab stock Lab stock E. faecalis FA2-2 Rifampicin/Fususidic acid resistant mutant derived from plasmid-free strain JH2 (30) FAZL1 fabi of E. faecalis FA2-2 (18) BHK260 spontaneous RBS mutant S1 of FAZL1, C -12 to G This work BHK261 spontaneous RBS mutant S2 of FAZL1, C -12 deletion This work BHK262 spontaneous RBS mutant S3 of FAZL1, A -9 to G This work BHK263 spontaneous RBS mutant S4 of FAZL1, T -19 to G This work FAZL6 FA2-2 fabk (disruption) This work FAZL7 FAZL1 fabk (disruption) This work FAZL8 S1 fabk (disruption) This work FAZL9 S2 fabk (disruption) This work FAZL10 S3 fabk (disruption) This work FAZL11 S4 fabk (disruption) This work
25 Plasmids ptrkl2 Erm r, low-copy-number E. coli/lactococcus shuttle vector (42) pcr2.1 Amp r, Kan r, TOPO TA cloning vector Invitrogen pzl52 pzl53 pzl54 pzl55 pzl56 pzl157 pzl158 pzl159 Amp r, Kan r, pcr2.1 carrying E. faecalis FAZL1 fabk plus 275 bp upstream and 288 bp downstream Amp r, Kan r, pcr2.1 carrying E. faecalis mutant S1 (strain BHK260) fabk plus 275 bp upstream and 288 bp downstream Amp r, Kan r, pcr2.1 carrying E. faecalis mutant S2 (strain BHK261) fabk plus 275 bp upstream and 288 bp downstream Amp r, Kan r, pcr2.1 carrying E. faecalis mutant S3 (strain BHK262) fabk plus 275 bp upstream and 288 bp downstream Amp r, Kan r, pcr2.1 carrying E. faecalis mutant S4 (strain BHK263) fabk plus 275 bp upstream and 288 bp downstream Erm r, EcoRV and BamHI digested fabk fragment of pzl52 inserted into the same sites of ptrkl2 Erm r, EcoRV and BamHI digested fabk fragment of pzl53 inserted into the same sites of ptrkl2, C -12 to G Erm r, EcoRV and BamHI digested fab fragment of pzl54 inserted into the same sites of ptrkl2, C -12 This work This work This work This work This work This work This work This work
26 pzl160 pzl161 deletion Erm r, EcoRV and BamHI digested fabk fragment of pzl55 inserted into the same sites of ptrkl2, A -9 to G Erm r, EcoRV and BamHI digested fabk fragment of pzl56 inserted into the same sites of ptrkl2, T -19 to G This work This work pbhk368 Erm r, fabk A -9 T of pzl157 This work pbhk369 Erm r, fabk A -9 C of pzl157 This work pbhk370 Erm r, fabk C -12 A of pzl157 This work pbhk371 Erm r, fabk C -12 T of pzl157 This work pbhk372 Erm r, fabk T -19 A of pzl157 This work pbhk373 Erm r, fabk T -19 C of pzl157 This work pmc1871 Tet r, a promoter-less lacz reporter plasmid (32) pbhk322 Erm r, lacz from pmc1871 cloned into the BamHI site of ptrkl2, p(lacz) This work pbhk323 Erm r, ptrkl2 P32::fabK (-136 to +35)::lacZ, p(wt) This work pbhk394 Erm r, ptrkl2 fabk (-275 to +35)::lacZ This work pbhk324 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, C -12 to G, p(s1) pbhk325 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, C -12 deletion, p(s2) pbhk326 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, A -9 to G, p(s3) This work This work This work
27 pbhk327 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, T -19 to G, p(s4) This work pbhk334 Erm r, ptrkl2 P32::fabK (-136 to +35)::lacZ, A -9 to T This work pbhk335 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, A -9 to C This work pbhk336 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ,C -12 to A This work pbhk337 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, C -12 to T This work pbhk338 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, T -19 to A This work pbhk339 Erm r, ptrkl2 P32:: fabk (-136 to +35)::lacZ, T -19 to C This work
28 Table 2. Oligonucleotide primers used in this study Oligonucleotides fabkup275 fabkdo288 P32-f fabkfusion-r P32FabK-f P32FabK-r fabkup275-psti fabk-random-f fabk-random-r Ef16S-f Ef16S-r lacz-f lacz-r fabk-rt-f fabk-rt-r c-12a-f c-12a-r Sequence ATTAGACGCGGACAGCTTAG TACGACATCAGGCGTTAGTC ACTCGCCTGCAGAGATTAATAG TTTTAGCTA ATACTTGTCGACTTGATTCCAAGCAACTCACA CTAGGTAGGTAAAAAAATATTACATGAAAATAGGGGGCT AGCCCCCTATTTTCATGTAATATTTTTTTACCTACCTAG AATATCTGCAGATTAGACGCGGACAGCTTAG TAGTGGATCCATTAGACGCGGACAGCTTAGA ATATCTGCAGTTCACTTAGCCCCAACGCTGAT CATCATGCCCCTTATGACCTGG CCTGCAATCCGAACTGAGAGA ATGAACGGTCTGGTCTTTGC ATCGCTATGACGGAACAGGT GGCAATCCAGCCAAATACAT ATGTCCACCAGCTTCCATTC GAAGTGTACTTATCTTAGAACTAAAGGAAGTATAAAATCAATGA ATCAACAGTTATG CATAACTGTTGATTCATTGATTTTATACTTCCTTTAGTTCTAAGAT
29 AAGTACACTTC c-12t-f c-12t-r a-9c-f a-9c-r a-9t-f a-9t-r t-19a-f t-19a-r t-19c-f t-19c-f fabk inside For fabkinside Rev pbvgh For (P1) GAAGTGTACTTATCTTAGAACTAAAGGATGTATAAAATCAATGAA TCAACAGTTATG CATAACTGTTGATTCATTGATTTTATACATCCTTTAGTTCTAAGAT AAGTACACTTC GTACTTATCTTAGAACTAAAGGACGTCTAAAATCAATGAATCAAC AGTTATGT ACATAACTGTTGATTCATTGATTTTAGACGTCCTTTAGTTCTAAGA TAAGTAC GTACTTATCTTAGAACTAAAGGACGTTTAAAATCAATGAATCAAC AGTTATGT ACATAACTGTTGATTCATTGATTTTAAACGTCCTTTAGTTCTAAGA TAAGTAC CTTGAATGAAGTGTACTTATCTTAGAACAAAAGGACGTATAAAAT CAATGAATC GATTCATTGATTTTATACGTCCTTTTGTTCTAAGATAAGTACACTT CATTCAAG CTTGAATGAAGTGTACTTATCTTAGAACCAAAGGACGTATAAAAT CAATGAATC GATTCATTGATTTTATACGTCCTTTGGTTCTAAGATAAGTACACTT CATTCAAG ATGCGAGCTCTGAAGAACAGGTTCCTGTCG ATCGCTGCAGCTGCATCTAAATCACGTGCC GTACCTACGTAGGATCGATC
30 pbvgh Rev (P2) fabk check UP (P3) fabk check DOWN (P4) GCTTCCAAGGAGCTAAAGAG ATTAGACGCGGACAGCTTAG TACGACATCAGGCGTTAGTC The underlined italic sequences are the introduced restriction sites.
31 Table 3. Results of error prone PCR mutagenesis of fabk and selection for complementation of the fabi strain FAZL1. Isolate Ran2 Ran9 Ran16 Ran26 Ran12 Ran13 Ran15 Ran17 FabK mutations A-9G A-9G, L9 (CUU to CUC) A-9G, D78G, L154I, I163V A-9G, I285V C-12G, A185V, I220V, I261V C-12G, V84I, K124R, L148 (UUA to CUA) C-12G, M281L C-12G, K250 Stop (AAA to UAA) Note that in Ran2 and Ran13, two synonymous mutations occurred for residues L9 and L148, respectively. In Ran17, a nonsense mutation truncates the protein at residue K250 suggesting that the last 69 FabK residues are not essential for ENR activity.
