Understanding Reporting Obligations to the IRB
|
|
|
- Ethelbert Jennings
- 5 years ago
- Views:
Transcription


1 Understanding Reporting Obligations to the IRB fully accredited since 2006 May 13 & 15, 2014 Mitchell E. Parrish, JD, RAC, CIP Regulatory Attorney
2 2

3 3
4 WEBINAR HOUSEKEEPING Questions & Answers Feel free to submit questions at any point during the webinar using the chat box on your webinar dashboard Responses will be sent by the presenters following the presentation 4
5 WEBINAR HOUSEKEEPING Recording & Slide Deck The webinar recording and slide deck will be posted on our website within 5 business days We will you a link to view the recording as soon as it is available Feel free to share the link with your staff and/or colleagues 5

6 ABOUT QUORUM REVIEW IRB Accredited Regulatory Leadership Fully accredited by AAHRPP through 2014 Fully compliant with FDA and OHRP requirements 6 in-house licensed attorneys providing guidance and thought-leadership Boards available for the review of U.S. and Canadian studies can review for GCP and IHC internationally International ABOUT QUORUM REVIEW IRB Strong Framework Certified IRB Professionals (CIP) One of the largest IRBs in the U.S. with ~180 employees Quorum Review is the ONLY equivalently sized IRB not owned by Venture Capital 60% of Affiliated IRB members, 40% of Regulatory staff and 20% of study management & study support positions 6
7 THE QUORUM ADVANTAGE 15 Board meetings each week 24-hour site turnaround, 36-hour amendment review, and same day site changes One time CV and audit documentation submission Support available 8am-8pm ET Dedicated Study Manager Industry leading legal team 7
8 THE QUORUM ADVANTAGE Secure portal with SmartForms, status reports, and approval documents Customized Phase I and Expeditable Research processes Flexible, customized process for AMCs Over 850 Institutions work with Quorum 100% Quality Control on all documents Commitment to 6 Sigma Process Analysis 8
9 ABOUT THE PRESENTER Quorum Review Regulatory Attorney Mitchell E. Parrish, JD, RAC,CIP IRB Experience Joined Quorum Review, Inc. in January 2010 CIP certification Regulatory Affairs Certification Member of Public Responsibility in Medicine & Research (PRIM&R) Legal Background Juris Doctor from University of Oregon Member of the Washington State Bar Association (WSBA) Member of the Association of Corporate Counsel (ACC) and Regulatory Affairs Professionals Society (RAPS) 9

10 Today s Webinar: Understanding Reporting Obligations To the IRB
11 Webinar Overview Topic Page Role of the IRB 11 Problems with Reporting 15 Regulatory Landscape 18 Obligations for Reporting Safety Information 26 Unanticipated problems that are Adverse Events SUSAR UADE Recommended practices for reporting Safety Information Obligations for Reporting Non-Safety Information 48 Unanticipated Problems that are not Adverse Events Recommended practices for reporting Non-Safety Information Key Take Aways 62 11

