Regulatory expectations on impurities in drug substances - Pavia, October 2, Luisa Torchio Euticals SpA
|
|
|
- Arleen Kathleen Warner
- 6 years ago
- Views:
Transcription
1 Regulatory expectations on impurities in drug substances - Pavia, October 2, 2015 Luisa Torchio Euticals SpA
2 An Impurity is defined as any substance or element present in a drug substance (DS) that is not the chemical entity defined as the drug substance. Impurities lower the quality of the DS in terms of purity and they can potentially affect the efficacy and/or safety of the final Drug Product. Impurities can be toxic, affect stability or efficacy of final product. Even if completely inert, they do not add any therapeutic benefit, so they are always considered as unwanted components.
3 Impurities come from the manufacturing process. No process (chemical synthesis, extraction, fermentation, biological/ biotechnological pathway) can assure the complete absence or removal of impurities. Theproductitselfcanbeasourceofimpurities,incaseofdegradationdue to improper packaging or storage conditions.
4
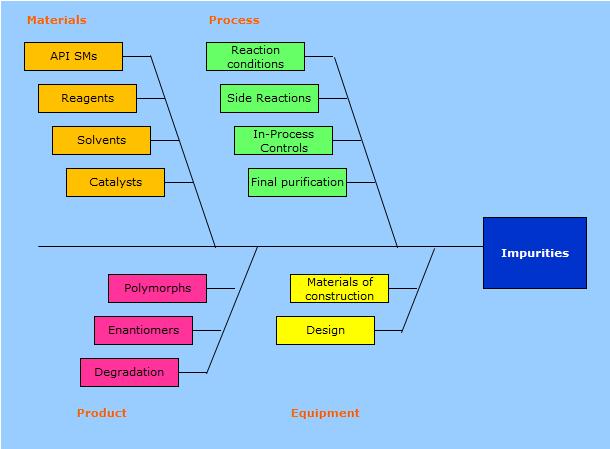
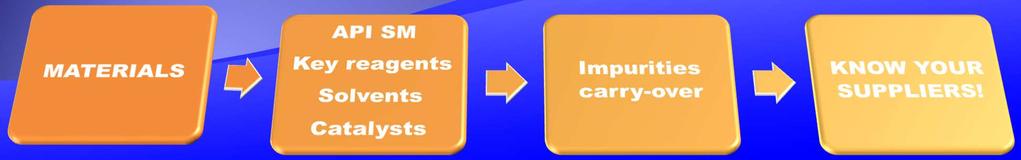
5 The control strategy is based on: A thorough knowledge of the product characteristics (enantiomers, polymorphic forms, isomers, temperature/light/moisture sensitivity...). Knowledge of all manufacturing steps(raw materials properties, process parameters, final purification capabilities). Availability of suitable analytical methods to ensure the appropriate control of all the impurities(including degradation products). A good knowledge helps also in evaluating changes to the original process.
6
7 A good knowledge of the process is necessary to determine the impurity profile of the product. Impurity profile is the list of any single and/ or total identified and unidentified impurities present in a standard batch of DS, obtained by a specific manufacturing process and tested by a suitable analytical method. It includes the identification of each impurity (e.g. chemical name or RRT) andthetypicallevel(asarea%orasotherunit). Being the impurity profile process-based, not always it corresponds to the compendial monograph one. Unexpected or different impurities can be found in the product if obtained by a route different from the historical one upon which the monograph was prepared.
8 Regulatory authorities expect to find in the ASMF or Application Dossier all the information needed to assess if the DS/DP represents a risk for the health of the patient. Data relevant to impurities are a critical point in this assessment. The US-FDA issued on 2014 a guidance for industry titled «ANDAs submissions- Refuse-to-receive for lack of proper justification for impurity limits». The International Conference On Harmonisation, ICH, founded on 1990, issued, starting from late 90 s, different specific guidelines on Impurities, identified as ICH Q3 series, replacing the different, several documents previously available, to harmonise expectations on this topic.
9 Being most APIs manufactured by chemical synthesis, the first guideline ICH Q3A- specifically addressed to Impurities in DS is focused only on synthetically obtained APIs, classifying impurities in 3 classes Organic impurities Inorganic impurities Residual solvents It gives recommendations on both chemical and safety aspect of impurities. In particular, three thresholds are defined for impurities identification, reporting and qualification, based on the typical daily dosage of DS. In case the proposed limits cannot be met, additional information and proper justification has to be provided.

10 General notes on setting specs for organic impurities Any impurity routinely observed in standard batches or long-term stability trials should be controlled by the impurity specifications. Impurities observed below the ICH identification threshold need not be individually specified in the specifications. They can be controlled under the limit for any unspecified impurity. Impurities above the ICH identification threshold need to be identified and individually specified in the specifications. A test for any unspecified impurity and total impurities should be included. General notes on setting limits for impurities The limit for any unspecified impurity should be at the ICH identification threshold. The limit for total impurity content should reflect batch data. Residual Solvents and Elemental impurities follows specific guidelines General notes on qualification Based on Toxicological data or Structure-activity studies Based on literature Based on limit in pre-approved applications
11
12 The attention on impurities continues to evolve because of scientific and technological advancements make impurity measurements and control strategies more effective. These changes, in combination with advancements in the field of toxicology, have contributed to the evolution of regulatory (and consequently compendial) standards for the control of impurities in drug substances and drug products. During time, it was considered necessary to have specific guidelines on Residual Solvents, considering their number and their different characteristics; on Elemental impurities, due to their potential ubiquitous nature; on Genotoxic impurities for safety concerns they pose. Moreover, after the issuance of ICH Q8, Q9 and Q10 that underlined the need of quality applied to all the DS/DP lifecycle, impurities have been considered as an important part of process development and, in general, of Quality Risk Management.
13 Source Title& Content ICH Q3A(R2) Impurities in new Drug Substances 2006 Year of issuance The Guideline addresses the chemistry and safety aspects of impurities, including the listing of impurities in specifications and defines the thresholds for reporting, identification and qualification. ICH Q3B(R2) Impurities in new Drug Products 2006 The Guideline specifically deals with those impurities which might arise as degradation products of the drug substance or arising from interactions between drug substance and excipients or components of primary packaging materials. ICH Q3C(R5) Impurities. Guideline on Residual Solvents 2011 It recommends the use of less toxic solvents in the manufacture of drug substances and dosage forms, and sets pharmaceutical limits for residual solvents (organic volatile impurities) in drug products. R6 available for comments. ICH Q3D Guideline on Elemental Impurities 2014 This guidance aims to provide a global policy for limiting metal impurities (included in ICH Q3A as inorganic ones) qualitatively and quantitatively in drug products and ingredients. Implementation on going.