32 Figure legends. Figure 1. The ENR reaction, the fabk and fabi genome neighborhoods and transcriptional analyses of the operon containing fabk. A. The enoyl-acyl-acp reductase reaction. B. Genetic organization of the E. faecalis fatty acid biosynthetic genes (fab). The numbered short lines (1, 2, 3) represent the specific PCR amplicons observed in the PCR and RT-PCR assays of Panel C. The second acpp gene (EF_3111 in strain V583) located downstream of plsx is conserved in all extant E. faecalis genome sequences. C. PCR and RT-PCR analyses of the fab operon. The results of the four genes at the 5 end of the operon are shown. The helix-turn-helix (HTH) encoding gene probably encodes a homologue of the FabT transcription factor found in other Firmacute bacteria (43, 44). The primer numbering system is that of Panel B. Both the PCR products obtained from genomic DNA and those obtained by RT-PCR were separated by electrophoresis on 1.5% agarose gel. The ck designation denotes two neighboring genes transcribed from the opposite strand that were included as controls. D. -Galactosidase activities of fabk::lacz translational fusions. The wild-type strain FA2-2 carrying each of the fusion plasmids was grown in GM17 medium to mid-log phase and galactosidase activities were measured in more than three independent experiments. The error bars indicate standard deviations. The plasmids encoded a promoterless lacz ( lacz), the -275 to +35 fragment which includes the first 35 bp of the fabk coding sequence fused to lacz (fabklacz), or the -136 to +35 fragment driven by the P32 promoter. The fusion plasmids were pbhk322, pbhk394 and pbhk323, respectively. Figure 2. Growth and sequences of the spontaneously arising mutant strains and complementation of the fabi strain by plasmid borne fragments that include the fabk mutant or wild type alleles. A. Growth of the four spontaneously arising mutant fabi strains (S1-S4) on GM17 medium in the absence of oleate supplementation. B. Complementation of the fabi strain FAZL1 by plasmids (Table 1) carrying a mutant fabk allele plus about 300 bp of upstream and downstream sequence. Each growth assay was carried out in triplicate in GM17 medium at 37 C and the average is given. Strain FAZL1 in panel A
33 and the strain carrying wild-type fabk in panel B were used as negative controls. Panels A and Panel B have the same color code for the mutant fabk alleles. C. Alignments of 5' untranslated regions of the wild-type gene fabk or of the four spontaneously arising mutant strains with the 3 end of the E. faecalis 16S rrna. The fabk genes of wild type strain FAZL1 and the four spontaneous mutants plus about 300 bp upstream and downstream were sequenced. Each point mutation expanded the fabk ribosome binding site. The underlined letters indicate bases capable of Watson-Crick base pairing with the 16S rrna. +1 indicates the A of the initiation ATG codon. Figure 3. Construction, characterization and growth of fabk disruption mutant strains. A. Strategy for isolation of erythromycin marked chromosomal E. faecalis fabk disruption mutant strains via plasmid insertion. P1, P2, P3 and P4 were the PCR primers used for characterization of the fabk disruption mutant strains. B. Characterization of the fabk disruption strains by PCR analyses. Lanes 1-6, the PCR products amplified from lane 1, the fabk disruption strain (FAZL6); lane 2, the fabk disruption fabi double mutant strain (FAZL7); and lanes 3-6, the S1-S4 fabk strains in which fabk was disrupted (strains FAZL8-11, respectively). Genomic DNA preparations were used as templates with primer sets pbvgh For (P1) and fabkcheck DOWN (P4). Lanes 9-14 are the PCR products amplified using the same template DNAs as lanes 1-6 but using primers pbvgh Rev (P2) and fabk check UP (P3). Lanes 7 and 15 are the PCR products formed from the genomic DNA of the wild type strain FA2-2. Lanes 8 and 16 are molecular size standards. C. Growth of E. faecalis mutant strains on GM17 medium plus 5 µg/ml of erythromycin at 42 C with (left) or without (right) oleic acid. Cell growth proceeded at 42 C for 36 h. The K designation indicates the disrupted fabk; the WT designation indicates E. faecalis wild-type strain the I designation indicates the E. faecalis fabi gene deletion strain; S1, S2, S3 and S4 designation indicates the spontaneously arising suppressors the of E. faecalis fabi deletion. The normal growth in the absence of fatty acid supplementation of the disrupted fabk derivative of the wild-type strain indicated that the disruption cassette is not polar on the downstream fatty acid synthesis genes.
34 Figure 4. The spontaneous mutations increase expression of the FabK ENR. A. ENR activities of the wild-type FA2-2, spontaneous mutants and the complementing plasmid bearing strains. The 100 µl ENR reaction mixtures contained 0.1 M sodium phosphate buffer (ph 7.0), 0.15 mm NADH, and 2 µg of cell-free extract protein and NADH-dependent ENR activity was assayed. The reactions were initiated by addition of 100 µm trans-2-decenoyl-acp. The data are from three independent experiments and are expressed in means ± standard deviation. B. Incorporation of [1-14 C]acetate into the membrane phospholipids of the wild-type strain FA2-2, the spontaneous mutant strains and the fabi strain FAZL1 transformed with the wild-type fabk gene or the mutant alleles. The strains used here are the same with those of Fig. 2. The fatty acid methyl esters were prepared and analyzed by argentation thin layer chromatography as described in Materials and Methods. The designations are: Sat, saturated fatty acids; 9C16:1, palmitoleic (cis-9-hexadecenoic) acid; 11C18:1, cis-vaccenic (cis-11-octadecenoic) acid. Figure 5. Effects of the spontaneous mutations on fabk transcription and translation. A. qrt-pcr analyses of the effects on fabk and lacz transcription. Transcription of fabk was analyzed in the wild type strain and the spontaneous mutant strains whereas lacz transcription was analyzed from the wild type strains carrying the translational fusion plasmid. Cells were grown in GM17 medium to mid-log phase and RNA was isolated as described in Materials and Methods. The qrt-pcr data were from no less than four independent tests and are expressed in means ± standard deviation. The p(lacz) designation denotes the promoterless lacz vector B. Effects of the spontaneous mutations on -galactosidase expression from a plasmid carrying a P32::fabK::lacZ translational fusion. The wild-type strain FA2-2 carrying the fusion plasmids was grown in GM17 medium to mid-log phase and -galactosidase activities were measured from more than three independent experiments. The error bars indicate standard deviations. p(wt), p(s1), p(s2), p(s3), p(s4) and p(lacz) indicate fusion plasmids pbhk323, pbhk324, pbhk325, pbhk326, pbhk327 and pbhk322 carrying the (-136 to +35 fragments) wild-type fabk or one of the four spontaneous fabk mutants or the empty lacz vector, respectively.
35 Figure 6. 16S rrna-mrna complementarity is required for growth of the spontaneous mutants. A. Growth was tested on GM17 plates incubated for 36 h at 37 C. In three of the spontaneous mutants the unique nucleotide of the mutation was changed to two other nucleotides. Each sitedirected fabk gene was cloned into the vector ptrkl2 and then transformed to the fabi strain, FAZL1. B. Effects of the nucleotide changes on -galactosidase expression from a plasmid carrying a P32::fabK::lacZ translational fusion. The wild-type strain FA2-2 carrying the fusion plasmids was grown in GM17 medium to mid-log phase and -galactosidase activities were measured from more than three independent experiments. The error bars indicate standard deviations. The designations p(wt), A-9T, A-9C, C-12A, C-12T, T-19A, T-19C and p(s4) indicate the fusion plasmids pbhk323, pbhk334, pbhk335, pbhk336, pbhk337, pbhk338, pbhk339 and pbhk327, respectively. The p(wt) and p(s4) plasmids were used as negative and positive controls, respectively.
