Klinisk kemisk diagnostik BIOINFORMATICS
|
|
|
- Amberlynn Shonda Hawkins
- 5 years ago
- Views:
Transcription
1 Klinisk kemisk diagnostik BIOINFORMATICS
2 What is bioinformatics? Bioinformatics: Research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, organize, archive, analyze, or visualize such data. (definition Committee, National Institute of Mental Health) In general, Bioinformatics is the sum of the computational approaches to analyze, manage, and store biological data. Includes the usage of statistical techniques, applied mathematics and the development of different algorithms. Bioinformatics is used in analyzing genomes, proteomes (protein sequences), three-dimensional modeling of biomolecules and biologic systems, etc.
 Atlas of protein sequences 1981 Smith-Waterman algorithm for sequence alignment 1982 GeneBank release 3 - public 1988 National Center for Biotechnology")
3 Milestones in Bioinformatics 1955 F. Sanger First protein sequence (insulin) 1965 M. Dayhoff ( mother of bioinformatics ) Atlas of protein sequences 1981 Smith-Waterman algorithm for sequence alignment 1982 GeneBank release 3 - public 1988 National Center for Biotechnology Information (NCBI) 1994 EMBL - European Bioinformatics Institute 1986 SWISS- PROT 1995 First bacterial genome 2001 Publication of the Human genome Needleman-Wunsch algorithm for sequence comparison 1977 DNA sequencing and software to analyze it (Staden) 1982 Phage lambda genome sequenced 1988 FASTA algorithm 1990 BLAST fast sequence similarity search 1999 First human chromosome sequenced 1996 Yeast genome 2003 Human Genome Project completed

4 Number of bioinformatics related publications in PubMed 18000" 16000" number'of'ar+cles'in'pubmed' 14000" 12000" 10000" 8000" 6000" 4000" 2000" 0" 1984" 1986" 1987" 1988" 1989" 1990" 1991" 1992" 1993" 1994" 1995" 1996" 1997" 1998" 1999" 2000" 2001" 2002" 2003" 2004" 2005" 2006" 2007" 2008" 2009" 2010" 2011" 2012" 2013" year'
5 The aims of bioinformatics The primary goal is to increase understanding of biological processes. Development of new algorithms, statistical measures and computer programs for the evaluation of large datasets. (tex. methods to locate genes within a sequence, predict protein structure from sequence, etc.) Implementation of the developed algorithms, programs in data evaluation and interpretation of the results. Construction and improvement of publicly available databases.
6 Types of biological information and bioinformatics methods Origin Size Bioinformatics areas DNA sequences Protein sequences Macromolecular structures Genomes Gene expression data 175 million sequences 180 billion bases 45 million sequences structures 9000 genomes (178 eukaryotic) different time points/ treatments for a number of genes of different organisms - sequence alignment, genome assembly - gene prediction, genome annotation - sequence alignment - identification of conserved sequence motifs - 3D structure alignment, prediction - molecule modeling - interaction prediction - phylogenetic analysis - genome-wide association studies - oncogenomics - expression pattern recognition, clustering, disease relations - correlation between gene and protein expression
7 Classification and homology Based on similarity a huge part of information can be sorted out into groups. This is the basis for several bioinformatics methods. Examples: Repetitive sequences in the genome Gene classification based on function Sequence similarity of different proteins A limited number of protein structures are exist Homolog general term, indicates genes or proteins that are evolutionary related (can be either orthologs or paralogs ) Ortholog for orthologs (ortho=exact), the homology is the result of speciation, i.e. same exact gene in different organisms Paralog for paralogs (para=in parallel), the homology is the result of a gene duplication, i.e. similar proteins, potentially within the same organism

8 Bioinformatics areas
9 Genomics shotgun sequencing, sequence assembly gene prediction phylogenetic analysis genome-wide association studies Gene expression analysis Proteomics structure prediction Biological networks