14 Source Title & Content Year of issuance ICH Q5A Q5E Quality of Biotechnological Products This group of guidelines specifically addresses quality aspects, including impurities, relevant to DS and DP obtained by biological sources and biotechnology ICH Q6B Test proceduresand acceptancecriteriafor Biological and Biotechnological Products 1999 It provides guidance on justifying and setting specifications for proteins and polypeptides which are derived from recombinant or non-recombinant cell cultures ICH Q11 Development and manufacturing of DS 2012 It addresses aspects of development and manufacture that pertain to drug substance, including the presence of steps designed to reduce impurities. ICH M7 Genotoxic impurities 2014 It offers guidance on analysis of SAR for genotoxicity. Also it is intended to resolve questions such as whether impurities with similar alerts that potentially have similar mechanism of action should not be combined in calculating a Threshold of Toxicological Concern (TTC) and whether the TTC may differ based on differences in the approved duration of use. Implementation on going. R1 distributed for comments.
15 Source Title & Content Year of issuance US-FDA ANDAs. Refuse-to-receive for lack of proper justification for impurity limits(draft) 2014 The guidance highlights deficiencies in relation to information about impurities that may cause FDA to refuse to receive an ANDA.A refuse-to-receive decision indicates that FDA determined that an ANDA is not sufficiently complete to permit a substantive review. EMA CHMP/CVMP/QWP/ Settingspecificationsfor related impurities in antibiotics It provides guidance in setting limits for impurities in antibiotics from fermentative or semi-synthetic route. EMA CPMP/QWP/2820/00 Rev.2 Guideline on specifications: test procedures and acceptance criteria for herbal substances( ) 2011 It provides guidance in setting limits for impurities in APIs from vegetal origin WHO Technical Report Series No 953 Annex 4 Suitability of Drug Substances for pharmaceutical products 2009
16 Basically, ICH concepts and proposed limits have been adopted by USP, EP and JP. Moreover, ICH recommendations have been described in general chapters or monographs that are complemental to single product monographs. For example: EP General Monograph Nr.2034 Substances for pharmaceutical use; EP General Chapter 5.10 Control of impurities in substances for pharmaceutical use; USP <1086> Impurities in Drug Substances and Drug Products; USP <476> Organic Impurities in DS and DP (new) Also in this case a continual improvement and revision program is ongoing.
17 Impurities is an always ongoing topic, for both manufacturers and regulators. Anumberofopenpointsarestillpresent,forexample Existing DSs. On which extent guidelines are applicable to old DSs? Guidance for other product type?. Toxicology and Genotoxicity. Is it possible to qualify impurities without the need to undertake additional studies? Are the currently proposed limits correct? What is the role of DS manufacturer? In the future new challenges will arise.

18
BRIEFING. . Over time, 466 may be used less frequently and may be withdrawn.
 Page 1 of 13 BRIEFING 1086 USP 37 page 828. As part of an ongoing monograph modernization initiative, the United States Pharmacopeial Convention (USP) is updating this general chapter, 1086 Impurities
Page 1 of 13 BRIEFING 1086 USP 37 page 828. As part of an ongoing monograph modernization initiative, the United States Pharmacopeial Convention (USP) is updating this general chapter, 1086 Impurities
A Generic Industry Perspective on Establishing Impurity Limits And a Corresponding Control Strategy
 A Generic Industry Perspective on Establishing Impurity Limits And a Corresponding Control Strategy Nicholas Cappuccino, PhD Vice-President, Head of Quality and Scientific Affairs Dr. Reddy s Laboratories,
A Generic Industry Perspective on Establishing Impurity Limits And a Corresponding Control Strategy Nicholas Cappuccino, PhD Vice-President, Head of Quality and Scientific Affairs Dr. Reddy s Laboratories,
IMPURITIES IN NEW DRUG PRODUCTS
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
Guidance for Industry Q3B(R2) Impurities in New Drug Products
 Impurities in New Drug Products") Guidance for Industry Q3B(R2) Impurities in New Drug Products U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics
Guidance for Industry Q3B(R2) Impurities in New Drug Products U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics
2017 AAM CMC Workshop
 2017 AAM CMC Workshop SETTING PROPER IMPURITIES LIMITS - INCLUDING GENOTOXIC IMPURITIES INDUSTRY PERSPECTIVE Janet Vaughn, Sr. Director Regulatory Affairs Teva Pharmaceuticals USA 23 May, 2017 Disclaimer
2017 AAM CMC Workshop SETTING PROPER IMPURITIES LIMITS - INCLUDING GENOTOXIC IMPURITIES INDUSTRY PERSPECTIVE Janet Vaughn, Sr. Director Regulatory Affairs Teva Pharmaceuticals USA 23 May, 2017 Disclaimer
Impurities from degradation of Drug Substances
 Impurities from degradation of Drug Substances REFERENCE 1. ICH - IMPURITIES IN NEW DRUG SUBSTANCES - Q3A(R2) 2. ICH - IMPURITIES IN NEW DRUG PRODUCTS - Q3B(R2) 3. ICH - VALIDATION OF ANALYTICAL PROCEDURES:
Impurities from degradation of Drug Substances REFERENCE 1. ICH - IMPURITIES IN NEW DRUG SUBSTANCES - Q3A(R2) 2. ICH - IMPURITIES IN NEW DRUG PRODUCTS - Q3B(R2) 3. ICH - VALIDATION OF ANALYTICAL PROCEDURES:
ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances
 The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit CPMP/ICH/142/95 ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances Step 5 NOTE
The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit CPMP/ICH/142/95 ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances Step 5 NOTE
ICH Topic Q 3 B Impurities in New Medicinal Products
 The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit ICH Topic Q 3 B Impurities in New Medicinal Products Step 4, Consensus Guideline, 6 November 1996 TE FOR GUIDANCE
The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit ICH Topic Q 3 B Impurities in New Medicinal Products Step 4, Consensus Guideline, 6 November 1996 TE FOR GUIDANCE
Guidance for Industry
 Draft-t for Implementation Guidance for Industry ANDAs: Impurities in Drug Substances DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding
Draft-t for Implementation Guidance for Industry ANDAs: Impurities in Drug Substances DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding
ICH guideline M7 on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk
 impurities in pharmaceuticals to limit potential carcinogenic risk") February 2013 EMA/CHMP/ICH/83812/2013 ICH guideline M7 on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk Step 3 Transmission to CHMP
February 2013 EMA/CHMP/ICH/83812/2013 ICH guideline M7 on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk Step 3 Transmission to CHMP
Guideline on setting specifications for related impurities in antibiotics
 1 2 3 4 14 July 2010 EMA/CHMP/CVMP/QWP/199250/2009 Committee for Medicinal Products for Human Use (CHMP) / Committee for Medicinal Products for Veterinary Use (CVMP) 5 6 7 Guideline on setting specifications
1 2 3 4 14 July 2010 EMA/CHMP/CVMP/QWP/199250/2009 Committee for Medicinal Products for Human Use (CHMP) / Committee for Medicinal Products for Veterinary Use (CVMP) 5 6 7 Guideline on setting specifications
New Drug Product Impurities