36
37
38
39
Design. Construction. Characterization
 Design Construction Characterization DNA mrna (messenger) A C C transcription translation C A C protein His A T G C T A C G Plasmids replicon copy number incompatibility selection marker origin of replication
Design Construction Characterization DNA mrna (messenger) A C C transcription translation C A C protein His A T G C T A C G Plasmids replicon copy number incompatibility selection marker origin of replication
Supplementary Information
 Supplementary Information Supplementary Figures Figure S1. Study of mgtl translation in vitro. (A) Detection of 5 LR RNA using wild-type and anti-sd (91-95) substituted templates in a transcription-translation
Supplementary Information Supplementary Figures Figure S1. Study of mgtl translation in vitro. (A) Detection of 5 LR RNA using wild-type and anti-sd (91-95) substituted templates in a transcription-translation
Figure 1. Map of cloning vector pgem T-Easy (bacterial plasmid DNA)
") Texas A&M University-Corpus Christi CHEM4402 Biochemistry II Laboratory Laboratory 6: Ligation & Bacterial Transformation (Bring your text and laptop to class if you wish to work on your assignment during
Texas A&M University-Corpus Christi CHEM4402 Biochemistry II Laboratory Laboratory 6: Ligation & Bacterial Transformation (Bring your text and laptop to class if you wish to work on your assignment during
Construction of a Mutant pbr322 Using Site-Directed Mutagenesis to Investigate the Exclusion Effects of pbr322 During Co-transformation with puc19
 Construction of a Mutant pbr322 Using Site-Directed Mutagenesis to Investigate the Exclusion Effects of pbr322 During Co-transformation with puc19 IVANA KOMLJENOVIC Department of Microbiology and Immunology,
Construction of a Mutant pbr322 Using Site-Directed Mutagenesis to Investigate the Exclusion Effects of pbr322 During Co-transformation with puc19 IVANA KOMLJENOVIC Department of Microbiology and Immunology,
XactEdit Cas9 Nuclease with NLS User Manual
 XactEdit Cas9 Nuclease with NLS User Manual An RNA-guided recombinant endonuclease for efficient targeted DNA cleavage Catalog Numbers CE1000-50K, CE1000-50, CE1000-250, CE1001-250, CE1001-1000 Table of
XactEdit Cas9 Nuclease with NLS User Manual An RNA-guided recombinant endonuclease for efficient targeted DNA cleavage Catalog Numbers CE1000-50K, CE1000-50, CE1000-250, CE1001-250, CE1001-1000 Table of
Generation of Non-typeable Haemophilus influenzae Directed Gene Deletion Mutants Jeroen D. Langereis *
 Generation of Non-typeable Haemophilus influenzae Directed Gene Deletion Mutants Jeroen D. Langereis * Laboratory of Pediatric Infectious Diseases, Department of Pediatrics and Laboratory of Medical Immunology,
Generation of Non-typeable Haemophilus influenzae Directed Gene Deletion Mutants Jeroen D. Langereis * Laboratory of Pediatric Infectious Diseases, Department of Pediatrics and Laboratory of Medical Immunology,
Designing and creating your gene knockout Background The rada gene was identified as a gene, that when mutated, caused cells to become hypersensitive
 Designing and creating your gene knockout Background The rada gene was identified as a gene, that when mutated, caused cells to become hypersensitive to ionizing radiation. However, why these mutants are
Designing and creating your gene knockout Background The rada gene was identified as a gene, that when mutated, caused cells to become hypersensitive to ionizing radiation. However, why these mutants are
Hetero-Stagger PCR Cloning Kit
 Product Name: Code No: Size: DynaExpress Hetero-Stagger PCR Cloning Kit DS150 20 reactions Kit Components: Box 1 (-20 ) phst-1 Vector, linearized Annealing Buffer Ligase Mixture phst Forward Sequence Primer
Product Name: Code No: Size: DynaExpress Hetero-Stagger PCR Cloning Kit DS150 20 reactions Kit Components: Box 1 (-20 ) phst-1 Vector, linearized Annealing Buffer Ligase Mixture phst Forward Sequence Primer
Biol/Chem 475 Spring 2007
 Biol/Chem 475 Spring 2007 Goal of lab: For most of the quarter, we will be exploring a gene family that was first discovered in fruitlfies and then found to be present in humans and worms and fish and
Biol/Chem 475 Spring 2007 Goal of lab: For most of the quarter, we will be exploring a gene family that was first discovered in fruitlfies and then found to be present in humans and worms and fish and
High Pure RNA Isolation Kit for isolation of total RNA from 50 samples Cat. No
 for isolation of total RNA from 50 samples Cat. No. 1 88 665 Principle A single reagent lyses the sample lysis and inactivates RNase. In the presence of a chaotropic salt (guanidine HCl), the released
for isolation of total RNA from 50 samples Cat. No. 1 88 665 Principle A single reagent lyses the sample lysis and inactivates RNase. In the presence of a chaotropic salt (guanidine HCl), the released
Cat # Box1 Box2. DH5a Competent E. coli cells CCK-20 (20 rxns) 40 µl 40 µl 50 µl x 20 tubes. Choo-Choo Cloning TM Enzyme Mix
 40 µl 40 µl 50 µl x 20 tubes. Choo-Choo Cloning TM Enzyme Mix") Molecular Cloning Laboratories User Manual Version 3.3 Product name: Choo-Choo Cloning Kits Cat #: CCK-10, CCK-20, CCK-096, CCK-384 Description: Choo-Choo Cloning is a highly efficient directional PCR
Molecular Cloning Laboratories User Manual Version 3.3 Product name: Choo-Choo Cloning Kits Cat #: CCK-10, CCK-20, CCK-096, CCK-384 Description: Choo-Choo Cloning is a highly efficient directional PCR
Data Sheet Quick PCR Cloning Kit
 Data Sheet Quick PCR Cloning Kit 6044 Cornerstone Ct. West, Ste. E DESCRIPTION: The Quick PCR Cloning Kit is a simple and highly efficient method to insert any gene or DNA fragment into a vector, without
Data Sheet Quick PCR Cloning Kit 6044 Cornerstone Ct. West, Ste. E DESCRIPTION: The Quick PCR Cloning Kit is a simple and highly efficient method to insert any gene or DNA fragment into a vector, without
CHAPTER 20 DNA TECHNOLOGY AND GENOMICS. Section A: DNA Cloning
 Section A: DNA Cloning 1. DNA technology makes it possible to clone genes for basic research and commercial applications: an overview 2. Restriction enzymes are used to make recombinant DNA 3. Genes can
Section A: DNA Cloning 1. DNA technology makes it possible to clone genes for basic research and commercial applications: an overview 2. Restriction enzymes are used to make recombinant DNA 3. Genes can
PLNT2530 (2018) Unit 6b Sequence Libraries
 Unit 6b Sequence Libraries") PLNT2530 (2018) Unit 6b Sequence Libraries Molecular Biotechnology (Ch 4) Analysis of Genes and Genomes (Ch 5) Unless otherwise cited or referenced, all content of this presenataion is licensed under the
PLNT2530 (2018) Unit 6b Sequence Libraries Molecular Biotechnology (Ch 4) Analysis of Genes and Genomes (Ch 5) Unless otherwise cited or referenced, all content of this presenataion is licensed under the
GenBuilder TM Plus Cloning Kit User Manual
 GenBuilder TM Plus Cloning Kit User Manual Cat. No. L00744 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2
GenBuilder TM Plus Cloning Kit User Manual Cat. No. L00744 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2
Lac Operon contains three structural genes and is controlled by the lac repressor: (1) LacY protein transports lactose into the cell.
 LacY protein transports lactose into the cell.") Regulation of gene expression a. Expression of most genes can be turned off and on, usually by controlling the initiation of transcription. b. Lactose degradation in E. coli (Negative Control) Lac Operon
Regulation of gene expression a. Expression of most genes can be turned off and on, usually by controlling the initiation of transcription. b. Lactose degradation in E. coli (Negative Control) Lac Operon
pgm-t Cloning Kit For direct cloning of PCR products generated by Taq DNA polymerases For research use only Cat. # : GVT202 Size : 20 Reactions
 pgm-t Cloning Kit For direct cloning of PCR products generated by Taq DNA polymerases Cat. # : GVT202 Size : 20 Reactions Store at -20 For research use only 1 pgm-t Cloning Kit Cat. No.: GVT202 Kit Contents
pgm-t Cloning Kit For direct cloning of PCR products generated by Taq DNA polymerases Cat. # : GVT202 Size : 20 Reactions Store at -20 For research use only 1 pgm-t Cloning Kit Cat. No.: GVT202 Kit Contents
PRODUCT INFORMATION. Composition of SOC medium supplied :
 Product Name : Competent Cell BL21(DE3)pLysS Code No. : DS260 Size : 100 μl 10 Competency : > 5 10 7 cfu/μg (puc19) Supplied product : SOC medium, 1 ml 10 This product is for research use only Description
Product Name : Competent Cell BL21(DE3)pLysS Code No. : DS260 Size : 100 μl 10 Competency : > 5 10 7 cfu/μg (puc19) Supplied product : SOC medium, 1 ml 10 This product is for research use only Description
Orthogonal Ribosome Biofirewall
 1 2 3 4 5 Supporting Information Orthogonal Ribosome Biofirewall Bin Jia,, Hao Qi,, Bing-Zhi Li,, Shuo pan,, Duo Liu,, Hong Liu,, Yizhi Cai, and Ying-Jin Yuan*, 6 7 8 9 10 11 12 Key Laboratory of Systems
1 2 3 4 5 Supporting Information Orthogonal Ribosome Biofirewall Bin Jia,, Hao Qi,, Bing-Zhi Li,, Shuo pan,, Duo Liu,, Hong Liu,, Yizhi Cai, and Ying-Jin Yuan*, 6 7 8 9 10 11 12 Key Laboratory of Systems
Journal of Experimental Microbiology and Immunology (JEMI) Vol. 6:20-25 Copyright December 2004, M&I UBC
 Vol. 6:20-25 Copyright December 2004, M&I UBC") Preparing Plasmid Constructs to Investigate the Characteristics of Thiol Reductase and Flavin Reductase With Regard to Solubilizing Insoluble Proteinase Inhibitor 2 in Bacterial Protein Overexpression
Preparing Plasmid Constructs to Investigate the Characteristics of Thiol Reductase and Flavin Reductase With Regard to Solubilizing Insoluble Proteinase Inhibitor 2 in Bacterial Protein Overexpression
GenBuilder TM Plus Cloning Kit User Manual
 GenBuilder TM Plus Cloning Kit User Manual Cat.no L00744 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2 Ⅱ.