12 The Role of the IRB 12
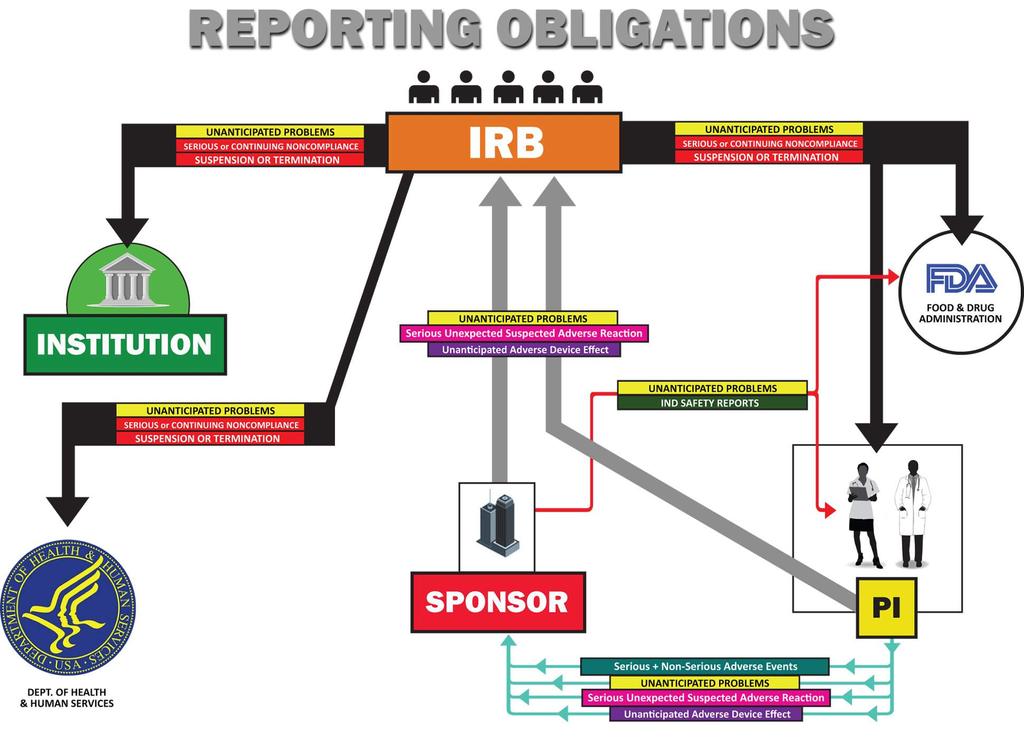
![Role of the IRB The primary purpose of both initial and continuing review of [a] study is](/docs-images/89/97487219/images/13-0.jpg "to assure the protection of the rights and welfare of the human subjects ( 56.109(f).")


13 Role of the IRB The primary purpose of both initial and continuing review of [a] study is to assure the protection of the rights and welfare of the human subjects ( (f). To fulfill its obligations an IRB must have, among other things, information concerning unanticipated problems involving risk to human subjects in the study, including adverse events that are considered unanticipated problems. FDA Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 13
14 14
15 15

16 16 Problems with Reporting

17 Problems with Reporting Unnecessary Reporting Much of the information that is being reported does not meet reporting requirements and therefore results in the unnecessary expenditure of resources by all stakeholders. Specifically, the way that unanticipated problem is interpreted does not yield information about adverse events that is useful to IRBs and thus hinders their ability to ensure protection of human subjects. FDA Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 17
 have complicated the reporting pathways for adverse event information described in the regulations.")
18 Problems with Reporting No Explanation Provided Not only is unnecessary information reported, but also reported information is not explained: In the years since the IRB and IND regulations issued, changes in the conduct of clinical trials (e.g., increased use of multi-center studies, international trials) have complicated the reporting pathways for adverse event information described in the regulations. IN particular the practice of local investigators reporting individual, unanalyzed events to IRBs, including reports of events from other study sites that the investigator receives from the sponsor of a multi-center study often with limited information no explanation of how the event represents an unanticipated problem has led to the submission of large numbers of reports to IRBs that are uninformative. FDA Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 18

