ICH GCP E6(R2): Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc.
|
|
|
- Laura Ellis
- 6 years ago
- Views:
Transcription


1 ICH GCP E6(R2): Changes? Yes Challenges? Not as Difficult as You May Fear Lorrie D. Divers, President QRCP Solutions, Inc. 25 October 2017: University of Rochester Achieving High Quality Clinical Research Seminar Series

2 2
3 OBJECTIVES ICH GCP E6(R2) Who does it impact? Regulations versus Guidance What Are Our Obligations? Major Changes Updated Why and How? What Do They Mean For You? Clinical Investigator / Study Site Sponsor What to Remember 3
4 ICH GCP E6(R2): What is it? International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) The objective of [the] ICH GCP Guideline is to provide a unified standard for the European Union (EU), Japan, and the United States to facilitate the mutual acceptance of clinical data by the regulatory authorities. This guideline should be followed when generating clinical trial data that are intended to be submitted to regulatory authorities. 4
!")
5 Oh good, this doesn t apply to me. It says, pharmaceuticals right in the name of the organization (and its definition of a clinical trial )! I do behavioral intervention trials! I am studying an intraocular lens! 5 This Photo by Unknown Author is licensed under CC BY-NC-SA
6 ICH GCP E6(R2) also says. The principles established in this guideline may also be applied to other clinical investigations that may have an impact on the safety and well-being of human subjects. 6

7 7

8 8
9 21 CFR Part 812 (IDE) says. Investigational device means a device, including a transitional device, that is the object of an investigation. Investigation means a clinical investigation or research involving one or more subjects to determine the safety or effectiveness of a device. 9
 to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes. 10 This Photo by Unknown Author is licensed under CC BY-NC-ND")
10 A clinical trial is defined by NIH as a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes. 10 This Photo by Unknown Author is licensed under CC BY-NC-ND


11 But my study will never be submitted to the FDA. Don t bet on it.. 11

12 It s just guidance, not a regulation! 12 This Photo by Unknown Author is licensed under CC BY-NC-SA
13 Regulations vs. Guidance Regulations are minimum standards You can (and most often) should do more You cannot do less You are obligated to comply with the protocol, IRB approval, and/or contracts which will often state the regulations and guidances 13


14 Speaking of Panic.. More or less? 14
15 Regulations vs. Guidance Exceeding minimum requirements: Standardization based on best practices Less challenges now Demonstrates compliance in the future Provides greater protection for research subjects rights, safety and welfare Strengthens data integrity 15
16 Regulations vs. Guidance Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible. 16

17 Regulations vs. Guidance Guidance documents are not legally binding but they are proven best practices 17
18 Regulations vs. Guidance U.S. Federal GCP regulations can seem like a (frustrating) exercise in the Socratic method Often have to consult multiple resources and arrange multiple passages in sequential order then ask a lot of questions What does this one say that this one does not? Where is this term defined? When was this regulation revised and why? 18
19 Regulations vs. Guidance Guidance documents can be extremely helpful for Untangling the legalese Clarifying the if-then and what-how debates Applying legal (and ethical) principles in practical ways 19

20 ICH GCP E6(R2) Major Changes Updated Why and How? What Do They Mean For You? 20 This Photo by Unknown Author is licensed under CC BY-NC-ND
21 ICH GCP E6(R2): Revision History Since the development of the ICH GCP Guideline, the scale, complexity, and cost of clinical trials have increased. Evolutions in technology and risk management processes offer new opportunities to increase efficiency and focus on relevant activities. When the original ICH E6(R1) text was prepared, clinical trials were performed in a largely paper-based process. Advances in use of electronic data recording and reporting facilitate implementation of other approaches. For example, centralized monitoring can now offer a greater advantage, to a broader range of trials Copyright, than QRCP Solutions, is Inc. suggested in the original text. 10/25/
22 ICH GCP E6(R2): Revision History Therefore, this guideline has been amended to encourage implementation of improved and more efficient approaches to clinical trial design, conduct, oversight, recording and reporting while continuing to ensure human subject protection and reliability of trial results. Standards regarding electronic records and essential documents intended to increase clinical trial quality and efficiency have also been updated. 22
23 23