GenBuilder TM Plus Cloning Kit User Manual Cat.no L00744 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2 Ⅱ.
Molecular Genetics Techniques. BIT 220 Chapter 20
 Molecular Genetics Techniques BIT 220 Chapter 20 What is Cloning? Recombinant DNA technologies 1. Producing Recombinant DNA molecule Incorporate gene of interest into plasmid (cloning vector) 2. Recombinant
Molecular Genetics Techniques BIT 220 Chapter 20 What is Cloning? Recombinant DNA technologies 1. Producing Recombinant DNA molecule Incorporate gene of interest into plasmid (cloning vector) 2. Recombinant
Influence of tlp5, che1p, tlp4a, and che4stas Promoters on Chemotaxis in Azospirillum brasilense
 University of Tennessee, Knoxville Trace: Tennessee Research and Creative Exchange University of Tennessee Honors Thesis Projects University of Tennessee Honors Program 5-2015 Influence of tlp5, che1p,
University of Tennessee, Knoxville Trace: Tennessee Research and Creative Exchange University of Tennessee Honors Thesis Projects University of Tennessee Honors Program 5-2015 Influence of tlp5, che1p,
M I C R O B I O L O G Y WITH DISEASES BY TAXONOMY, THIRD EDITION
 M I C R O B I O L O G Y WITH DISEASES BY TAXONOMY, THIRD EDITION Chapter 7 Microbial Genetics Lecture prepared by Mindy Miller-Kittrell, University of Tennessee, Knoxville The Structure and Replication
M I C R O B I O L O G Y WITH DISEASES BY TAXONOMY, THIRD EDITION Chapter 7 Microbial Genetics Lecture prepared by Mindy Miller-Kittrell, University of Tennessee, Knoxville The Structure and Replication
Guide-it sgrna In Vitro Transcription and Screening Systems User Manual
 Clontech Laboratories, Inc. Guide-it sgrna In Vitro Transcription and Screening Systems User Manual Cat. Nos. 631438, 631439 & 631440 (042114) Clontech Laboratories, Inc. A Takara Bio Company 1290 Terra
Clontech Laboratories, Inc. Guide-it sgrna In Vitro Transcription and Screening Systems User Manual Cat. Nos. 631438, 631439 & 631440 (042114) Clontech Laboratories, Inc. A Takara Bio Company 1290 Terra
Non-Organic-Based Isolation of Mammalian microrna using Norgen s microrna Purification Kit
 Application Note 13 RNA Sample Preparation Non-Organic-Based Isolation of Mammalian microrna using Norgen s microrna Purification Kit B. Lam, PhD 1, P. Roberts, MSc 1 Y. Haj-Ahmad, M.Sc., Ph.D 1,2 1 Norgen
Application Note 13 RNA Sample Preparation Non-Organic-Based Isolation of Mammalian microrna using Norgen s microrna Purification Kit B. Lam, PhD 1, P. Roberts, MSc 1 Y. Haj-Ahmad, M.Sc., Ph.D 1,2 1 Norgen
The GeneEditor TM in vitro Mutagenesis System: Site- Directed Mutagenesis Using Altered Beta-Lactamase Specificity
 Promega Notes Magazine Number 62, 1997, p. 02 The GeneEditor TM in vitro Mutagenesis System: Site- Directed Mutagenesis Using Altered Beta-Lactamase Specificity By Christine Andrews and Scott Lesley Promega
Promega Notes Magazine Number 62, 1997, p. 02 The GeneEditor TM in vitro Mutagenesis System: Site- Directed Mutagenesis Using Altered Beta-Lactamase Specificity By Christine Andrews and Scott Lesley Promega
peco TM -T7-nGST, Eco cloning Kit User Manual (Patent pending)
") peco TM -T7-nGST, Eco cloning Kit User Manual (Patent pending) Cloning PCR products for E Coli expression of N-term GST-tagged protein Cat# Contents Amounts Application IC-1004 peco-t7-ngst vector built-in
peco TM -T7-nGST, Eco cloning Kit User Manual (Patent pending) Cloning PCR products for E Coli expression of N-term GST-tagged protein Cat# Contents Amounts Application IC-1004 peco-t7-ngst vector built-in
TD-P Revision 3.0 Creation Date: 6/9/2016 Revision Date: 8/16/2018
 Protocol TD-P Revision 3.0 Creation Date: 6/9/2016 Revision Date: 8/16/2018 Electrotransformation of Agrobacterium tumefaciens Modified from Methods in Molecular Biology Vol. 47 Introduction Agrobacterium
Protocol TD-P Revision 3.0 Creation Date: 6/9/2016 Revision Date: 8/16/2018 Electrotransformation of Agrobacterium tumefaciens Modified from Methods in Molecular Biology Vol. 47 Introduction Agrobacterium
Oligonucleotides containing either a single cyclo-da lesion or a single CPD in an
 Materials and Methods Preparation of Constructs Oligonucleotides containing either a single cyclo-da lesion or a single CPD in an unique MfeI site were prepared as described previously (Brooks et al.,
Materials and Methods Preparation of Constructs Oligonucleotides containing either a single cyclo-da lesion or a single CPD in an unique MfeI site were prepared as described previously (Brooks et al.,
NZYGene Synthesis kit
 Kit components Component Concentration Amount NZYGene Synthesis kit Catalogue number: MB33901, 10 reactions GS DNA Polymerase 1U/ μl 30 μl Reaction Buffer for GS DNA Polymerase 10 150 μl dntp mix 2 mm
Kit components Component Concentration Amount NZYGene Synthesis kit Catalogue number: MB33901, 10 reactions GS DNA Polymerase 1U/ μl 30 μl Reaction Buffer for GS DNA Polymerase 10 150 μl dntp mix 2 mm
Creating pentr vectors by BP reaction
 Creating pentr vectors by BP reaction Tanya Lepikhova and Rafael Martinez 15072011 Overview: 1. Design primers to add the attb sites to gene of interest 2. Perform PCR with a high fidelity DNA polymerase
Creating pentr vectors by BP reaction Tanya Lepikhova and Rafael Martinez 15072011 Overview: 1. Design primers to add the attb sites to gene of interest 2. Perform PCR with a high fidelity DNA polymerase
7. (10pts.) The diagram below shows the immunity region of phage l. The genes are:
 The diagram below shows the immunity region of phage l. The genes are:") 7. (10pts.) The diagram below shows the immunity region of phage l. The genes are: ci codes for ci repressor. The ci857 allele is temperature sensitive. cro codes for Cro protein. N codes for the transcription
7. (10pts.) The diagram below shows the immunity region of phage l. The genes are: ci codes for ci repressor. The ci857 allele is temperature sensitive. cro codes for Cro protein. N codes for the transcription
Genetics Lecture Notes Lectures 13 16
 Genetics Lecture Notes 7.03 2005 Lectures 13 16 Lecture 13 Transposable elements Transposons are usually from 10 3 to 10 4 base pairs in length, depending on the transposon type. The key property of transposons
Genetics Lecture Notes 7.03 2005 Lectures 13 16 Lecture 13 Transposable elements Transposons are usually from 10 3 to 10 4 base pairs in length, depending on the transposon type. The key property of transposons
pgm-t Cloning Kit Cat. # : GVT202 Size : 20 Reactions Store at -20
 pgm-t Cloning Kit Cat. # : GVT202 Size : 20 Reactions Store at -20 1 Kit Contents Contents pgm-t Cloning Kit pgm-t Vector (50 ng/μl) 20 μl T4 DNA Ligase (3 U/μl) 20 μl 10X T4 DNA Ligation Buffer 30 μl