19 19 Regulatory The Landscape
 21 CFR 812 (Investigational Device Exemptions) Guidance (HHS/FDA) HHS, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to")
20 Regulatory Landscape Regulations (HHS/FDA) 45 CFR 46 (Protection of Human Subjects) 21 CFR 56 (Institutional Review Boards) 21 CFR 312 (Investigational New Drug Application) 21 CFR 320 (Bioavailability and Bioequivalence Requirements) 21 CFR 812 (Investigational Device Exemptions) Guidance (HHS/FDA) HHS, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events, January 15, 2007 FDA, Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection, January, 2009 FDA, Guidance for Industry and Investigators, Safety Reporting Requirements for INDs and BA/BE Studies, December,
21 Regulatory Landscape From Where is the Obligation to Report Safety Information to the IRB derived? FDA and HHS regulations require the IRB to receive safety and other information throughout study and during continuing review to ensure the criteria for approval is still met and to ensure the safety, rights, and welfare of subjects are protected 45 CFR and ; 21 CFR and
 There is a lot of safety data and other information in clinical trials, so where in the regulations does it say exactly what to report to the IRB?")
22 Regulatory Landscape From Where is the Obligation to Report Safety Information to the IRB derived? (continued) There is a lot of safety data and other information in clinical trials, so where in the regulations does it say exactly what to report to the IRB? While the regulations do not contain specifics, they do provide the term Unanticipated Problem (UP) 22
23 Regulatory Landscape Unanticipated Problem 21 CFR The investigator shall... Promptly report to the IRB... All unanticipated problems involving risk to human subjects or others. 21 CFR (b)(1) [E]ach IRB shall: Follow written procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the Food and Drug Administration of: (1) Any unanticipated problem CFR (b) (4) An IRB must have [W]ritten procedures for ensuring prompt reporting to the IRB, appropriate institutional officials, and the department or agency head of (i) any unanticipated problems involving risks to subjects or others
24 Regulatory Landscape Unanticipated Problem Unanticipated Problem = Any incident, experience, or outcome that meets all of the following criteria: Unexpected (in terms of nature severity or frequency) given(a) the research procedures that are described in the protocol-related documents, and (b) the characteristics of the subject population being studied Related or possibly related to participation in the research Suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized HHS Guidance, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events (January 15, 2007) *Note: This general criteria is essentially the same for unanticipated problems under FDA regulations as well. 24

25 Regulatory Landscape Unanticipated Problems & Adverse Events In addition to defining Unanticipated Problem, the HHS Guidance also explains that there are UPs that stem from adverse events, essentially safety related UPs, and those that do not stem from adverse events, essentially non-safety related UPs. Adverse Event: Any untoward or unfavorable medical occurrence in a human subject, including any abnormal sign (for example abnormal physical exam or laboratory finding), symptom, or disease, temporally associated with the subject's participation in the research, whether or not considered related to the subject s participation in the research. This encompasses both physical and psychological harms and can be categorized as internal (happening at the site) or external (happening at other locations). FDA Guidance, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) Note: The terms adverse effect and adverse experience are interchangeable with adverse event. 25

26 Regulatory Landscape Unanticipated Problems and Adverse Events Under 45 CFR 46 do not report A, do report B + C. Safety Related: Do not report adverse events that are not UPs Safety Related: Must report adverse events that are UPs Non-Safety Related: Must report UPs that are not adverse events Adapted from HHS Guidance, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events (January 15, 2007); See also FDA Guidance, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 26


27 27 Obligations for Reporting Safety Information to the IRB


; See also FDA")
28 Regulatory Landscape Unanticipated Problems and Adverse Events Under 45 CFR 46 do not report A, do report B + C. Safety Related: Do not report adverse events that are not UPs Safety Related: Must report adverse events that are UPs Non-Safety Related: Must report UPs that are not adverse events Adapted from HHS Guidance, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events (January 15, 2007); See also FDA Guidance, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 28
29 Reporting Safety Information to the IRB SUSAR: SERIOUS & UNEXPECTED SUSPECTED ADVERSE REACTIONS UP: UNANTICIPATED PROBLEM UADE: UNANTICIPATED ADVERSE DEVICE EFFECT By its very nature, if an event is an SUSAR or a UADE then it is a UP and must be reported to the IRB 29
30 Reporting Safety Information - SUSAR Serious and unexpected suspected adverse reaction (SUSAR) originates from 21 CFR 312 and is designed to guide sponsors on when to submit IND safety reports to the FDA and investigators Must report to the IRB all SUSARs that satisfy these three criteria: 1. Serious (Death, life-threatening, inpatient hospitalization or prolongation of existing hospitalization, persistent or significant incapacity, substantial disruption of the ability to conduct normal life functions, and congenital anomaly/birth defect) 2. Unexpected (an event not listed in the investigator brochure or not listed at the specificity or severity observed; or an event not consistent with the risk information described in the investigational plan) 3. Suspected Adverse Reaction ( a reasonable possibility that the drug caused the adverse event ) Evidence to suggest a causal relationship between the drug and event 30
 31")
31 Reporting Safety Information - SUSAR Individual Occurrences A single occurrence of an event that is uncommon and known to be strongly associated with drug exposure (e.g., angioedema, hepatic injury, Stevens-Johnson Syndrome) 31
32 Reporting Safety Information - SUSAR Example: In a phase II study testing an investigational drug for Hepatitis C, a subject experiences hepatic injury. In addition to the investigational drug, the subject was continuing her standard Hepatitis C therapy at the time of hepatic injury. Is this individual occurrence a suspected adverse reaction? 32