 Title Page Image - with courtesy of the FDA CDER original web site at www. cder.fda.gov/ New Drug Product Impurities IAGIM Scientific Committee Block JD; Holmann E ; West P NEW DRAFT GUIDANCE FDA Viewpoint
Title Page Image - with courtesy of the FDA CDER original web site at www. cder.fda.gov/ New Drug Product Impurities IAGIM Scientific Committee Block JD; Holmann E ; West P NEW DRAFT GUIDANCE FDA Viewpoint
COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) NOTE FOR GUIDANCE ON SUMMARY OF REQUIREMENTS FOR ACTIVE SUBSTANCES IN PART II OF THE DOSSIER
 NOTE FOR GUIDANCE ON SUMMARY OF REQUIREMENTS FOR ACTIVE SUBSTANCES IN PART II OF THE DOSSIER") The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit London, 28 January 1998 CPMP/QWP/297/97 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) NOTE FOR GUIDANCE
The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit London, 28 January 1998 CPMP/QWP/297/97 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) NOTE FOR GUIDANCE
Questions and answers on the 'Guideline on the limits of genotoxic impurities'
 23 September 2010 EMA/CHMP/SWP/431994/2007 Rev. 3 Committee for Medicinal Products for Human Use (CHMP) Safety Working Party (SWP) Questions and answers on the 'Guideline on the limits of genotoxic impurities'
23 September 2010 EMA/CHMP/SWP/431994/2007 Rev. 3 Committee for Medicinal Products for Human Use (CHMP) Safety Working Party (SWP) Questions and answers on the 'Guideline on the limits of genotoxic impurities'
European Medicines Agency Evaluation of Medicines for Human Use
 European Medicines Agency Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BWP/49348/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL
European Medicines Agency Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BWP/49348/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL
M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk
 Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk") M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk This draft guidance, when finalized, will represent the Food and Drug Administration's
M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk This draft guidance, when finalized, will represent the Food and Drug Administration's
Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review
 Review") Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review Presented by Erika E. Englund, Ph.D. Slides courtesy of Dorota Matecka, Ph.D. Office of Pharmaceutical
Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review Presented by Erika E. Englund, Ph.D. Slides courtesy of Dorota Matecka, Ph.D. Office of Pharmaceutical
Guidance for Industry
 Guidance for Industry ANDAs: Impurities in Drug Products DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should
Guidance for Industry ANDAs: Impurities in Drug Products DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should
Overview of Impurities
 Overview of Impurities Regulatory and GMP aspects presented by Dr Clive Simon The SPD Company 2 September 2014 Definition Substances that provide no therapeutic benefit but have potential to cause adverse
Overview of Impurities Regulatory and GMP aspects presented by Dr Clive Simon The SPD Company 2 September 2014 Definition Substances that provide no therapeutic benefit but have potential to cause adverse
A.1 Contents file 4 to 5 A.1 (1)
") Contents file 4 to 5 Contents file 4 to 5 A Information Contents file 4 to 5 A.2 Index file 4 to 5 A.3 List of Abbreviations A.4 Glossary A.5 Adress-Register A.6 References B Japanese Regulations B.1 MHW
Contents file 4 to 5 Contents file 4 to 5 A Information Contents file 4 to 5 A.2 Index file 4 to 5 A.3 List of Abbreviations A.4 Glossary A.5 Adress-Register A.6 References B Japanese Regulations B.1 MHW
VICH Topic GL10. Step 7 (after revision at step 9) GUIDELINE ON IMPURITIES IN NEW VETERINARY DRUG SUBSTANCES
 GUIDELINE ON IMPURITIES IN NEW VETERINARY DRUG SUBSTANCES") European Medicines Agency Veterinary Medicines and Inspections London, 19 February 2007 Doc. Ref.EMEA/CVMP/VICH/837/99-Rev.1 VICH Topic GL10 Step 7 (after revision at step 9) GUIDELINE ON IMPURITIES IN
European Medicines Agency Veterinary Medicines and Inspections London, 19 February 2007 Doc. Ref.EMEA/CVMP/VICH/837/99-Rev.1 VICH Topic GL10 Step 7 (after revision at step 9) GUIDELINE ON IMPURITIES IN
Elemental impurities impact on APIs
 Regulatory Expectations on impurities in Drug Substances: Authority and Industry perspective Pavia, 2nd October 2015 Elemental impurities impact on APIs October 2 nd 2015 Annalisa Scali RegulatoryAffairsManager
Regulatory Expectations on impurities in Drug Substances: Authority and Industry perspective Pavia, 2nd October 2015 Elemental impurities impact on APIs October 2 nd 2015 Annalisa Scali RegulatoryAffairsManager
ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological entities)
") May 2011 EMA/CHMP/ICH/425213/2011 ICH/ Committee for medicinal products for human use (CHMP) ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological
May 2011 EMA/CHMP/ICH/425213/2011 ICH/ Committee for medicinal products for human use (CHMP) ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological
CEP submission: How to prepare a New Application?
 CEP submission: How to prepare a New Application? Nathalie Vicente Certification of Substances Division Summary How to prepare a New Application? Requirements for a new CEP application Content of the dossier
CEP submission: How to prepare a New Application? Nathalie Vicente Certification of Substances Division Summary How to prepare a New Application? Requirements for a new CEP application Content of the dossier
Guideline on the Requirements for Quality Documentation Concerning Biological Investigational Medicinal Products in Clinical Trials
 1 2 3 18 February 2010 EMA/CHMP/BWP/534898/2008 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 7 Guideline on the Requirements for Quality Documentation Concerning Biological Investigational
1 2 3 18 February 2010 EMA/CHMP/BWP/534898/2008 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 7 Guideline on the Requirements for Quality Documentation Concerning Biological Investigational
Impact factor: 3.958/ICV: 4.10 ISSN:
 Impact factor: 3.958/ICV: 4.10 ISSN: 0976-7908 24 Pharma Science Monitor 8(2), Apr-Jun 2017 PHARMA SCIENCE MONITOR AN INTERNATIONAL JOURNAL OF PHARMACEUTICAL SCIENCES Journal home page: http://www.pharmasm.com
Impact factor: 3.958/ICV: 4.10 ISSN: 0976-7908 24 Pharma Science Monitor 8(2), Apr-Jun 2017 PHARMA SCIENCE MONITOR AN INTERNATIONAL JOURNAL OF PHARMACEUTICAL SCIENCES Journal home page: http://www.pharmasm.com
Reflection paper on the requirements for selection and justification of starting materials for the manufacture of chemical active substances
 3 July 2017 EMA/CHMP/CVMP/QWP/826771/2016 Corr. 1 Committee for Medicinal Products for Human Use (CHMP) Committee for Medicinal Products for Veterinary Use (CVMP) Reflection paper on the requirements for
3 July 2017 EMA/CHMP/CVMP/QWP/826771/2016 Corr. 1 Committee for Medicinal Products for Human Use (CHMP) Committee for Medicinal Products for Veterinary Use (CVMP) Reflection paper on the requirements for
Examples of regulatory expectations for analytical characterization and testing
 Examples of regulatory expectations for analytical characterization and testing AT Europe 2016, 18 March 2016 Vicki Venizelos Quality RA B.V. Leiden, the Netherlands Overview What are the regulatory expectations?