pgm-t Cloning Kit Cat. # : GVT202 Size : 20 Reactions Store at -20 1 Kit Contents Contents pgm-t Cloning Kit pgm-t Vector (50 ng/μl) 20 μl T4 DNA Ligase (3 U/μl) 20 μl 10X T4 DNA Ligation Buffer 30 μl
GeNei TM Transformation Teaching Kit Manual
 Teaching Kit Manual Cat No. New Cat No. KT07 107385 KT07A 106220 Revision No.: 00060505 CONTENTS Page No. Objective 3 Principle 3 Kit Description 6 Materials Provided 7 Procedure 9 Observation & Interpretation
Teaching Kit Manual Cat No. New Cat No. KT07 107385 KT07A 106220 Revision No.: 00060505 CONTENTS Page No. Objective 3 Principle 3 Kit Description 6 Materials Provided 7 Procedure 9 Observation & Interpretation
Hurricane Miniprep Kit PROTOCOL
 Hurricane Miniprep Kit PROTOCOL Description: The Hurricane Miniprep Kit is designed for purification of up to 25 ug of high purity plasmid DNA from a starting volume of 2-5 ml of bacterial culture. The
Hurricane Miniprep Kit PROTOCOL Description: The Hurricane Miniprep Kit is designed for purification of up to 25 ug of high purity plasmid DNA from a starting volume of 2-5 ml of bacterial culture. The
Construction of Glycine Oxidase Mutant Libraries by Random Mutagenesis, Site Directed Mutagenesis and DNA Shuffling Tao Zhan1, 2*
 Construction of Glycine Oxidase Mutant Libraries by Random Mutagenesis, Site Directed Mutagenesis and DNA Shuffling Tao Zhan1, 2* 1 Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences,
Construction of Glycine Oxidase Mutant Libraries by Random Mutagenesis, Site Directed Mutagenesis and DNA Shuffling Tao Zhan1, 2* 1 Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences,
Table of Contents. i. Insertion of DNA fragments into plasmid vector...4. ii. Self-circularization of linear blunt-ended DNA...4
 Table of Contents I. Description...2 II. Kit Components...2 III. Storage...2 IIV. Notes...2 V. Reference...3 VI. PROCEDURES A. Dephosphorylation of vector DNA...3 B. Blunting reaction...3 C. Ligation reaction
Table of Contents I. Description...2 II. Kit Components...2 III. Storage...2 IIV. Notes...2 V. Reference...3 VI. PROCEDURES A. Dephosphorylation of vector DNA...3 B. Blunting reaction...3 C. Ligation reaction
NEBNext RNase III RNA Fragmentation Module
 SAMPLE PREPARATION NEBNext RNase III RNA Fragmentation Module Instruction Manual NEB #E6146S 100 reactions NEBNext RNase III RNA Fragmentation Module Table of Contents: Description....2 Applications....2
SAMPLE PREPARATION NEBNext RNase III RNA Fragmentation Module Instruction Manual NEB #E6146S 100 reactions NEBNext RNase III RNA Fragmentation Module Table of Contents: Description....2 Applications....2
Regulation of enzyme synthesis
 Regulation of enzyme synthesis The lac operon is an example of an inducible operon - it is normally off, but when a molecule called an inducer is present, the operon turns on. The trp operon is an example
Regulation of enzyme synthesis The lac operon is an example of an inducible operon - it is normally off, but when a molecule called an inducer is present, the operon turns on. The trp operon is an example
Roche Molecular Biochemicals Technical Note No. LC 10/2000
 Roche Molecular Biochemicals Technical Note No. LC 10/2000 LightCycler Overview of LightCycler Quantification Methods 1. General Introduction Introduction Content Definitions This Technical Note will introduce
Roche Molecular Biochemicals Technical Note No. LC 10/2000 LightCycler Overview of LightCycler Quantification Methods 1. General Introduction Introduction Content Definitions This Technical Note will introduce
Reading Lecture 3: 24-25, 45, Lecture 4: 66-71, Lecture 3. Vectors. Definition Properties Types. Transformation
 Lecture 3 Reading Lecture 3: 24-25, 45, 55-66 Lecture 4: 66-71, 75-79 Vectors Definition Properties Types Transformation 56 VECTORS- Definition Vectors are carriers of a DNA fragment of interest Insert
Lecture 3 Reading Lecture 3: 24-25, 45, 55-66 Lecture 4: 66-71, 75-79 Vectors Definition Properties Types Transformation 56 VECTORS- Definition Vectors are carriers of a DNA fragment of interest Insert
GenBuilder TM Cloning Kit User Manual
 GenBuilder TM Cloning Kit User Manual Cat.no L00701 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2 Ⅱ. DNA
GenBuilder TM Cloning Kit User Manual Cat.no L00701 Version 11242017 Ⅰ. Introduction... 2 I.1 Product Information... 2 I.2 Kit Contents and Storage... 2 I.3 GenBuilder Cloning Kit Workflow... 2 Ⅱ. DNA
ENDEXT Technology. Instruction manual for protein synthesis. with wheat germ cell-free system
 ENDEXT Technology Instruction manual for protein synthesis with wheat germ cell-free system 1 Protocol Overview Plasmid DNA construction (see Section 3.1) Preparation of plasmid DNA for transcription (see
ENDEXT Technology Instruction manual for protein synthesis with wheat germ cell-free system 1 Protocol Overview Plasmid DNA construction (see Section 3.1) Preparation of plasmid DNA for transcription (see
Presto Mini Plasmid Kit
 Instruction Manual Ver. 03.06.17 For Research Use Only Presto Mini Plasmid Kit PDH004 (4 Preparation Sample Kit) PDH100 (100 Preparation Kit) PDH300 (300 Preparation Kit) Advantages Sample: 1-7 ml of cultured
Instruction Manual Ver. 03.06.17 For Research Use Only Presto Mini Plasmid Kit PDH004 (4 Preparation Sample Kit) PDH100 (100 Preparation Kit) PDH300 (300 Preparation Kit) Advantages Sample: 1-7 ml of cultured
High-Purity Bacterial RNA Isolated with the Agilent Total RNA Isolation Mini Kit Application
 High-Purity Bacterial RNA Isolated with the Total RNA Isolation Mini Kit Application Gene Expression Author Christopher Rizzo Claudia A. Robbins Rhonda R. Taylor Technologies, Inc. 285 Centerville Road
High-Purity Bacterial RNA Isolated with the Total RNA Isolation Mini Kit Application Gene Expression Author Christopher Rizzo Claudia A. Robbins Rhonda R. Taylor Technologies, Inc. 285 Centerville Road
CopyCutter EPI400 Electrocompetent E. coli CopyCutter EPI400 Chemically Competent E. coli CopyCutter Induction Solution
 CopyCutter EPI400 Electrocompetent E. coli CopyCutter EPI400 Chemically Competent E. coli CopyCutter Induction Solution Cat. Nos. C400EL10, C400CH10, and CIS40025 Available exclusively thru Lucigen. lucigen.com/epibio
CopyCutter EPI400 Electrocompetent E. coli CopyCutter EPI400 Chemically Competent E. coli CopyCutter Induction Solution Cat. Nos. C400EL10, C400CH10, and CIS40025 Available exclusively thru Lucigen. lucigen.com/epibio
Mcbio 316: Exam 3. (10) 2. Compare and contrast operon vs gene fusions.
