Alignment & Variant Discovery. J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014
|
|
|
- Roderick McKinney
- 5 years ago
- Views:
Transcription
1 Alignment & Variant Discovery J Fass UCD Genome Center Bioinformatics Core Tuesday June 17, 2014
2 From reads to molecules

3 Why align? Individual A Individual B ATGATAGCATCGTCGGGTGTCTGCTCAATAATAGTGCCGTATCATGCTGGTGTTATAATCGCCGCATGACATGATCAATGG CAATAAAAGTGCCGTATCATGCTGGTGTTACAATCGCCGCA CGTATCATGCTGGTGTTACAATCGCCGCATGACATGATCAATGG TGTCTGCTCAATAAAAGTGCCGTATCATGCTGGTGTTACAATC ATCGTCGGGTGTCTGCTCAATAAAAGTGCCGTATCATG--GGTGTTATAA CTCAATAAGAGTGCCGTATCATG--GGTGTTATAATCGCCGCA GTTATAATCGCCGCATGACATGATCAATGG To measure variation.
4 Why align?
5 Why align?

6 Short Read Aligners: choices... Fall '12 - Apr '13:... now Gbp / day!* *
7 Burrows-Wheeler Aligners Burrows-Wheeler Transform used in bzip2 file compression tool; FM-index (Ferragina & Manzini) allow efficient finding of substring matches within compressed text algorithm is sub-linear with respect to time and storage space required for a certain set of input data (reference 'ome, essentially). Reduced memory footprint, faster execution.
8 BWA BWA is fast, and can do gapped alignments. When run without seeding, it will find all hits within a given edit distance. Long read aligner is also fast, and can perform well for 454, Ion Torrent, Sanger, and PacBio reads. BWA is actively developed and has a strong user / developer community. bio-bwa.sourceforge.net Short reads under 200 bp Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25: [PMID: ] Long reads over 200 bp chimeric alignments built-in Li H. and Durbin R. (2010) Fast and accurate long read alignment with Burrows-Wheeler Transform. Bioinformatics, 26: [PMID: ] don't forget to join the mailing groups!
9 Bowtie Bowtie (now Bowtie 2) is probably faster than BWA for some types of alignment, but it may not find the best alignments (see discussions on sensitivity, accuracy on SeqAnswers.com). Bowtie is part of a suite of tools (Bowtie, Tophat, Cufflinks, CummeRbund) that address RNAseq experiments. Langmead B., Trapnell C., Pop M., and Salzberg S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome Genome Biology 10:R25 [PMID: ] don't forget to join the mailing groups!
10 Alignment concepts / parameters Paired-End reads Mate-Paired reads
11 Alignment concepts / parameters 454 "Paired-End" reads Single End Construct

12 Alignment concepts / parameters

13 Alignment concepts / parameters

14 Alignment concepts / parameters
15 Alignment concepts / parameters
16 File Format: SAM / BAM Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, [PMID: ] SAM specification (currently v1.4) samtools man page FAQ mailing list!

17 File Format: SAM / BAM
18 File Format: SAM / BAM SAM Format Specification v1.4-r985 7,8 - formerly MRNM, MPOS (mate reference name, mate position) 9 - formerly ISIZE ("insert" size)