Examples of regulatory expectations for analytical characterization and testing AT Europe 2016, 18 March 2016 Vicki Venizelos Quality RA B.V. Leiden, the Netherlands Overview What are the regulatory expectations?
Clinical qualification of specifications - a Regulator s view
 Clinical qualification of specifications - a Regulator s view Mats Welin Medical Products Agency, Uppsala, Sweden Disclaimer: The opinions expressed are my own and do not necessarily represent those of
Clinical qualification of specifications - a Regulator s view Mats Welin Medical Products Agency, Uppsala, Sweden Disclaimer: The opinions expressed are my own and do not necessarily represent those of
Adopted by CHMP for release for consultation 15 December Start of public consultation 15 December 2016
 1 September 2017 EMA/CHMP/ICH/809509/2016 Committee for Human Medicinal Products ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological / biological
1 September 2017 EMA/CHMP/ICH/809509/2016 Committee for Human Medicinal Products ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological / biological
How to Avoid Common Deficiencies in INDs and NDAs. Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER
 How to Avoid Common Deficiencies in INDs and NDAs Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER 1 Structure of FDA Office of Commissioner Chief Scientist FOODS Medical Products & Tobacco
How to Avoid Common Deficiencies in INDs and NDAs Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER 1 Structure of FDA Office of Commissioner Chief Scientist FOODS Medical Products & Tobacco
Derivation and Justification of Safety Thresholds
 Derivation and Justification of Safety Thresholds Douglas J. Ball, MS, DABT Chair, PQRI L&E Toxicology Subgroup Research Fellow, Safety Sciences - Pfizer, Inc. Agenda Basic Definitions Current Regulatory
Derivation and Justification of Safety Thresholds Douglas J. Ball, MS, DABT Chair, PQRI L&E Toxicology Subgroup Research Fellow, Safety Sciences - Pfizer, Inc. Agenda Basic Definitions Current Regulatory
Starting Material Selection for Type II Drug Master Files
 Starting Material Selection for Type II Drug Master Files Ronald S. Michalak Quality Assessment Lead (Acting), Division of Lifecycle API Office of New Drug Products, OPQ/CDER/ FDA CDER Reorganization Office
Starting Material Selection for Type II Drug Master Files Ronald S. Michalak Quality Assessment Lead (Acting), Division of Lifecycle API Office of New Drug Products, OPQ/CDER/ FDA CDER Reorganization Office
Excipients Facing Increased Scrutiny How to Use Secondary Reference Standards to Help Maintain Regulatory Compliance
 Excipients Facing Increased Scrutiny How to Use Secondary Reference Standards to Help Maintain Regulatory Compliance by Tom Savage, NSF International Since 2008, when patient deaths were first linked to
Excipients Facing Increased Scrutiny How to Use Secondary Reference Standards to Help Maintain Regulatory Compliance by Tom Savage, NSF International Since 2008, when patient deaths were first linked to
Regulation of Active Pharmaceutical Ingredients (API)
") Regulation of Active Pharmaceutical Ingredients (API) Stephan Rönninger Head External Affairs Europe, International Quality June 21, 2013 SINDUSFARMA / ANVISA / FIP-IPS Conference, Brasilia Agenda Regulations
Regulation of Active Pharmaceutical Ingredients (API) Stephan Rönninger Head External Affairs Europe, International Quality June 21, 2013 SINDUSFARMA / ANVISA / FIP-IPS Conference, Brasilia Agenda Regulations
Guideline on assessment and control of DNA reactive (mutagenic) impurities in veterinary medicinal products
 impurities in veterinary medicinal products") 1 2 3 24 February 2017 EMA/CVMP/SWP/377245/2016 Committee for Medicinal Products for Veterinary Use (CVMP) 4 5 6 Guideline on assessment and control of DNA reactive (mutagenic) impurities in veterinary
1 2 3 24 February 2017 EMA/CVMP/SWP/377245/2016 Committee for Medicinal Products for Veterinary Use (CVMP) 4 5 6 Guideline on assessment and control of DNA reactive (mutagenic) impurities in veterinary
GPhA Fall Technical Conference Nov 2-5, 2015 Bethesda, MD ICH M7 Guidance Overview and Current FDA Perspectives
 GPhA Fall Technical Conference Nov 2-5, 2015 Bethesda, MD ICH M7 Guidance Overview and Current FDA Perspectives Stephen Miller, Ph.D. CMC-Lead; Office of New Drug Products Office of Pharmaceutical Quality
GPhA Fall Technical Conference Nov 2-5, 2015 Bethesda, MD ICH M7 Guidance Overview and Current FDA Perspectives Stephen Miller, Ph.D. CMC-Lead; Office of New Drug Products Office of Pharmaceutical Quality
Wirkstoffdokumentation & CEP- Verfahren
 Wirkstoffdokumentation & CEP- Verfahren Dr. Wolfgang Herzog AGES PharmMed, Institut Zulassung & Lifecycle Management AGES-Gespräch Am Pulsschlag des Arzneimittelwesens Das Institut LCM 25.11.2010 www.basg.at
Wirkstoffdokumentation & CEP- Verfahren Dr. Wolfgang Herzog AGES PharmMed, Institut Zulassung & Lifecycle Management AGES-Gespräch Am Pulsschlag des Arzneimittelwesens Das Institut LCM 25.11.2010 www.basg.at
Quality Control in the Synthesis of Atorvastatin Calcium Secondary Reference Standard Kits Help Generic Manufacturers Minimize Impurities
 Quality Control in the Synthesis of Atorvastatin Calcium Secondary Reference Standard Kits Help Generic Manufacturers Minimize Impurities by Steve Lane, NSF International When patent protection for Lipitor
Quality Control in the Synthesis of Atorvastatin Calcium Secondary Reference Standard Kits Help Generic Manufacturers Minimize Impurities by Steve Lane, NSF International When patent protection for Lipitor
CMC Considerations for 505(b)(2) Applications. Monica Cooper, Ph.D. FDA/CDER/OPS/ONDQA AAPS Annual Meeting Washington, D.C.