 2. Compare and contrast operon vs gene fusions.") Mcbio 316: Exam 3 Name (15) 1. Transposons provide useful tools for genetic analysis. List 5 different uses of transposon insertions. ANSWER: Many answers are possible, however, if multiple items on the
Mcbio 316: Exam 3 Name (15) 1. Transposons provide useful tools for genetic analysis. List 5 different uses of transposon insertions. ANSWER: Many answers are possible, however, if multiple items on the
Chapter 4. Recombinant DNA Technology
 Chapter 4 Recombinant DNA Technology 5. Plasmid Cloning Vectors Plasmid Plasmids Self replicating Double-stranded Mostly circular DNA ( 500 kb) Linear : Streptomyces, Borrelia burgdorferi Replicon
Chapter 4 Recombinant DNA Technology 5. Plasmid Cloning Vectors Plasmid Plasmids Self replicating Double-stranded Mostly circular DNA ( 500 kb) Linear : Streptomyces, Borrelia burgdorferi Replicon
MCB 421 Second Exam October 27, 2004
 MCB 421 Second Exam October 27, 2004 1. (10 pts) As discussed in class in complementation studies using F plasmids complementation can be confused with the products of homologous recombination between
MCB 421 Second Exam October 27, 2004 1. (10 pts) As discussed in class in complementation studies using F plasmids complementation can be confused with the products of homologous recombination between
Capsule deletion via a λ-red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH
 Capsule deletion via a λ-red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH 78578 Tzu-Wen Huang 1,2, Irene Lam 2, Hwan-You Chang 3, Shih-Feng Tsai 1, Bernhard
Capsule deletion via a λ-red knockout system perturbs biofilm formation and fimbriae expression in Klebsiella pneumoniae MGH 78578 Tzu-Wen Huang 1,2, Irene Lam 2, Hwan-You Chang 3, Shih-Feng Tsai 1, Bernhard
Lecture Four. Molecular Approaches I: Nucleic Acids
 Lecture Four. Molecular Approaches I: Nucleic Acids I. Recombinant DNA and Gene Cloning Recombinant DNA is DNA that has been created artificially. DNA from two or more sources is incorporated into a single
Lecture Four. Molecular Approaches I: Nucleic Acids I. Recombinant DNA and Gene Cloning Recombinant DNA is DNA that has been created artificially. DNA from two or more sources is incorporated into a single
Step 1: Digest vector with Reason for Step 1. Step 2: Digest T4 genomic DNA with Reason for Step 2: Step 3: Reason for Step 3:
 Biol/Chem 475 Spring 2007 Study Problems for Quiz 2 Quiz 2 (~50 pts) is scheduled for Monday May 14 It will cover all handouts and lab exercises to date except the handout/worksheet (yet to be distributed)
Biol/Chem 475 Spring 2007 Study Problems for Quiz 2 Quiz 2 (~50 pts) is scheduled for Monday May 14 It will cover all handouts and lab exercises to date except the handout/worksheet (yet to be distributed)
RayBio Genomic DNA Magnetic Beads Kit
 RayBio Genomic DNA Magnetic Beads Kit Catalog #: 801-112 User Manual Last revised January 4 th, 2017 Caution: Extraordinarily useful information enclosed ISO 13485 Certified 3607 Parkway Lane, Suite 100
RayBio Genomic DNA Magnetic Beads Kit Catalog #: 801-112 User Manual Last revised January 4 th, 2017 Caution: Extraordinarily useful information enclosed ISO 13485 Certified 3607 Parkway Lane, Suite 100
CHAPTER 9 DNA Technologies
 CHAPTER 9 DNA Technologies Recombinant DNA Artificially created DNA that combines sequences that do not occur together in the nature Basis of much of the modern molecular biology Molecular cloning of genes
CHAPTER 9 DNA Technologies Recombinant DNA Artificially created DNA that combines sequences that do not occur together in the nature Basis of much of the modern molecular biology Molecular cloning of genes
BL21(DE3) expression competent cell pack DS265. Component Code No. Contents Competent cell BL21(DE3) DS250 5 tubes (100 μl/tube)
 expression competent cell pack DS265. Component Code No. Contents Competent cell BL21(DE3) DS250 5 tubes (100 μl/tube)") Product Name : Code No. : Kit Component : BL21(DE3) expression competent cell pack DS265 Component Code No. Contents Competent cell BL21(DE3) DS250 5 tubes (100 μl/tube) transfromation efficiency: 5 10
Product Name : Code No. : Kit Component : BL21(DE3) expression competent cell pack DS265 Component Code No. Contents Competent cell BL21(DE3) DS250 5 tubes (100 μl/tube) transfromation efficiency: 5 10
Supporting Information
 Supporting Information Development of a 2,4-Dinitrotoluene-Responsive Synthetic Riboswitch in E. coli cells Molly E. Davidson, Svetlana V. Harbaugh, Yaroslav G. Chushak, Morley O. Stone, Nancy Kelley-
Supporting Information Development of a 2,4-Dinitrotoluene-Responsive Synthetic Riboswitch in E. coli cells Molly E. Davidson, Svetlana V. Harbaugh, Yaroslav G. Chushak, Morley O. Stone, Nancy Kelley-
Determination of Exclusion Effect in Wild Type and Rop Deficient Mutated pbr322 Co-transformations
 Determination of Exclusion Effect in Wild Type and Rop Deficient Mutated pbr322 Co-transformations Suzana Sabaiduc and Andy Lo Microbiology and Immunology University of British Columbia pbr322 is an expression
Determination of Exclusion Effect in Wild Type and Rop Deficient Mutated pbr322 Co-transformations Suzana Sabaiduc and Andy Lo Microbiology and Immunology University of British Columbia pbr322 is an expression
Guide-it Indel Identification Kit User Manual
 Clontech Laboratories, Inc. Guide-it Indel Identification Kit User Manual Cat. No. 631444 (120114) Clontech Laboratories, Inc. A Takara Bio Company 1290 Terra Bella Avenue, Mountain View, CA 94043, USA
Clontech Laboratories, Inc. Guide-it Indel Identification Kit User Manual Cat. No. 631444 (120114) Clontech Laboratories, Inc. A Takara Bio Company 1290 Terra Bella Avenue, Mountain View, CA 94043, USA
Transformation (The method of CaCl 2 )
") PROTOCOLS E. coli Transformation (The method of CaCl 2 ) Ligation PRODUCTION competent cells of E. COLI. (Rubidium cells). Gel DNA Recovery Kit Electroporation Digestiones Plasmid purification (The method
PROTOCOLS E. coli Transformation (The method of CaCl 2 ) Ligation PRODUCTION competent cells of E. COLI. (Rubidium cells). Gel DNA Recovery Kit Electroporation Digestiones Plasmid purification (The method
ml recombinant E. coli cultures (at a density of A 600 units per ml)
") for purification of plasmid DNA, from bacterial cultures Cat. No. 1 754 777 (50 purifications) Cat. No. 1 754 785 (50 purifications) Principle Alkaline lysis releases plasmid DNA from bacteria and RNase
for purification of plasmid DNA, from bacterial cultures Cat. No. 1 754 777 (50 purifications) Cat. No. 1 754 785 (50 purifications) Principle Alkaline lysis releases plasmid DNA from bacteria and RNase
TECHNICAL BULLETIN. In Vitro Bacterial Split Fluorescent Protein Fold n Glow Solubility Assay Kits
 In Vitro Bacterial Split Fluorescent Protein Fold n Glow Solubility Assay Kits Catalog Numbers APPA001 In Vitro Bacterial Split GFP "Fold 'n' Glow" Solubility Assay Kit (Green) APPA008 In Vitro Bacterial
In Vitro Bacterial Split Fluorescent Protein Fold n Glow Solubility Assay Kits Catalog Numbers APPA001 In Vitro Bacterial Split GFP "Fold 'n' Glow" Solubility Assay Kit (Green) APPA008 In Vitro Bacterial
Solid Phase cdna Synthesis Kit
 #6123 v.02.09 Table of Contents I. Description... 2 II. Kit components... 2 III. Storage... 2 IV. Principle... 3 V. Protocol V-1. Preparation of immobilized mrna... 4 Protocol A: Starting from Tissue or
#6123 v.02.09 Table of Contents I. Description... 2 II. Kit components... 2 III. Storage... 2 IV. Principle... 3 V. Protocol V-1. Preparation of immobilized mrna... 4 Protocol A: Starting from Tissue or
Phosphate buffered saline (PBS) for washing the cells TE buffer (nuclease-free) ph 7.5 for resuspending the SingleShot RNA control template