10 Genomics I. shotgun sequencing, sequence assembly Shotgun sequencing is used for sequencing long DNA strands. DNA is broken up randomly into numerous small segments, which are sequenced. After several rounds of fragmentation and sequencing computer programs are used to assemble the overlapping ends of different reads into a continuous sequence. Genome assembly is a difficult computational problem, it works by taking all the pieces and aligning them to one another, and detecting all places where two of the short sequences, or reads, overlap. These overlapping reads can be merged, and the process continues. Repeats (large numbers of identical sequences) in the genomes make gene assembly more difficult. Shotgun sequencing was one of the technologies that was responsible for enabling full genome sequencing.
11 Genomics II. gene prediction The process of identifying the regions of genomic DNA that encode genes. (includes protein-coding genes, RNA genes and other functional elements such as regulatory regions) Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced. Important steps: filtering out of non-coding regions and repeats detection of functional places (pattern recognition) like initiation, termination etc. detection of open reading frame Methods: empirical methods ab initio methods combined methods Gene prediction software: GLIMMER (for prokaryotes) ( GeneMark ( GENSCAN ( Augustus ( mgene ( StarORF (
12 ab initio gene prediction is an intrinsic method based on gene content and signal detection. Prokaryotes Eukaryotes genes have specific and wellunderstood promoter sequences the sequence coding for a protein occurs as one contiguous open reading frame (ORF) with a lengths of many hundred or thousands of base pairs protein-coding DNA has certain periodicities and other statistical properties promoter and other regulatory signals are more complex and less well-understood (two classic examples are CpG islands and binding sites for a poly(a) tail) a particular protein-coding sequence is divided into several parts (exons), separated by noncoding sequences (introns) (splicing)
13 Genomics III. genome-wide association studies Genome-wide association studies (GWAS) are a relatively new way to identify genes involved in human disease. GWAS typically focuses on single nucleotide polymorphisms (SNPs) that occur more frequently in people with a particular disease than in people without the disease. It is a non-candidate driven approach, since it investigates the entire genome. compares two large groups of individuals, one healthy control group and one case group affected by a disease all individuals are genotyped for the majority of common known SNPs the odds ratio is calculated (ratio of the odds of disease for individuals having a specific allele and the odds of disease for individuals who do not have that same allele) p-value for the significance of the odds ratio is calculated (chi-squared test) Odds ratio that is significantly differ from 1 shows that a SNP is associated with the disease.

14 The graphical interpretation of the GWAS results is Manhattan plot. The plot shows the negative logarithm of the P-value as a function of genomic location. GWA studies focus only on common genetic variants, since their assumption is that common genetic variation plays a large role in explaining the heritable variation of common disease. GWA studies typically perform the first analysis in a discovery cohort, followed by validation of the most significant SNPs in an independent validation cohort.
15 Gene expression analysis Gene expression profiling is the measurement of the expression of thousands of genes simultaneously, to create a global picture of cellular function. The sequence tells us what the cell could possibly do, while the expression profile tells us what it is actually doing at a particular time point. Techniques for gene expression measurement DNA microarray - measures the relative activity of previously identified target genes serial analysis of gene expression (SAGE) - produce a snapshot of the mrna population in the sample in the form of small tags that correspond to fragments of those transcripts RNA-seq (RNA sequencing) - uses the capabilities of nextgeneration sequencing to reveal a snapshot of RNA presence and quantity at a given time point
 attached to a solid surface.")


16 DNA microarrays are used to measure the expression levels of large numbers of genes simultaneously. A DNA chip is a collection of microscopic DNA spots (short gene sections) attached to a solid surface. Each spot contains a specific DNA sequence (probes). The probes are used to hybridize with a labeled cdna sample. Probe-target hybridization is detected and quantified by detection of the labeled targets. Comparing gene expression of two samples mrna present only in the control sample mrna present only in the treated sample mrna equally expressed in both samples genes transcribed in control cells genes transcribed equally in both cells low gene expression genes transcribed in treated cells