19
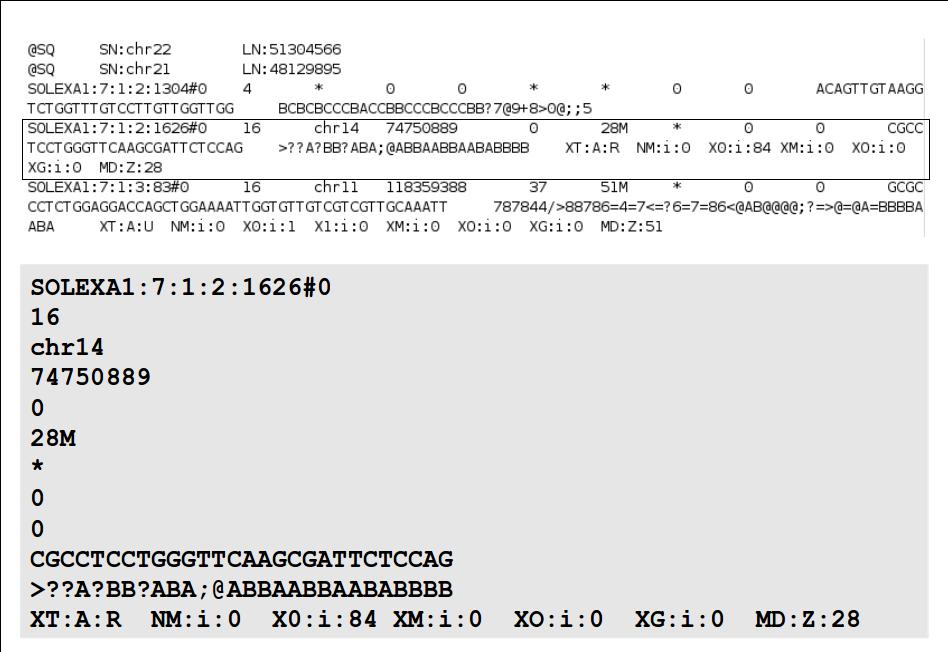
20

21

22

23

24

25

26

27

28
29
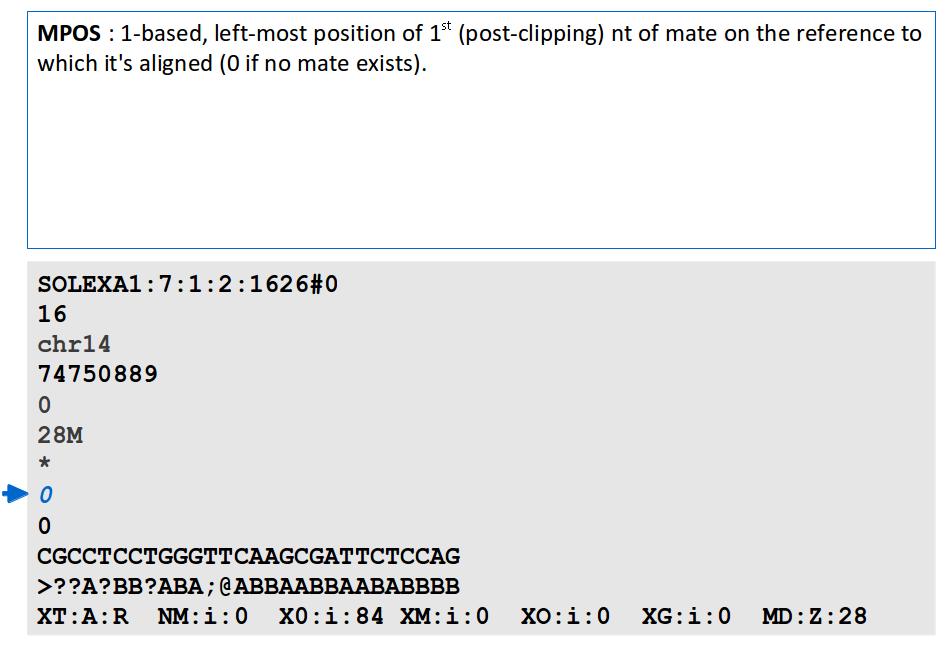
30
31
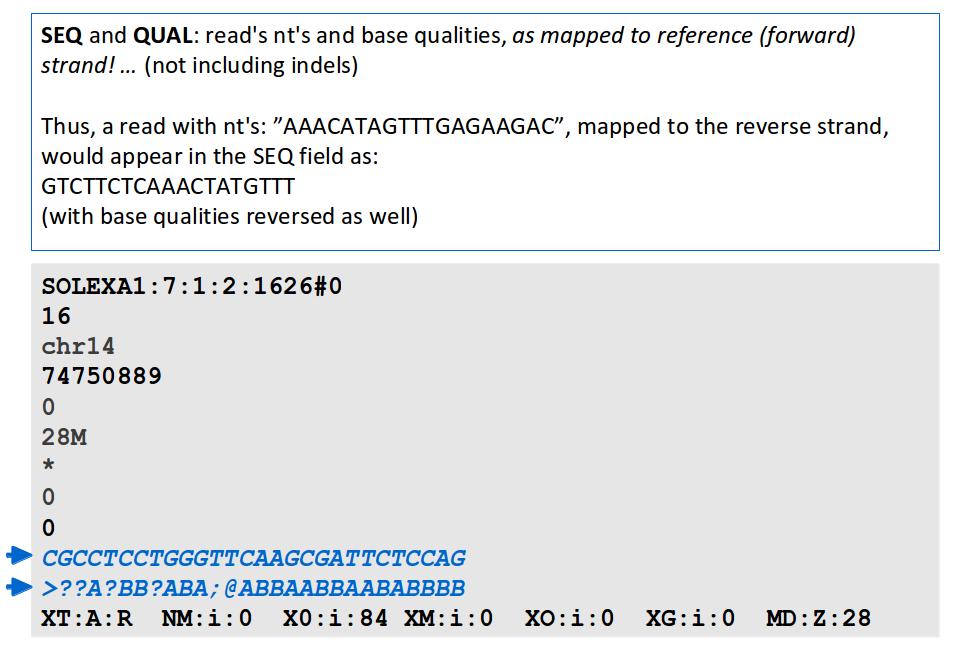
32