33 Reporting Safety Information - SUSAR Example: In a phase II study testing an investigational drug for Hepatitis C, a subject experiences hepatic injury. In addition to the investigational drug, the subject was continuing her standard Hepatitis C therapy at the time of hepatic injury. Is this individual occurrence a suspected adverse reaction? YES 33
 34")
34 Reporting Safety Information - SUSAR One or more Occurrences One or more occurrences of an event that is not commonly associated with drug exposure, but is otherwise uncommon in the population exposed to the drug (e.g., tendon rupture, progressive multifocal leukoencephalopathy) 34
35 Reporting Safety Information - SUSAR Example: A subject with extrapulmonary small-cell carcinoma receiving an investigational chemotherapy agent experiences a bowel perforation during his second cycle of chemotherapy. Is this one occurrence a suspected adverse reaction? 35

36 Reporting Safety Information - SUSAR Example: A subject with extrapulmonary small-cell carcinoma receiving an investigational chemotherapy agent experiences a bowel perforation during his second cycle of chemotherapy. Is this one occurrence a suspected adverse reaction? NO 36
 that indicates those events occur more frequently in the drug treatment group")
37 Reporting Safety Information - SUSAR Aggregate Analysis of Specific Events An aggregate analysis of specific events observed in a clinical trial (such as known consequences of the underlying disease or condition under investigation or other events that commonly occur in the study population independent of drug therapy) that indicates those events occur more frequently in the drug treatment group than in a concurrent or historical control group. 37
38 Reporting Safety Information - SUSAR Example: 17 oncology sites are participating in a research study comparing an investigational chemotherapy agent to standard therapy in subjects ages Of those subjects receiving the investigational drug, an average of 35% of subjects across the 17 sites experience deep vein thrombosis. Does this aggregate analysis indicate a suspected adverse reaction? 38

39 Reporting Safety Information - SUSAR Example: 17 oncology sites are participating in a research study comparing an investigational chemotherapy agent to standard therapy in subjects ages Of those subjects receiving the investigational drug, an average of 35% of subjects across the 17 sites experience deep vein thrombosis. Does this aggregate analysis indicate a suspected adverse reaction? YES 39

40 Reporting Safety Information - SUSAR Critical to understand the definition of Suspected Adverse Reaction coupled with the examples the FDA provides: Individual occurrence One or more occurrences Aggregate analysis 40