17
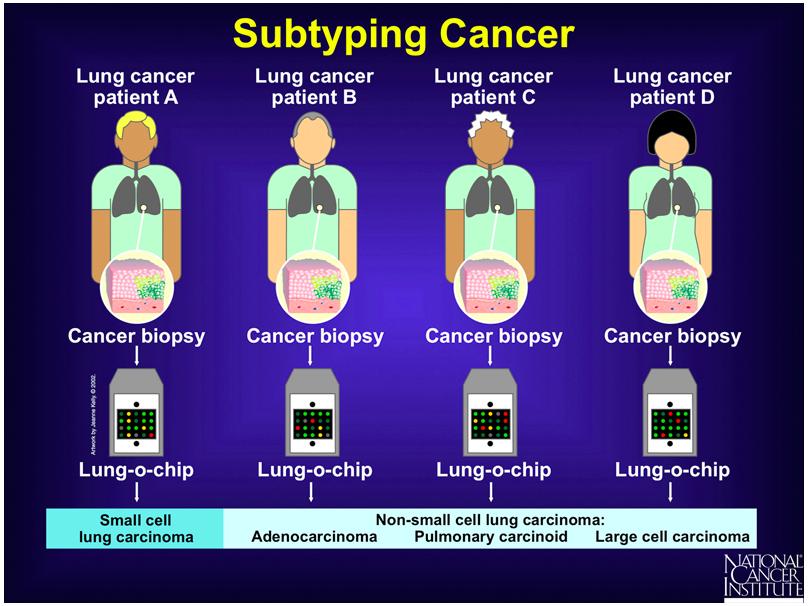
18
. The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded.")
19 Proteomics structure prediction Each protein exists as an unfolded polypeptide or random coil when translated. Then it folds into a characteristic and functional threedimensional structure. 3D structure is determined by the AA sequence (Anfinsen's dogma). The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded. Failure to fold into the intended shape usually produces inactive proteins. Neurodegenerative diseases are resulted from the accumulation of misfolded (incorrectly folded) proteins. (Alzheimer s, Parkinson s diseases) Many allergies are caused by the folding of the proteins, for the immune system does not produce antibodies. Folding

20 Levels of protein structure level description stabilized by primary amino acid sequence peptide bonds secondary formation of αhelices and βsheets in a polypeptide hydrogen bonds between groups along the peptide backbone tertiary overall threedimensional shape of a polypeptide interactions between R-groups, and Rgroups and peptide backbone quaternary shape produced by combinations of polypeptides interactions between R-groups and between peptide backbones of different polypeptides
21 Secondary structure prediction is aimed to predict the local secondary structures of proteins based only on knowledge of their amino acid sequence. The prediction consists of assigning regions of the amino acid sequence as likely alpha helices, beta strands, or turns. Specialized algorithms have been developed for the detection of specific well-defined patterns such as transmembrane helices and coiled coils in proteins. The best modern methods of secondary structure prediction in proteins reach about 80% accuracy. Online prediction tools: PsiPred server CFSSP server
22 Tertiary structure prediction is even more challenging and remains extremely difficult. 1. Comparative protein modeling It uses previously solved structures as starting points, or templates. a. Homology modeling a method, where a known homologous is used to predict the structure of a new protein. b. Protein threading (fold recognition) - a method to model those proteins which have the same fold as proteins of known structures, but do not have a known homologous. 2. De novo physics-based modeling It is an algorithmic process by which protein tertiary structure is predicted from the amino acid sequence (primary structure). Prediction is based on general principles that direct protein folding energetics and/or statistical tendencies of conformational features, without the use of explicit templates. Database: Software tools: Protein Data Bank (PDB) SWISS-MODEL homology modeling RAPTOR protein threading software I-TASSER fold recognition method ROBETTA ab initio modeling
 Modeling biological systems is a significant task of systems biology.")
23 Biological networks Complex biological systems can be represented and analyzed as computable networks. (ecosystems can be modeled as networks of interacting species or a protein can be modeled as a network of amino acids) Modeling biological systems is a significant task of systems biology. Computational systems biology aims to develop and use efficient algorithms, data structures, visualization and communication tools for modeling of biological systems. Basic components of a network: nodes: units in the network edges: interactions between the units edge node Important properties of a network: degree (or connectivity): the number of edges that connect a node betweenness: a measure of how central a node is in a network
24 Interactome Molecular interactions can occur between molecules belonging to different biochemical families (proteins, nucleic acids, carbohydrates, lipids, etc.) and also within a given family. Whenever such molecules are connected by physical interactions, they form molecular interaction networks. protein protein interaction network gene-regulatory network (protein DNA interaction) - formed by transcription factors, chromatin regulatory proteins, and their target genes metabolic networks - metabolites, i.e. chemical compounds in a cell, are converted into each other by enzymes signaling networks Interactome mapping Experimental methods from experimental data such as affinity purification Predicting PPIs - interactome from one organism are used to predict interactions among homologous proteins in another organism Text mining of PPIs systematic extraction of interaction networks directly from the scientific literature
 KEGG PATHWAY Database (Univ.")
, New York University Medical Centre")
25 Network and pathway databases STRING - a database of known and predicted protein-protein interactions (EMBL) KEGG PATHWAY Database (Univ. of Kyoto) Reactome - human biological pathways, ranging from metabolic processes to hormonal signalling (Ontario Institute for Cancer Research (OICR), New York University Medical Centre (NYUMC), European Bioinformatics Institute (EBI))