(2) Applications. Monica Cooper, Ph.D. FDA/CDER/OPS/ONDQA AAPS Annual Meeting Washington, D.C.") CMC Considerations for 505(b)(2) Applications Monica Cooper, Ph.D. FDA/CDER/OPS/ONDQA AAPS Annual Meeting Washington, D.C. October 2011 1 Introduction Outline Brief overview of FDA drug approval pathways
CMC Considerations for 505(b)(2) Applications Monica Cooper, Ph.D. FDA/CDER/OPS/ONDQA AAPS Annual Meeting Washington, D.C. October 2011 1 Introduction Outline Brief overview of FDA drug approval pathways
Workshop Summaries. Discussions of the Global Stability Workshop are summarized for the following topics:
 Workshop Summaries Discussions of the Global Stability Workshop are summarized for the following topics: QbD and Stability Program Dr. Alasandro Setting Specifications Dr. Gentry Challenges of Different
Workshop Summaries Discussions of the Global Stability Workshop are summarized for the following topics: QbD and Stability Program Dr. Alasandro Setting Specifications Dr. Gentry Challenges of Different
EMPROVE For Raw and Starting Materials & For Filtration Devices and Single Use Systems. Jan Thomsen Warsaw, November 15 th, 2016
 EMPROVE For Raw and Starting Materials & For Filtration Devices and Single Use Systems Jan Thomsen Warsaw, November 15 th, 2016 2 Content Emprove - An Introduction Emprove for Raw and Starting Materials
EMPROVE For Raw and Starting Materials & For Filtration Devices and Single Use Systems Jan Thomsen Warsaw, November 15 th, 2016 2 Content Emprove - An Introduction Emprove for Raw and Starting Materials
Validation/Verification of Test Methods An FDA Perspective. Laure H. Kairawicz, Ph.D. Senior Scientist Expert Witness
 Validation/Verification of Test Methods An FDA Perspective Laure H. Kairawicz, Ph.D. Senior Scientist Expert Witness FD & C Act Overview Definition of drugs What cgmps are Finished Pharmaceuticals cgmp
Validation/Verification of Test Methods An FDA Perspective Laure H. Kairawicz, Ph.D. Senior Scientist Expert Witness FD & C Act Overview Definition of drugs What cgmps are Finished Pharmaceuticals cgmp
ICH Q11 Questions & Answers Selection & Justification of Starting Materials. Q11 Implementation Working Group 22 May 2018
 IC Q11 Questions & Answers Selection & Justification of. Training Material Q11 Implementation Working Group 22 May 2018 International Council for armonisation of Technical Requirements for Pharmaceuticals
IC Q11 Questions & Answers Selection & Justification of. Training Material Q11 Implementation Working Group 22 May 2018 International Council for armonisation of Technical Requirements for Pharmaceuticals
Reference Standard Characterization. Steve Lane. General Manager, NSF Reference Standards.
 Reference Standard Characterization Steve Lane General Manager, NSF Reference Standards www.nsf-rs.org Regulatory Bodies will require that an Excipient: Be safe in the amount or dose used Meet applicable
Reference Standard Characterization Steve Lane General Manager, NSF Reference Standards www.nsf-rs.org Regulatory Bodies will require that an Excipient: Be safe in the amount or dose used Meet applicable
Experience with Health Canada s Approach for Post-Approval Changes. Kiran Krishnan Vice President US Regulatory Affairs September 2014
 Experience with Health Canada s Approach for Post-Approval Changes Kiran Krishnan Vice President US Regulatory Affairs September 2014 Important Quotes to consider Dr. Janet Woodcock on desired state: A
Experience with Health Canada s Approach for Post-Approval Changes Kiran Krishnan Vice President US Regulatory Affairs September 2014 Important Quotes to consider Dr. Janet Woodcock on desired state: A
Safety assessment of pharmaceutical impurities A reflection paper
 Safety assessment of pharmaceutical impurities A reflection paper Benoît Ruot, Ph.D., European Registered Toxicologist Associate Director, Non Clinical Safety Program Management All drugs contain more
Safety assessment of pharmaceutical impurities A reflection paper Benoît Ruot, Ph.D., European Registered Toxicologist Associate Director, Non Clinical Safety Program Management All drugs contain more
Question-based Review (QbR)
") Question-based Review (QbR) Rebecca L. Owen, Ph.D. Team Leader, Feed/Topical Team Division of Manufacturing Technologies ONADE/CVM/FDA Outline Background on CMC Filing Requirements What is QbR? QbR at
Question-based Review (QbR) Rebecca L. Owen, Ph.D. Team Leader, Feed/Topical Team Division of Manufacturing Technologies ONADE/CVM/FDA Outline Background on CMC Filing Requirements What is QbR? QbR at
Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials
 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE- GENERAL Health systems and products Medicinal products quality, safety and efficacy 18 February 2010 EMA/CHMP/BWP/534898/2008 Committee for Medicinal
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE- GENERAL Health systems and products Medicinal products quality, safety and efficacy 18 February 2010 EMA/CHMP/BWP/534898/2008 Committee for Medicinal
An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications
 An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications On behalf of Specifications Team Kirby Wong-Moon, Ph.D. Amgen Inc. Best Practices and Application of GMPs for
An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications On behalf of Specifications Team Kirby Wong-Moon, Ph.D. Amgen Inc. Best Practices and Application of GMPs for
ICH 교육가이드라인 [ 안전성 & 품질 ]
![ICH 교육가이드라인 [ 안전성 & 품질 ]](/thumbs/96/128946468.jpg "ICH 교육가이드라인 [ 안전성 & 품질 ]") ICH 교육가이드라인 [ 안전성 & 품질 ] 1 SAFETY ( 안전성 ) Safety Series / 11 월 27 일 ( 화 ) ICH S9 guideline is focused on anticancer drugs. However, for better understanding, the presentation will give the comparison with
ICH 교육가이드라인 [ 안전성 & 품질 ] 1 SAFETY ( 안전성 ) Safety Series / 11 월 27 일 ( 화 ) ICH S9 guideline is focused on anticancer drugs. However, for better understanding, the presentation will give the comparison with
GUIDANCE FOR QUALITY ASSESSORS DRUG SUBSTANCE. IGDRP Quality Working Group
 GUIDANCE FOR QUALITY ASSESSORS DRUG SUBSTANCE IGDRP Quality Working Group Version Description of Change Author Effective Date v 1.0 Original publication Quality WG June 8, 2017 Foreword In order to achieve
GUIDANCE FOR QUALITY ASSESSORS DRUG SUBSTANCE IGDRP Quality Working Group Version Description of Change Author Effective Date v 1.0 Original publication Quality WG June 8, 2017 Foreword In order to achieve
Documentation requirements for an initial consultation
 Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
European reflections on reviewing NDAs and ANDAs for ICH Q3D elemental impurity compliance
 European reflections on reviewing NDAs and ANDAs for ICH Q3D elemental impurity compliance Diana van Riet-Nales, Medicines Evaluation Board/NL NL CHMP/CVMP Quality Working Party delegate (humans) Former
European reflections on reviewing NDAs and ANDAs for ICH Q3D elemental impurity compliance Diana van Riet-Nales, Medicines Evaluation Board/NL NL CHMP/CVMP Quality Working Party delegate (humans) Former
Pharmacist Rana Musa Al-ali (Malkawi) MSc (Pharmaceutical Quality Assurance) Registration Department/JFDA