41 Reporting Safety Information - SUSAR Without this understanding, the FDA explains that the following problem occurs: Reporting of individual events even though unlikely that the event was caused by the drug. Examples: Serious adverse experiences (e.g. mortality or major morbidity) that are unlikely to have been manifestations of the underlying disease Serious adverse experiences that commonly occurred in the study population independent of drug exposure (e.g. strokes or acute myocardial infarction in an elderly population) Serious adverse experiences that were study endpoints (i.e. the study was evaluating whether the drug reduced the rate of these events) Such reporting causes a drain on resources when the FDA, and IRBs are inundated with generally uninformative Safety Information especially when reported as single events without any context 41
 Not previously identified (an effect not previously identified in nature, severity,")
42 Reporting Safety Information - UADE Unanticipated Adverse Device Effect (UADE) originates from 21 CFR 812 and is designed to guide sponsors on when to submit reports to the FDA and investigators Must report to the IRB all UADEs that satisfy the following criteria: Serious (death, life-threatening, serious problem) Not previously identified (an effect not previously identified in nature, severity, or degree of incidence in the investigational plan or application) Caused by, or associated with, a device 42
43 Reporting Safety Information - UADE FDA Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) Essentially states that reporting UADEs to the IRB should be treated the same as reporting SUSARs to the IRB While not in the device regulations and while there are different considerations between safety information for drugs and devices, take into account whether the effect is a: Individual occurrence One or more occurrences Multiple occurrences determined through an aggregate analysis 43
44 Recommended Practices for Reporting Safety Information For Multi-center studies: Even though the FDA regulations indicate it is the investigator s responsibility to notify the IRB of unanticipated problems (21 CFR ) FDA guidance states: Investigators must rely on the sponsor to provide them information about AEs occurring at other study sites Although the investigator s view of the causal relationship between an adverse event and the investigational drug is important, FDA believes that the sponsor is better positioned than the individual investigator to assess the overall safety of the investigational drug because the sponsor has access to serious adverse event reports from multiple study sites and is able to aggregate and analyze these reports. The sponsor is in a better position to process and analyze the significance of AE information from multiple sites and to make a determination about whether an AE is an unanticipated problem FDA supports an arrangement in which the sponsor prepares UP reports and submits to the IRB when the sponsor, investigator, and IRB have made an explicit agreement for the sponsor to report directly to the IRB FDA Guidance for Clinical Investigators, Sponsors, and IRBs, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009); FDA Guidance for Industry and Investigators, Safety Reporting Requirements for INDs and BA/BE Studies (December 2012) 44
45 Recommended Practices for Reporting Safety Information (continued) While Investigators may report SUSARs and UADEs to the IRB, Quorum recommends that Sponsors/CROs submit SUSAR and UADE information to the IRB on behalf of investigators. This means... The Sponsor/CRO should inform investigators and arrange with the IRB that it will report on behalf of investigators (arrangement with IRB should include in what format to report) The protocol should not state generally that all adverse events require reporting the IRB or include similar language because then if all adverse events are subsequently not reported, investigators run the risk of being out of compliance with the protocol Also it is not appropriate to submit all AEs to the IRB, just AEs that are SUSARs or UADEs! 45

46 Recommended Practices for Reporting Safety Information For single-center or investigator initiated studies: The investigator is likely the one reporting to the IRB This still means: The investigator can arrange with IRB in what format to report The protocol should still not state generally that all adverse events require reporting the IRB or include similar language because then if all adverse events are subsequently not reported, investigators run the risk of being out of compliance with the protocol Also it is not appropriate to submit all AEs to the IRB, just AEs that are SUSARs or UADEs! 46

47 Recommended Practices for Reporting Safety Information TIMING of Reporting to the IRB (continued) Report SUSARs and UADEs promptly to the IRB Promptly is generally considered 10 business days, which is supported in the FDA s guidance on reporting AEs to the IRB This 10-day timeframe exists whether there is an arrangement for the sponsor to submit on behalf of the IRB or whether the investigator is responsible for submitting to the IRB 47
48 Recommended Practices for Reporting Safety Information QUESTIONS ABOUT: What to report Whether to report How to report When to report (continued) CONTACT the 48

49 49 Obligations for Reporting Non-Safety Information to the IRB

; See also FDA Guidance, Adverse Event Reporting to IRBs Improving Human Subject Protection (January")
50 Reporting Non-Safety Information to the IRB Unanticipated Problems and Adverse Events Safety Related: Do not report adverse events that are not UPs Safety Related: Must report adverse events that are UPs Non-Safety Related: Must report UPs that are not adverse events Adapted from HHS Guidance, Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events (January 15, 2007); See also FDA Guidance, Adverse Event Reporting to IRBs Improving Human Subject Protection (January 2009) 50

51 Reporting Non-Safety Information to the IRB UNEXPECTED RELATED OR POSSIBLY RELATED UP: UNANTICIPATED PROBLEM GREATER RISK OF HARM You MUST report ALL UP s to the IRB 51
52 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: Failure to obtain informed consent Major protocol deviations (e.g. inclusion/exclusion violation; omitting a study procedure) Study personnel misconduct that adversely impacts the study Adverse findings by a regulatory agency, medical board, or other relevant body Are these UPs? * Whether these are UPs depends on the type of study and the specific factors surrounding each event or incident 52
53 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: An investigator is conducting behavioral research and collects individually identifiable sensitive information about illicit drug use by surveying college students. The data is stored on a laptop computer that is password protected. The laptop is stolen from the investigator s car. Is this reportable to the IRB as a UP? 53