26 Bioinformatics in practice
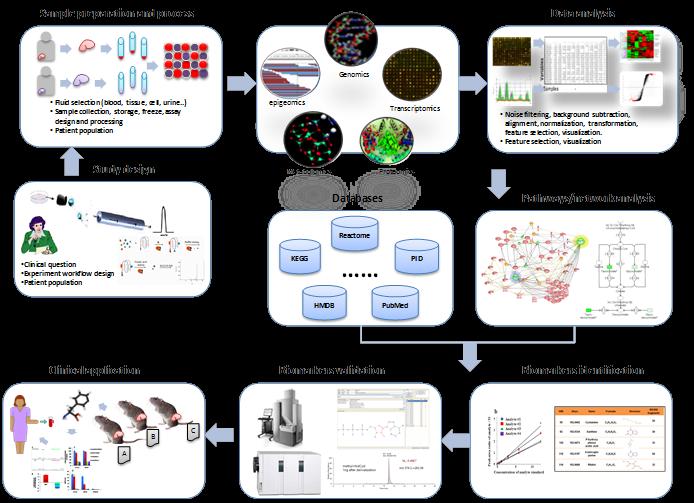
27


28 Databases Databases are essential for bioinformatics research and applications. There are a huge number of available databases covering almost everything from DNA and protein sequences, molecular structures, to phenotypes and biodiversity. There are meta-databases that incorporate data compiled from multiple other databases. Some others are specialized, such as those specific to an organism. Interconnectivity in between the different databases is essential. Bioinformatics organizations NCBI National Center for Biotechnology Information EMBL-EBI European Molecular Biology Laboratory European Bioinformatics Institute SIB Swiss Institute of Bioinformatics These centers host a number of publicly open, free to use life science resources, including biomedical databases and analysis tools.

29 Bibliographic database MEDLINE PubMed free search engine comprises more than 24 million citations for biomedical literature

30 Databases

31 GeneBank 5.00E+08' GeneBank'0'Sequences' 5.00E+07' 5.00E+06' 5.00E+05' 5.00E+04' 5.00E+03' 5.00E+02' Dec082' Apr084' Aug085' Dec086' Apr088' Aug089' Dec090' Apr092' Aug093' Dec094' Apr096' Aug097' Dec098' Apr000' Aug001' Dec002' Apr004' Aug005' Dec006' Apr008' Aug009' Dec010' Apr012' Aug013'