33
34 File Format: SAM / BAM google "Heng Li slides" - Challenges and Solutions in the Analysis of Next Generation Sequencing Data (2010)
35 File Format: SAM / BAM BAMs are compressed SAMs (so, binary, not human-readable text don't look directly at them!). They can be indexed to allow rapid extraction of information, so alignment viewers do not need to uncompress the whole BAM file in order to look at information for a particular read or coordinate range, somewhere in the file. Indexing your BAM file, mycoolbamfile.bam, will create an index file, mycoolbamfile.bam.bai, which is needed (in addition to the BAM file) by viewers and other downstream tools. An occasional downstream tool will require an index called mycoolbamfile.bai (notice that the.bai replaces the.bam, instead of being appended after it).
, IGB, GenomeView, SAMscope.")
36 Alignment Viewers IGV (Integrated Genomics Viewer) BAMview, tview (in SAMtools), IGB, GenomeView, SAMscope... UCSC Genome Browser, GBrowse

37 IGV red box indicates region of reference in view below coverage track: read coverage depth plot read alignments: (various view styles - squished shown here) read positions, orientations, pairing, sequence that disagrees with reference highlighted, improper pairs highlighted, etc. annotation tracks (GTF, BED, etc.)
 improper pairs (mate aligns far away, in wrong orientation, or on another chromosome) reference")
38 IGV colored bases where they disagree with reference (substitution, indel, etc.) improper pairs (mate aligns far away, in wrong orientation, or on another chromosome) reference sequence, reading frames, etc.
 is now the standard format for variant reporting. http://vcftools.sourceforge.net/specs.html.")
39 Variant Calling - VCF format One main application of read alignment. A.k.a. "resequencing", SNP / indel discovery. VCF (variant call format) is now the standard format for variant reporting. VCF poster
40 Variant Call Format ##fileformat=vcfv4.1 ##filedate= ##source=freebayes v gfbf46fc-dirty ##reference=../results/8/8.fa ##phasing=none ##commandline="../tools/freebayes/bin/freebayes -f../results/8/8.fa --min-alternate-fraction minmapping-quality 20 --min-base-quality 20 --ploidy 1 --pooled-continuous --use-best-n-alleles 4 --usemapping-quality --min-alternate-fraction min-alternate-count 1../results/8/8.bam" ##INFO=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description="Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description="Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description="Sum of quality of the alternate observations"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB1 26. TGTTACGCG GCTTTTGC,TGTTTCTAC AO=1,2;RO=0;TYPE=complex, complex GT:DP:RO:QR:AO:QA:GL 2:3:0:0:1,2:31,70:-4.46,-1.65,0 8_PB1 38. TCA ACG,TA,AGA AO=1,1,1;RO=3;TYPE=complex,del,mnp GT:DP:RO:QR:AO:QA:GL 2:6:3:101:1,1,1:31,37,34:0,-4.556,-4.004, _PB1 42. G A e-14. AO=8;RO=128;TYPE=snp GT:DP:RO:QR:AO:QA: GL
41 Variant Call Format ##fileformat=vcfv4.1 ##filedate= ##source=freebayes v gfbf46fc-dirty ##reference=../results/8/8.fa ##phasing=none ##commandline="../tools/freebayes/bin/freebayes -f../results/8/8.fa --min-alternate-fraction minmapping-quality 20 --min-base-quality 20 --ploidy 1 --pooled-continuous --use-best-n-alleles 4 --usemapping-quality --min-alternate-fraction min-alternate-count 1../results/8/8.bam" ##INFO=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description="Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description="Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth"> ##FORMAT=<ID=RO,Number=1,Type=Integer,Description="Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description="Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description="Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description="Sum of quality of the alternate observations"> #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB1 26. TGTTACGCG GCTTTTGC,TGTTTCTAC AO=1,2;RO=0;TYPE=complex, complex GT:DP:RO:QR:AO:QA:GL 2:3:0:0:1,2:31,70:-4.46,-1.65,0 8_PB1 38. TCA ACG,TA,AGA AO=1,1,1;RO=3;TYPE=complex,del,mnp GT:DP:RO:QR:AO:QA:GL 2:6:3:101:1,1,1:31,37,34:0,-4.556,-4.004, _PB1 42. G A e-14. AO=8;RO=128;TYPE=snp GT:DP:RO:QR:AO:QA: GL
42 Variant Call Format #CHROM POS ID REF 8_PB A 170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = ALT G QUAL FILTER INFO FORMAT 8 AO=149;RO=21;TYPE=snp GT:DP:RO:QR:AO:QA:GL 1: FILTER =. INFO = AO=149;RO=21;TYPE=snp FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0