 MSc (Pharmaceutical Quality Assurance) Registration Department/JFDA") Pharmacist Rana Musa Al-ali (Malkawi) MSc (Pharmaceutical Quality Assurance) Registration Department/JFDA 1 2 ND MENA Regulatory Conference On Bioequivalence, Biowaivers, Bioanalysis, Dissolution & Biosimilars
Pharmacist Rana Musa Al-ali (Malkawi) MSc (Pharmaceutical Quality Assurance) Registration Department/JFDA 1 2 ND MENA Regulatory Conference On Bioequivalence, Biowaivers, Bioanalysis, Dissolution & Biosimilars
Analytical Procedures and Methods Validation for Drugs and Biologics
 Final Guidance for Industry Analytical Procedures and Methods Validation for Drugs and Biologics Analytical procedures and Method Validation June 21, 2016 Lokesh Bhattacharyya Chief, LACBRP/DBSQC OCBQ/CBER/FDA
Final Guidance for Industry Analytical Procedures and Methods Validation for Drugs and Biologics Analytical procedures and Method Validation June 21, 2016 Lokesh Bhattacharyya Chief, LACBRP/DBSQC OCBQ/CBER/FDA
Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials
 1 2 3 23 June 2016 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 7 Guideline on the requirements for quality documentation concerning biological investigational
1 2 3 23 June 2016 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 7 Guideline on the requirements for quality documentation concerning biological investigational
WHO guidelines on variations to a prequalified product
 Annex 3 WHO guidelines on variations to a prequalified product Introduction 96 1. Background 97 1.1 Objectives 97 1.2 Scope and application 98 2. Guidance for implementation 99 2.1 s 99 2.1.1 Notifications
Annex 3 WHO guidelines on variations to a prequalified product Introduction 96 1. Background 97 1.1 Objectives 97 1.2 Scope and application 98 2. Guidance for implementation 99 2.1 s 99 2.1.1 Notifications
API Stability Protocols and. Chris Byrne Tasmanian Alkaloids
 API Stability Protocols and Evaluations Chris Byrne Tasmanian Alkaloids API Stability Overview APIs = 100% pure Limited (if any) degradation No interactions with other agents in drug products Less likelihood
API Stability Protocols and Evaluations Chris Byrne Tasmanian Alkaloids API Stability Overview APIs = 100% pure Limited (if any) degradation No interactions with other agents in drug products Less likelihood
Annex 1. Good pharmacopoeial practices
 Annex 1 Good pharmacopoeial practices 1. Background 68 2. Purpose and scope of good pharmacopoeial practices 69 3. Glossary 69 4. Benefits of good pharmacopoeial practices 70 5. Implementation 70 6. Monograph
Annex 1 Good pharmacopoeial practices 1. Background 68 2. Purpose and scope of good pharmacopoeial practices 69 3. Glossary 69 4. Benefits of good pharmacopoeial practices 70 5. Implementation 70 6. Monograph
Implementation strategy of ICH Q3D guideline
 1 2 3 1 July 2016 EMA/404489/2016 Committee for Medicinal Products for Human use (CHMP) 4 5 Draft Draft agreed by QWP and BWP June 2016 Adopted by CHMP for release for consultation June 2016 Start of public
1 2 3 1 July 2016 EMA/404489/2016 Committee for Medicinal Products for Human use (CHMP) 4 5 Draft Draft agreed by QWP and BWP June 2016 Adopted by CHMP for release for consultation June 2016 Start of public
International Pharmaceutical Excipients Council Of The Americas
 October 6, 2008 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Subject: FDA Draft Guidance for Industry, Residual Solvents in Drug
October 6, 2008 Division of Dockets Management (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Subject: FDA Draft Guidance for Industry, Residual Solvents in Drug
5. Changes to a CEP or to a confirmation of API-prequalification document 106
 Annex 3 WHO guidelines on variations to a prequalified product Introduction 96 1. Background 97 1.1 Objectives 97 1.2 Scope and application 98 2. Guidance for implementation 99 2.1 s 99 2.1.1 Notifications
Annex 3 WHO guidelines on variations to a prequalified product Introduction 96 1. Background 97 1.1 Objectives 97 1.2 Scope and application 98 2. Guidance for implementation 99 2.1 s 99 2.1.1 Notifications
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX BATCH Q: Quality Q1A(R2) Stability Testing of New Drug Substances
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX BATCH Q: Quality Q1A(R2) Stability Testing of New Drug Substances
Interested parties (organisations or individuals) that commented on the draft document as released for consultation.
 that commented on the draft document as released for consultation.") 20 September 2017 EMA/CHMP/QWP/546045/2017 Overview of comments received on 'Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal
20 September 2017 EMA/CHMP/QWP/546045/2017 Overview of comments received on 'Guideline on the requirements for the chemical and pharmaceutical quality documentation concerning investigational medicinal
How we set specifications for impurities (including Genotoxic impurities) 24 May 2017 Elisabeth Kovacs, Apotex CSO Chemistry and Analytical Sci.
 24 May 2017 Elisabeth Kovacs, Apotex CSO Chemistry and Analytical Sci.") 2017 AAM CMC Workshop How we set specifications for impurities (including Genotoxic impurities) 24 May 2017 Elisabeth Kovacs, Apotex CSO Chemistry and Analytical Sci. The information within this presentation
2017 AAM CMC Workshop How we set specifications for impurities (including Genotoxic impurities) 24 May 2017 Elisabeth Kovacs, Apotex CSO Chemistry and Analytical Sci. The information within this presentation
PHARMACEUTICAL TESTING
 WHITEHOUSE, NJ PHARMACEUTICAL TESTING Pharmaceutical Expertise for GMP & CMC Testing Our Pharmaceutical Expertise With more than 20 years of experience in a variety of industries, our Whitehouse, New Jersey
WHITEHOUSE, NJ PHARMACEUTICAL TESTING Pharmaceutical Expertise for GMP & CMC Testing Our Pharmaceutical Expertise With more than 20 years of experience in a variety of industries, our Whitehouse, New Jersey
ICH Quality Implementation Working Group POINTS TO CONSIDER
 ICH Quality Implementation Working Group POINTS TO CONSIDER ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation Document date: 16 June 2011 International Conference on Harmonisation of Technical Requirements
ICH Quality Implementation Working Group POINTS TO CONSIDER ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation Document date: 16 June 2011 International Conference on Harmonisation of Technical Requirements
Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials
 14 September 2017 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) Guideline on the requirements for quality documentation concerning biological investigational medicinal
14 September 2017 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) Guideline on the requirements for quality documentation concerning biological investigational medicinal
The European Pharmacopoeia
 The European Pharmacopoeia General methods, Control of impurities, FP monographs, Pharmacopoeial harmonisation International workshop on the Chinese and the European Pharmacopoeias The new editions Dr.
The European Pharmacopoeia General methods, Control of impurities, FP monographs, Pharmacopoeial harmonisation International workshop on the Chinese and the European Pharmacopoeias The new editions Dr.