 for washing the cells TE buffer (nuclease-free) ph 7.5 for resuspending the SingleShot RNA control template") Catalog # Description 172-5085 SingleShot SYBR Green Kit, 100 x 50 µl reactions For research purposes only. Introduction The SingleShot SYBR Green Kit prepares genomic DNA (gdna) free RNA directly from
Catalog # Description 172-5085 SingleShot SYBR Green Kit, 100 x 50 µl reactions For research purposes only. Introduction The SingleShot SYBR Green Kit prepares genomic DNA (gdna) free RNA directly from
FROM EXPERIMENTS IN BACTERIAL GENETICS AND GENE TECHNIQUE
 Uppsala 2001-04-01 REPORT FROM EXPERIMENTS IN BACTERIAL GENETICS AND GENE TECHNIQUE Laboratory assistants: Maria Jönsson Amera Gibreel Students: Contents ASSIGNMENT:... 3 INTRODUCTION:... 3 MATERIAL AND
Uppsala 2001-04-01 REPORT FROM EXPERIMENTS IN BACTERIAL GENETICS AND GENE TECHNIQUE Laboratory assistants: Maria Jönsson Amera Gibreel Students: Contents ASSIGNMENT:... 3 INTRODUCTION:... 3 MATERIAL AND
Roche Molecular Biochemicals Technical Note No. LC 12/2000
 Roche Molecular Biochemicals Technical Note No. LC 12/2000 LightCycler Absolute Quantification with External Standards and an Internal Control 1. General Introduction Purpose of this Note Overview of Method
Roche Molecular Biochemicals Technical Note No. LC 12/2000 LightCycler Absolute Quantification with External Standards and an Internal Control 1. General Introduction Purpose of this Note Overview of Method
TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli
 TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli Cat. Nos. EC300102, EC300110, EC300150, and C300C105 Available exclusively thru Lucigen. lucigen.com/epibio www.lucigen.com
TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli Cat. Nos. EC300102, EC300110, EC300150, and C300C105 Available exclusively thru Lucigen. lucigen.com/epibio www.lucigen.com
ITS Sequencing in Millepora. 10/09 Subcloning DNA Fragments into pbluescript Preparation of pbluescript Vector
 Page 1 of 5 10/09 Subcloning DNA Fragments into pbluescript Preparation of pbluescript Vector 1. Digest 1 µg of pbluescript with Eco RI 2. Following digestion, add 0.1 volumes of 3M sodium acetate (ph
Page 1 of 5 10/09 Subcloning DNA Fragments into pbluescript Preparation of pbluescript Vector 1. Digest 1 µg of pbluescript with Eco RI 2. Following digestion, add 0.1 volumes of 3M sodium acetate (ph
Molecular Techniques Third-year Biology
 PLANNING Genetics Lab practices Molecular Techniques. Genetics Lab practices protocol. 2015-16 PCR-DIRECTED MUTAGENESIS, MOLECULAR CLONING AND RESTRICTION ANALYSIS Sessions 1 & 2 (2x3 hours): PCR-directed
PLANNING Genetics Lab practices Molecular Techniques. Genetics Lab practices protocol. 2015-16 PCR-DIRECTED MUTAGENESIS, MOLECULAR CLONING AND RESTRICTION ANALYSIS Sessions 1 & 2 (2x3 hours): PCR-directed
Module Code: BIO00007C
 Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Genetics Time Allowed: 1 Hour 30 Minutes Marking Scheme: Total marks available for
Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Genetics Time Allowed: 1 Hour 30 Minutes Marking Scheme: Total marks available for
Gateway Cloning Protocol (Clough Lab Edition) This document is a modification of the Gateway cloning protocol developed by Manju in Chris Taylor's lab
 This document is a modification of the Gateway cloning protocol developed by Manju in Chris Taylor's lab") Gateway Cloning Protocol (Clough Lab Edition) This document is a modification of the Gateway cloning protocol developed by Manju in Chris Taylor's lab With the Gateway cloning system, a PCR fragment is
Gateway Cloning Protocol (Clough Lab Edition) This document is a modification of the Gateway cloning protocol developed by Manju in Chris Taylor's lab With the Gateway cloning system, a PCR fragment is
Recitation CHAPTER 9 DNA Technologies
 Recitation CHAPTER 9 DNA Technologies DNA Cloning: General Scheme A cloning vector and eukaryotic chromosomes are separately cleaved with the same restriction endonuclease. (A single chromosome is shown
Recitation CHAPTER 9 DNA Technologies DNA Cloning: General Scheme A cloning vector and eukaryotic chromosomes are separately cleaved with the same restriction endonuclease. (A single chromosome is shown
THE INSTITUTE FOR GENOMIC RESEARCH Standard Operating Procedure SOP #: M022 REVISION LEVEL:.1 EFFECTIVE DATE: 04/12/04
 PAGE: 1 of 13 SOP #: M022 REVISION LEVEL:.1 EFFECTIVE DATE: 04/12/04 AUTHOR: Nicholas Marko PRIMARY REVIEWERS: Renee Rubio, Bryan Frank 1. PURPOSE This protocol describes the procedure for amplifying RNA
PAGE: 1 of 13 SOP #: M022 REVISION LEVEL:.1 EFFECTIVE DATE: 04/12/04 AUTHOR: Nicholas Marko PRIMARY REVIEWERS: Renee Rubio, Bryan Frank 1. PURPOSE This protocol describes the procedure for amplifying RNA
ENDEXT TM Technology. Wheat Germ Premium Expression Kit. Ver 1.7. CellFree Sciences Co., Ltd.
 ENDEXT TM Technology Wheat Germ Premium Expression Kit Ver 1.7 CellFree Sciences Co., Ltd. 1. Purpose Wheat Germ Premium Expression Kit is a starter kit to ascertain if the Wheat Germ Cell-Free System
ENDEXT TM Technology Wheat Germ Premium Expression Kit Ver 1.7 CellFree Sciences Co., Ltd. 1. Purpose Wheat Germ Premium Expression Kit is a starter kit to ascertain if the Wheat Germ Cell-Free System
Protocol for in vitro transcription
 Protocol for in vitro transcription Assemble the reaction at room temperature in the following order: Component 10xTranscription Buffer rntp T7 RNA Polymerase Mix grna PCR DEPC H 2 O volume 2μl 2μl 2μl
Protocol for in vitro transcription Assemble the reaction at room temperature in the following order: Component 10xTranscription Buffer rntp T7 RNA Polymerase Mix grna PCR DEPC H 2 O volume 2μl 2μl 2μl
Chapter 20 Recombinant DNA Technology. Copyright 2009 Pearson Education, Inc.
 Chapter 20 Recombinant DNA Technology Copyright 2009 Pearson Education, Inc. 20.1 Recombinant DNA Technology Began with Two Key Tools: Restriction Enzymes and DNA Cloning Vectors Recombinant DNA refers
Chapter 20 Recombinant DNA Technology Copyright 2009 Pearson Education, Inc. 20.1 Recombinant DNA Technology Began with Two Key Tools: Restriction Enzymes and DNA Cloning Vectors Recombinant DNA refers
3 Designing Primers for Site-Directed Mutagenesis
 3 Designing Primers for Site-Directed Mutagenesis 3.1 Learning Objectives During the next two labs you will learn the basics of site-directed mutagenesis: you will design primers for the mutants you designed
3 Designing Primers for Site-Directed Mutagenesis 3.1 Learning Objectives During the next two labs you will learn the basics of site-directed mutagenesis: you will design primers for the mutants you designed
GENETIC ENGINEERING worksheet
 Section A: Genetic Engineering Overview 1. What is genetic engineering? 2. Put the steps of genetic engineering in order. Recombinant product is isolated, purified and analyzed before marketing. The DNA
Section A: Genetic Engineering Overview 1. What is genetic engineering? 2. Put the steps of genetic engineering in order. Recombinant product is isolated, purified and analyzed before marketing. The DNA
BIOLOGY 205 Midterm II - 19 February Each of the following statements are correct regarding Eukaryotic genes and genomes EXCEPT?
 BIOLOGY 205 Midterm II - 19 February 1999 Name Multiple choice questions 4 points each (Best 12 out of 13). 1. Each of the following statements are correct regarding Eukaryotic genes and genomes EXCEPT?
BIOLOGY 205 Midterm II - 19 February 1999 Name Multiple choice questions 4 points each (Best 12 out of 13). 1. Each of the following statements are correct regarding Eukaryotic genes and genomes EXCEPT?