43 Variant Call Format

44 Variant Call Format

45 Variant Call Format
46 Variant Call Format #CHROM POS ID REF 8_PB A 170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = ALT G QUAL FILTER INFO FORMAT 8 AO=149;RO=21;TYPE=snp GT:DP:RO:QR:AO:QA:GL 1: ##FORMAT=<ID=DP,Number=1,Type=Integer, Description="Read Depth"> FILTER =. INFO = AO=149;RO=21;TYPE=snp FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0
47 Variant Call Format ##INFO=<ID=RO,Number=1,Type=Integer,Description=" Reference allele observation count, with partial observations recorded fractionally"> ##INFO=<ID=AO,Number=A,Type=Integer,Description=" Alternate allele observations, with partial observations recorded fractionally"> ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex.">
48 Variant Call Format ##FORMAT=<ID=GT,Number=1,Type=String,Description=" Genotype"> ##FORMAT=<ID=GQ,Number=1,Type=Float,Description=" Genotype Quality, the Phred-scaled marginal (or unconditional) probability of the called genotype"> ##FORMAT=<ID=GL,Number=G,Type=Float,Description=" Genotype Likelihood, log10-scaled likelihoods of the data given the called genotype for each possible genotype generated from the reference and alternate alleles given the sample ploidy"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description=" Read Depth">
49 Variant Call Format ##FORMAT=<ID=RO,Number=1,Type=Integer,Description=" Reference allele observation count"> ##FORMAT=<ID=QR,Number=1,Type=Integer,Description=" Sum of quality of the reference observations"> ##FORMAT=<ID=AO,Number=A,Type=Integer,Description=" Alternate allele observation count"> ##FORMAT=<ID=QA,Number=A,Type=Integer,Description=" Sum of quality of the alternate observations">
50 Variant Effect Prediction snpeff Variant Effect Predictor (EMBL) SIFT
51 VCF after Effect Prediction #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 8 8_PB A G AO=149;RO=21;TYPE=snp;EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1) GT:DP:RO:QR:AO:QA:GL 1:170:21:788:149:5579:-5,0 CHROM = 8_PB2 POS = 407 ID =. REF = A ALT = G QUAL = FILTER =. INFO = AO=149;RO=21;TYPE=snp;EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1) FORMAT = GT:DP:RO:QR:AO:QA:GL 8 = 1:170:21:788:149:5579:-5,0
52 VCF after Effect Prediction ##INFO=<ID=TYPE,Number=A,Type=String,Description="The type of allele, either snp, mnp, ins, del, or complex."> ##INFO=<ID=EFF,Number=.,Type=String,Description="Predicted effects for this variant.format: 'Effect ( Effect_Impact Functional_Class Codon_Change Amino_Acid_change Amino_Acid_length Gene_Name Transcript_BioType Gene_Coding Transcript_ID Exon GenotypeNum [ ERRORS WARNINGS ] )' "> INFO = AO=149;RO=21;TYPE=snp; EFF=SYNONYMOUS_CODING (LOW SILENT gaa/gag E PB2 CODING Tr_PB2 1 1)