Guidance Document 01 January 2016 CONTENTS. 1. Introduction Background 1.2. Objectives 1.3. Scope and application 1.4 APIMF holder obligations
 GUIDANCE ON AMENDMENTS TO AN ACTIVE PHARMACEUTICAL INGREDIENT MASTER FILE (APIMF) SUBMITTED IN SUPPORT OF A PREQUALIFIED PHARMACEUTICAL PRODUCT (FPP) OR PREQUALIFIED ACTIVE PHARMACEUTICAL INGREDIENT (API)
GUIDANCE ON AMENDMENTS TO AN ACTIVE PHARMACEUTICAL INGREDIENT MASTER FILE (APIMF) SUBMITTED IN SUPPORT OF A PREQUALIFIED PHARMACEUTICAL PRODUCT (FPP) OR PREQUALIFIED ACTIVE PHARMACEUTICAL INGREDIENT (API)
Setting Specifications for Biotech Products
 Setting Specifications for Biotech Products Session 1: What to Control? Presentation by an EU Regulator Nanna Aaby Kruse, Senior Biological Assessor, member of BWP and BMWP WHAT TO CONTROL? Control of
Setting Specifications for Biotech Products Session 1: What to Control? Presentation by an EU Regulator Nanna Aaby Kruse, Senior Biological Assessor, member of BWP and BMWP WHAT TO CONTROL? Control of
Strategies for IND Filing Success: Chemistry, Manufacturing and Controls
 Strategies for IND Filing Success: Chemistry, Manufacturing and Controls October 21, 2016 Presented by: Sharon Ayd, Ph.D., MBA Chief Scientific Officer and SVP, Pharmaceuticals document contains proprietary
Strategies for IND Filing Success: Chemistry, Manufacturing and Controls October 21, 2016 Presented by: Sharon Ayd, Ph.D., MBA Chief Scientific Officer and SVP, Pharmaceuticals document contains proprietary
COMMON DEFICIENCIES IN FINISHED PHARMACEUTICAL PRODUCT (FPP) DOSSIERS
 DOSSIERS") COMMON DEFICIENCIES IN FINISHED PHARMACEUTICAL PRODUCT (FPP) DOSSIERS Additional guidance for manufacturers This note identifies the most common quality related deficiencies in recently assessed dossiers
COMMON DEFICIENCIES IN FINISHED PHARMACEUTICAL PRODUCT (FPP) DOSSIERS Additional guidance for manufacturers This note identifies the most common quality related deficiencies in recently assessed dossiers
Regulatory Perspective on Analytical Method Validation During Product Development
 Regulatory Perspective on Analytical Method Validation During Product Development CASSS CMC Strategy Forum 2018 Jacek Cieslak CDER/OPQ/OBP FDA Disclaimer This presentation reflects the views of the author
Regulatory Perspective on Analytical Method Validation During Product Development CASSS CMC Strategy Forum 2018 Jacek Cieslak CDER/OPQ/OBP FDA Disclaimer This presentation reflects the views of the author
Future of Question-based Review and Regulatory Submissions
 Future of Question-based Review and Regulatory Submissions Robert Iser Associate Director for Policy Development (Acting) Office of Pharmaceutical Science / CDER / FDA FDA/PQRI Conference on Evolving Product
Future of Question-based Review and Regulatory Submissions Robert Iser Associate Director for Policy Development (Acting) Office of Pharmaceutical Science / CDER / FDA FDA/PQRI Conference on Evolving Product
Impurity Control in the European Pharmacopoeia
 Impurity Control in the European Pharmacopoeia Training Session Zagreb, Croatia, 24-25 May, 2018 Dr Ulrich Rose Head of Division European Pharmacopoeia Department, EDQM Agenda Which impurities are controlled?
Impurity Control in the European Pharmacopoeia Training Session Zagreb, Croatia, 24-25 May, 2018 Dr Ulrich Rose Head of Division European Pharmacopoeia Department, EDQM Agenda Which impurities are controlled?
Consultation by a notified body on an ancillary medicinal substance integrated in a medical device
 Consultation by a notified body on an ancillary medicinal substance integrated in a medical device Documentation requirements for an initial consultation Language : French or English Because of the wide
Consultation by a notified body on an ancillary medicinal substance integrated in a medical device Documentation requirements for an initial consultation Language : French or English Because of the wide
ICH Q3D. Questions & Answers Q1: Q2: U n d e r s ta n d i n g the Risk Assessment Requirements. Knowing the Options
 ICH Q3D Questions & Answers Understanding the Risk Assessment Requirements Knowing the Options Controlling for Elemental Impurities Responsibilities U n d e r s ta n d i n g the Risk Assessment Requirements
ICH Q3D Questions & Answers Understanding the Risk Assessment Requirements Knowing the Options Controlling for Elemental Impurities Responsibilities U n d e r s ta n d i n g the Risk Assessment Requirements
Recent experience in scientific advice and marketing authorisations
 Recent experience in scientific advice and marketing authorisations Presented by Brigitte Brake on 16 April 2015 BfArM & BWP, Germany An agency of the European Union Introduction Short introduction to
Recent experience in scientific advice and marketing authorisations Presented by Brigitte Brake on 16 April 2015 BfArM & BWP, Germany An agency of the European Union Introduction Short introduction to
Japanese Pharmacopoeia s Challenge to the Globalization
 Japanese Pharmacopoeia s Challenge to the Globalization Naoyuki Yabana, Ph.D. Division of Pharmacopoeia and Standards for Drugs, Office of Standards and Guidelines Development, Pharmaceuticals and Agency
Japanese Pharmacopoeia s Challenge to the Globalization Naoyuki Yabana, Ph.D. Division of Pharmacopoeia and Standards for Drugs, Office of Standards and Guidelines Development, Pharmaceuticals and Agency
Introduction to CMC Regulatory Affairs
 Introduction to CMC Regulatory Affairs Bharathi Mamidipudi Regulatory Affairs Consultant II Syner-G Pharma Consulting, LLC Northeastern University, Boston November 10, 2016 My Background Experience ~4
Introduction to CMC Regulatory Affairs Bharathi Mamidipudi Regulatory Affairs Consultant II Syner-G Pharma Consulting, LLC Northeastern University, Boston November 10, 2016 My Background Experience ~4
Acceptable Intake (AI) and Permitted Daily Exposure (PDE) Data Sharing Project for Pharmaceutical Impurities
 and Permitted Daily Exposure (PDE) Data Sharing Project for Pharmaceutical Impurities") Acceptable Intake (AI) and Permitted Daily Exposure (PDE) Data Sharing Project for Pharmaceutical Impurities Dr. William Drewe Business Development Manager David Wilkinson Senior Data Scientist Agenda
Acceptable Intake (AI) and Permitted Daily Exposure (PDE) Data Sharing Project for Pharmaceutical Impurities Dr. William Drewe Business Development Manager David Wilkinson Senior Data Scientist Agenda
Drug Impurities: The Good, Bad and Ugly
 Drug Impurities: The Good, Bad and Ugly Joel Bercu, PhD, DABT, MPH Associate Director, Environmental and Occupational Toxicology Gilead Sciences jbercu@gilead.com Medicines Impurities 2015-2016 GrimsrudBerkowitz