54 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: An investigator is conducting behavioral research and collects individually identifiable sensitive information about illicit drug use by surveying college students. The data is stored on a laptop computer that is password protected. The laptop is stolen from the investigator s car. Is this reportable to the IRB as a UP? YES 54
55 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: An Investigator is conducting a psychology study evaluating decision making and response times when persons are listening to music at various decibel levels. In order to perform the study, participants are placed in a small, windowless, soundproof booth. The IRB-approved protocol and consent form describe claustrophobic reactions as one of the research risks. The 12 th subject enrolled in the research experiences significant claustrophobia, resulting in the subject withdrawing from the study. Is this reportable to the IRB as a UP? 55

56 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: An Investigator is conducting a psychology study evaluating decision making and response times when persons are listening to music at various decibel levels. In order to perform the study, participants are placed in a small, windowless, soundproof booth. The IRB-approved protocol and consent form describe claustrophobic reactions as one of the research risks. The 12 th subject enrolled in the research experiences significant claustrophobia, resulting in the subject withdrawing from the study. Is this reportable to the IRB as a UP? NO 56
57 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: As a result of a processing error by a pharmacy technician, a subject enrolled in a multi-center clinical trial receives a dose of an experimental agent that is 10-times higher than the dose dictated by the IRB-approved protocol. While the dosing error increased the risk of toxic manifestations of the experimental agent, the subject experienced no detectable harm or adverse effect after an appropriate period of careful observation. Is this reportable to the IRB as a UP? 57

58 Reporting Non-Safety Information to the IRB Unanticipated Problems - Examples: As a result of a processing error by a pharmacy technician, a subject enrolled in a multi-center clinical trial receives a dose of an experimental agent that is 10-times higher than the dose dictated by the IRB-approved protocol. While the dosing error increased the risk of toxic manifestations of the experimental agent, the subject experienced no detectable harm or adverse effect after an appropriate period of careful observation. Is this reportable to the IRB as a UP? YES 58

59 Recommended Practices for Reporting Non-Safety Information For multi-center or single-center: Generally, UPs that are not an adverse event (i.e. non-safety related) are typically site or investigator specific Therefore, it makes more sense for the investigator, not the sponsor, to report the UP to the IRB The investigator can arrange with IRB in what format to report 59


60 Recommended Practices for Reporting Non-Safety Information TIMING of Reporting to the IRB (continued) Report UPs promptly to the IRB Promptly is generally considered 10 business days, which is supported in the FDA s guidance on reporting to the IRB This 10-day timeframe exists whether there is an arrangement for the sponsor to submit on behalf of the IRB or whether the investigator is responsible for submitting to the IRB 60
61 Recommended Practices for Reporting Safety Information QUESTIONS ABOUT: What to report Whether to report How to report When to report (continued) CONTACT the 61

62 62 KEY TAKE AWAYS
63 Key Take Aways Know where the obligations to report to the IRB originate Understand the term Unanticipated Problem (UP) and its three criteria: Unexpected, Related, Greater Risk of Harm Understand the terms SUSAR and UADE, know their criteria, and know that these are UPs that require reporting to the IRB Understand that there are UPs that are not safety related that must be reported to the IRB Know how and in what timeframe to report to the IRB If there are ever any questions relating to reporting to the IRB, talk to your IRB. The IRB is a resource! 63
64 QUESTIONS? You may submit questions through our webinar survey Mitchell E. Parrish, JD, RAC,CIP Quorum Client Relations We will do our best to follow-up individually or answer your questions in the Q&A we post on our website 64
65 Webinar Follow-Up The webinar Recording, Slide Deck, and Q&A will be posted on our website We will you a link to view these items as they become available We value your opinion please take our SURVEY and provide us with feedback 65

66 Connect with us! linkedin.com/company/quorum review youtube.com/quorumreview 66
67 67 Thank You for Attending!
68 Understanding Reporting Obligations to the IRB fully accredited since 2006