E. cloni EXPRESS Electrocompetent Cells
 E. cloni EXPRESS Electrocompetent Cells FOR RESEARCH USE ONLY. NOT FOR HUMAN OR DIAGNOSTIC USE Lucigen Corporation 2905 Parmenter St, Middleton, WI 53562 USA Toll Free: (888) 575-9695 (608) 831-9011 FAX:
E. cloni EXPRESS Electrocompetent Cells FOR RESEARCH USE ONLY. NOT FOR HUMAN OR DIAGNOSTIC USE Lucigen Corporation 2905 Parmenter St, Middleton, WI 53562 USA Toll Free: (888) 575-9695 (608) 831-9011 FAX:
Polymerase Chain Reaction
 Polymerase Chain Reaction Amplify your insert or verify its presence 3H Taq platinum PCR mix primers Ultrapure Water PCR tubes PCR machine A. Insert amplification For insert amplification, use the Taq
Polymerase Chain Reaction Amplify your insert or verify its presence 3H Taq platinum PCR mix primers Ultrapure Water PCR tubes PCR machine A. Insert amplification For insert amplification, use the Taq
2014 Pearson Education, Inc. CH 8: Recombinant DNA Technology
 CH 8: Recombinant DNA Technology Biotechnology the use of microorganisms to make practical products Recombinant DNA = DNA from 2 different sources What is Recombinant DNA Technology? modifying genomes
CH 8: Recombinant DNA Technology Biotechnology the use of microorganisms to make practical products Recombinant DNA = DNA from 2 different sources What is Recombinant DNA Technology? modifying genomes
Figure S1. Biosynthetic pathway of GDP-PerNAc. Mass spectrum of purified GDP-PerNAc. Nature Protocols: doi: /nprot
 Synthesis of GDP-PerNAc In E. coli O157, the biosynthesis of GDP- -N-acetyl-D-perosamine (GDP-PerNAc) involves three enzymes (Fig. S1). GDP-D-Mannose is converted by GDP-mannose-4,6-dehydratase (GMD) into
Synthesis of GDP-PerNAc In E. coli O157, the biosynthesis of GDP- -N-acetyl-D-perosamine (GDP-PerNAc) involves three enzymes (Fig. S1). GDP-D-Mannose is converted by GDP-mannose-4,6-dehydratase (GMD) into
7 Synthesizing the ykkcd Mutant Toxin Sensor RNA in vitro
 7 Synthesizing the ykkcd Mutant Toxin Sensor RNA in vitro 7.1 Learning Objective In the quest toward understanding how the ykkcd toxin sensor recognizes the antibiotic tetracycline you thus far designed
7 Synthesizing the ykkcd Mutant Toxin Sensor RNA in vitro 7.1 Learning Objective In the quest toward understanding how the ykkcd toxin sensor recognizes the antibiotic tetracycline you thus far designed
BRED: Bacteriophage Recombineering with Electroporated DNA
 Phagehunting Program BRED: Bacteriophage Recombineering with Electroporated DNA Introduction We have developed a system for generating mutations in lytically replicating mycobacteriophages that we have
Phagehunting Program BRED: Bacteriophage Recombineering with Electroporated DNA Introduction We have developed a system for generating mutations in lytically replicating mycobacteriophages that we have
Different Upstream Activators Stimulate Transcription Through Distinct Coactivator Complexes
 Supplemental Material for Different Upstream Activators Stimulate Transcription Through Distinct Coactivator Complexes Dong-ki Lee, Soyoun Kim, and John T. Lis Due to be published as a Research Communication
Supplemental Material for Different Upstream Activators Stimulate Transcription Through Distinct Coactivator Complexes Dong-ki Lee, Soyoun Kim, and John T. Lis Due to be published as a Research Communication
ElectroTen-Blue Electroporation Competent Cells
 ElectroTen-Blue Electroporation Competent Cells Instruction Manual Catalog #200159 Revision C.0 For Research Use Only. Not for use in diagnostic procedures. 200159-12 LIMITED PRODUCT WARRANTY This warranty
ElectroTen-Blue Electroporation Competent Cells Instruction Manual Catalog #200159 Revision C.0 For Research Use Only. Not for use in diagnostic procedures. 200159-12 LIMITED PRODUCT WARRANTY This warranty
7.1 Techniques for Producing and Analyzing DNA. SBI4U Ms. Ho-Lau
 7.1 Techniques for Producing and Analyzing DNA SBI4U Ms. Ho-Lau What is Biotechnology? From Merriam-Webster: the manipulation of living organisms or their components to produce useful usually commercial
7.1 Techniques for Producing and Analyzing DNA SBI4U Ms. Ho-Lau What is Biotechnology? From Merriam-Webster: the manipulation of living organisms or their components to produce useful usually commercial
PeriPreps Periplasting Kit
 Cat. No. PS81100 The PeriPreps Periplasting Kit facilitates the release of proteins contained within the periplasmic space of E. coli cells by combining digestion of the cell wall using Ready-Lyse Lysozyme
Cat. No. PS81100 The PeriPreps Periplasting Kit facilitates the release of proteins contained within the periplasmic space of E. coli cells by combining digestion of the cell wall using Ready-Lyse Lysozyme
Rapid amplification of cdna ends (RACE)
") Rapid amplification of cdna ends (RACE) Rapid amplification of cdna ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE
Rapid amplification of cdna ends (RACE) Rapid amplification of cdna ends (RACE) is a technique used in molecular biology to obtain the full length sequence of an RNA transcript found within a cell. RACE
Biotechnology. Chapter 20. Biology Eighth Edition Neil Campbell and Jane Reece. PowerPoint Lecture Presentations for
 Chapter 20 Biotechnology PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp Copyright
Chapter 20 Biotechnology PowerPoint Lecture Presentations for Biology Eighth Edition Neil Campbell and Jane Reece Lectures by Chris Romero, updated by Erin Barley with contributions from Joan Sharp Copyright
Genetic Engineering & Recombinant DNA
 Genetic Engineering & Recombinant DNA Chapter 10 Copyright The McGraw-Hill Companies, Inc) Permission required for reproduction or display. Applications of Genetic Engineering Basic science vs. Applied
Genetic Engineering & Recombinant DNA Chapter 10 Copyright The McGraw-Hill Companies, Inc) Permission required for reproduction or display. Applications of Genetic Engineering Basic science vs. Applied
Guide-it sgrna In Vitro Transcription and Screening Systems User Manual
 Guide-it sgrna In Vitro Transcription and Screening Systems User Manual Cat. Nos. 632638, 632639, 632635, 632636, 632637 (040618) 1290 Terra Bella Avenue, Mountain View, CA 94043, USA U.S. Technical Support:
Guide-it sgrna In Vitro Transcription and Screening Systems User Manual Cat. Nos. 632638, 632639, 632635, 632636, 632637 (040618) 1290 Terra Bella Avenue, Mountain View, CA 94043, USA U.S. Technical Support:
TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli
 TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli Cat. Nos. EC300105, EC300110, EC300150, and C300C105 Connect with Epicentre on our blog (epicentral.blogspot.com),
TransforMax EPI300 Electrocompetent E. coli TransforMax EPI300 Chemically Competent E. coli Cat. Nos. EC300105, EC300110, EC300150, and C300C105 Connect with Epicentre on our blog (epicentral.blogspot.com),
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/315/5819/1709/dc1 Supporting Online Material for RISPR Provides Acquired Resistance Against Viruses in Prokaryotes Rodolphe Barrangou, Christophe Fremaux, Hélène Deveau,
www.sciencemag.org/cgi/content/full/315/5819/1709/dc1 Supporting Online Material for RISPR Provides Acquired Resistance Against Viruses in Prokaryotes Rodolphe Barrangou, Christophe Fremaux, Hélène Deveau,
Understanding the Cellular Mechanism of the Excess Microsporocytes I (EMSI) Gene. Andrew ElBardissi, The Pennsylvania State University
 Gene. Andrew ElBardissi, The Pennsylvania State University") Understanding the Cellular Mechanism of the Excess Microsporocytes I (EMSI) Gene Andrew ElBardissi, The Pennsylvania State University Abstract: Hong Ma, The Pennsylvania State University The Excess Microsporocytes
Understanding the Cellular Mechanism of the Excess Microsporocytes I (EMSI) Gene Andrew ElBardissi, The Pennsylvania State University Abstract: Hong Ma, The Pennsylvania State University The Excess Microsporocytes
CH 8: Recombinant DNA Technology
 CH 8: Recombinant DNA Technology Biotechnology the use of microorganisms to make practical products Recombinant DNA = DNA from 2 different sources What is Recombinant DNA Technology? modifying genomes
CH 8: Recombinant DNA Technology Biotechnology the use of microorganisms to make practical products Recombinant DNA = DNA from 2 different sources What is Recombinant DNA Technology? modifying genomes
Highly efficient one-step PCR-based mutagenesis technique for large plasmids using high-fidelity DNA polymerase
 Highly efficient one-step PCR-based mutagenesis technique for large plasmids using high-fidelity DNA polymerase H. Liu, R. Ye and Y.Y. Wang Department of Medical Microbiology and Parasitology, School of
Highly efficient one-step PCR-based mutagenesis technique for large plasmids using high-fidelity DNA polymerase H. Liu, R. Ye and Y.Y. Wang Department of Medical Microbiology and Parasitology, School of