Drug Impurities: The Good, Bad and Ugly Joel Bercu, PhD, DABT, MPH Associate Director, Environmental and Occupational Toxicology Gilead Sciences jbercu@gilead.com Medicines Impurities 2015-2016 GrimsrudBerkowitz
GUIDANCE ON VARIATIONS TO A PREQUALIFIED DOSSIER
 RESTRICTED GUIDANCE ON VARIATIONS TO A PREQUALIFIED DOSSIER Since being presented to the Fortieth meeting of the WHO Expert Committee on Specifications for Pharmaceutical Preparations in October 2005 this
RESTRICTED GUIDANCE ON VARIATIONS TO A PREQUALIFIED DOSSIER Since being presented to the Fortieth meeting of the WHO Expert Committee on Specifications for Pharmaceutical Preparations in October 2005 this
EU Regulatory Perspective and Expectations. Sven-Erik Hillver Senior expert, QWP delegate Medical Products Agency, Sweden
 EU Regulatory Perspective and Expectations Sven-Erik Hillver Senior expert, QWP delegate Medical Products Agency, Sweden Disclaimer EU regulators still have to build up an experience of applications based
EU Regulatory Perspective and Expectations Sven-Erik Hillver Senior expert, QWP delegate Medical Products Agency, Sweden Disclaimer EU regulators still have to build up an experience of applications based
Current version 13 October 2016
 Implementation Working Group ICH Q11 Guideline: DEVELOPMENT AND MANUFACTURE OF DRUG SUBSTANCES (CHEMICAL ENTITIES AND BIOTECHNOLOGICAL/BIOLOGICAL ENTITIES) and Current version 13 October 2016 In order
Implementation Working Group ICH Q11 Guideline: DEVELOPMENT AND MANUFACTURE OF DRUG SUBSTANCES (CHEMICAL ENTITIES AND BIOTECHNOLOGICAL/BIOLOGICAL ENTITIES) and Current version 13 October 2016 In order
Content of the dossier for chemical purity and microbiological quality
 Division Certification of Substances CP/CB PUBLIC DOCUMENT (Level 1) English only/anglais seulement PA/PH/ CEP (0) 1 R February 0 Certification of suitability of Monographs of the European Pharmacopoeia
Division Certification of Substances CP/CB PUBLIC DOCUMENT (Level 1) English only/anglais seulement PA/PH/ CEP (0) 1 R February 0 Certification of suitability of Monographs of the European Pharmacopoeia
Harmonized Quality Requirements for Biotech Investigational Medicinal Products - A Regulator's View
 Harmonized Quality Requirements for Biotech Investigational Medicinal Products - A Regulator's View Brigitte Brake Pharmaceutical Biotechnology BfArM, Bonn Evaluation Criteria National vs. EU Clinical
Harmonized Quality Requirements for Biotech Investigational Medicinal Products - A Regulator's View Brigitte Brake Pharmaceutical Biotechnology BfArM, Bonn Evaluation Criteria National vs. EU Clinical
Control strategy and validation. Emanuela Lacana PhD Office of Biotechnology Products CDER/FDA
 Control strategy and validation Emanuela Lacana PhD Office of Biotechnology Products CDER/FDA 1 Disclaimer The views and opinions expressed in this presentation are those of the speaker and should not
Control strategy and validation Emanuela Lacana PhD Office of Biotechnology Products CDER/FDA 1 Disclaimer The views and opinions expressed in this presentation are those of the speaker and should not
PHARMA KNOWLEDGE PARK
 GUIDELINES 2013 WWW.NOORMD.COM A COMPILATION BY ON PHARMACEUTICAL REGULATORY GUIDELINES INTRODUCED / REVISED DURING THE YEAR 2013. Page 2 ICH Assessment and control of DNA reactive (mutagenic) impurities
GUIDELINES 2013 WWW.NOORMD.COM A COMPILATION BY ON PHARMACEUTICAL REGULATORY GUIDELINES INTRODUCED / REVISED DURING THE YEAR 2013. Page 2 ICH Assessment and control of DNA reactive (mutagenic) impurities
Pharmacopoeial Reference Standards
 Pharmacopoeial Reference Standards Industry view point Antony Raj Gomes Head- Quality Management 1 Presentation Overview Indian Industry an update Available guidelines Industry practices Current challenges
Pharmacopoeial Reference Standards Industry view point Antony Raj Gomes Head- Quality Management 1 Presentation Overview Indian Industry an update Available guidelines Industry practices Current challenges
Regulatory Review Considerations of Drug-Linker Quality in ADCs
 Regulatory Review Considerations of Drug-Linker Quality in ADCs Xiao Hong Chen, Ph.D. Acting Quality Assessment Lead Division of New Drug Products I, Branch II ONDP/OPQ/CDER/FDA Outlines ADC IND submissions
Regulatory Review Considerations of Drug-Linker Quality in ADCs Xiao Hong Chen, Ph.D. Acting Quality Assessment Lead Division of New Drug Products I, Branch II ONDP/OPQ/CDER/FDA Outlines ADC IND submissions
On the Q&A to the Guideline for Common Technical Documents
 To: Public Health Bureau Prefectural Government Letter from PFSB/ELD 22 nd October 2001 From: Evaluation & Licensing Division, Pharmaceutical & Food Safety Bureau, The Ministry of Health, Labour and Welfare
To: Public Health Bureau Prefectural Government Letter from PFSB/ELD 22 nd October 2001 From: Evaluation & Licensing Division, Pharmaceutical & Food Safety Bureau, The Ministry of Health, Labour and Welfare
Implementation of the ICH Q3D guideline in the Ph. Eur.
 Implementation of the ICH Q3D guideline in the Ph. Eur. PQRI/USP Workshop, USP Meeting center 2-3 November 2017 Mr Bruno Spieldenner, European Pharmacopoeia, EDQM Elemental impurities in the Ph. Eur. A
Implementation of the ICH Q3D guideline in the Ph. Eur. PQRI/USP Workshop, USP Meeting center 2-3 November 2017 Mr Bruno Spieldenner, European Pharmacopoeia, EDQM Elemental impurities in the Ph. Eur. A
ICH Q11 Development & manufacture of drug substances
 ICH Q11 Development & manufacture of drug substances Keith McDonald Group Manager Medicines & Healthcare products Regulatory Agency (UK) 2011 ICH International Conference on Harmonisation of Technical
ICH Q11 Development & manufacture of drug substances Keith McDonald Group Manager Medicines & Healthcare products Regulatory Agency (UK) 2011 ICH International Conference on Harmonisation of Technical
Implementation of the ICH Q3D guideline in the Ph. Eur.
 Implementation of the ICH Q3D guideline in the Ph. Eur. PQRI/USP Workshop, USP Meeting center 9-10 November 2016 Bruno Spieldenner, Ph. Eur. division, EDQM Elemental impurities in the Ph. Eur. A (r)evolution
Implementation of the ICH Q3D guideline in the Ph. Eur. PQRI/USP Workshop, USP Meeting center 9-10 November 2016 Bruno Spieldenner, Ph. Eur. division, EDQM Elemental impurities in the Ph. Eur. A (r)evolution
Commonly Seen Drug Product Related Quality Deficiencies
 Commonly Seen Drug Product Related Quality Deficiencies 2016 GPhA CMC Workshop Bethesda, MD; May 18, 2016 Geoffrey Wu, PhD, CPH Lieutenant, US Public Health Service Associate Director for Science & Communication
Commonly Seen Drug Product Related Quality Deficiencies 2016 GPhA CMC Workshop Bethesda, MD; May 18, 2016 Geoffrey Wu, PhD, CPH Lieutenant, US Public Health Service Associate Director for Science & Communication