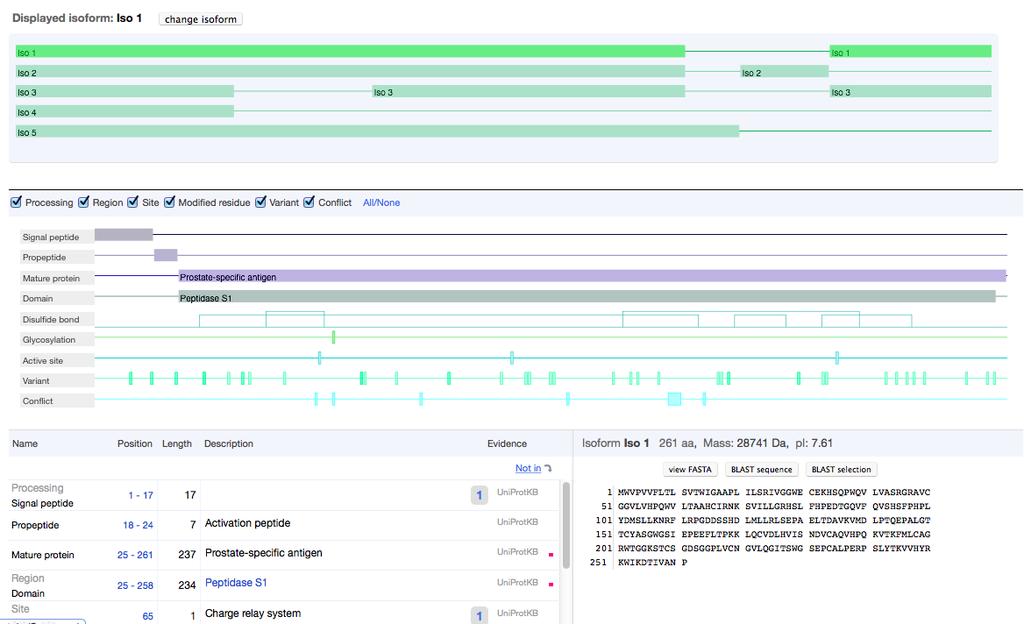
32 UniProtKB The mission of UniProt is to provide the scientific community with a comprehensive, high-quality and freely accessible resource of protein sequence and functional information.
33

34 nextprot nextprot is developed in collaboration between the SIB Swiss Institute of Bioinformatics and Geneva Bioinformatics (GeneBio) SA. nextprot will be a comprehensive human-centric discovery platform, offering its users a perfect integration of protein-related data.
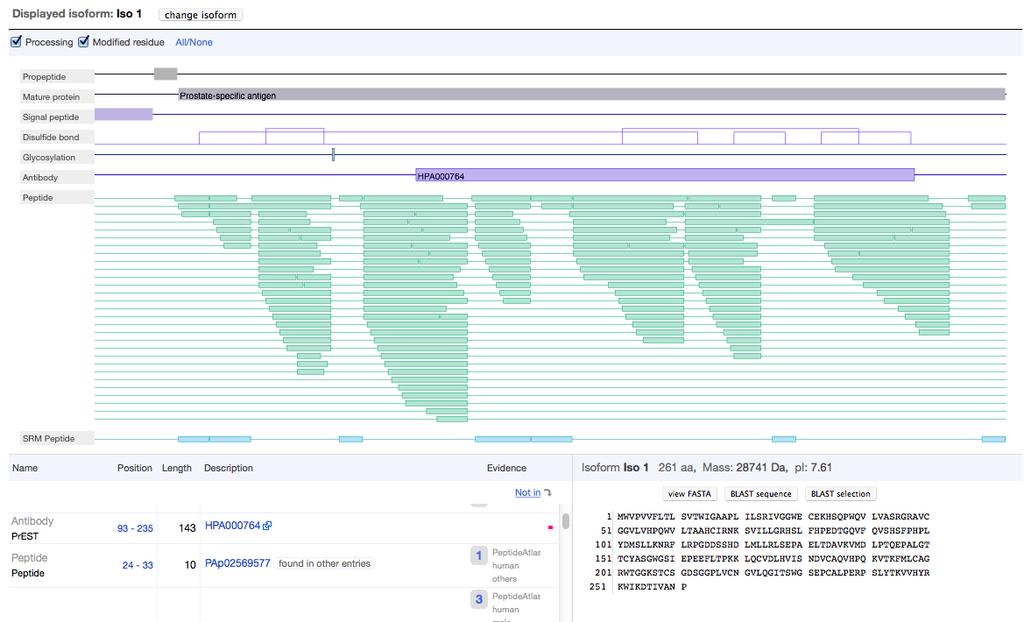
35


36 Thank You!