24 24
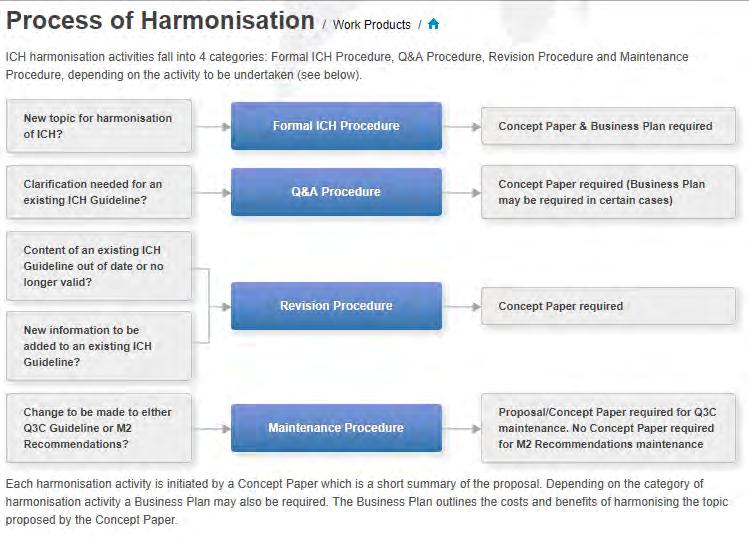
25 25

26 26
27 ICH GCP E6(R2): ICH Status Published Document: ICH Website: Implementation Step 5 27
: FDA")
28 ICH GCP E6(R2): FDA Status 28

29 ICH GCP E6(R2): NIH Status Sec NIH Policy on Good Clinical Practice Training for NIH Awardees Involved in NIH-funded Clinical Trials: Establishes the expectation that all NIH-funded investigators and staff who are involved in the conduct, oversight, or management of clinical trials should be trained in Good Clinical Practice (GCP), consistent with principles of the International Conference on Harmonisation (ICH) E6 (R2). Implements provisions announced in NOT-OD

30 ICH GCP E6(R2): What It Means For You This Photo by Unknown Author is licensed under CC BY-NC-SA 30

31 ICH GCP E6(R2): Revisions Identified by the word ADDENDUM prior to each added or revised section or text within a section New? Not really 31
32 ICH GCP E6(R2): Revisions Four general areas with the most significant changes: Increased clarity in the language regarding investigator oversight NOT new More comprehensive language on sponsor oversight of vendors NOT new Inclusion of risk-based monitoring NOT new Enhanced language on Electronic Systems and Data Handling - Bringing this into the 21st century 32


33 ICH GCP E6(R2): Section 1 - GLOSSARY 1.51 Source Data has always stated: All information in original records and certified copies of original records. 33
 and date or by generation through a validated process If source documents will be scanned (e.g., laboratory requisitions), validate the scanning software and procedures If transferring data from one database/software program to another one (i.")
34 Certified Copy Tips and Techniques Whenever the original document cannot (or will not) be retained Process driven or rational reason Initial (or sign) and date and simply note Complete Copy of Original Stamp Certified Copy and initial (or sign) and date or by generation through a validated process If source documents will be scanned (e.g., laboratory requisitions), validate the scanning software and procedures If transferring data from one database/software program to another one (i.e., new EHR implementation) 34
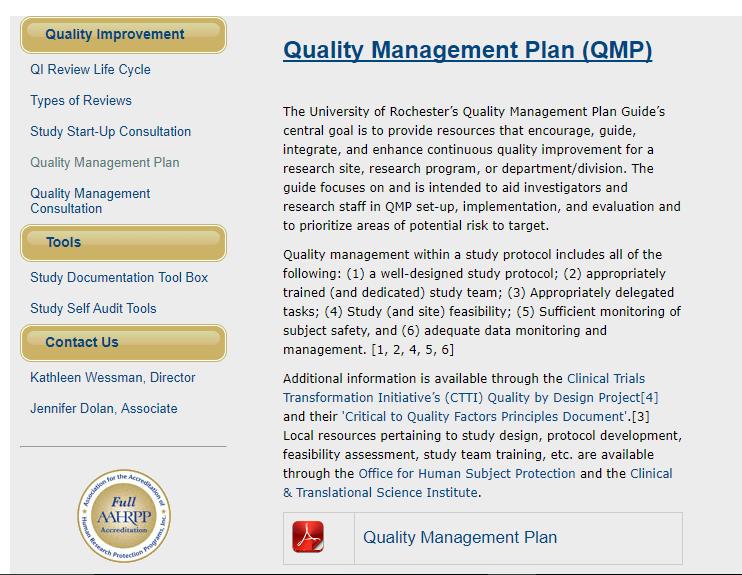
35 Section 2 - The Principles of ICH GCP 35
36 Section 4 - INVESTIGATOR *More consistent with U.S. regulatory language* 36

37 Investigator Oversight: How NOT to htm 37
38 Investigator Oversight Tips and Techniques Be involved Actively engage in the informed consent process Assess AEs and SAEs, inclusion/exclusion, study procedure results Document your involvement READ, initial/date IRB and sponsor correspondence Don t sign blank checks Hire qualified personnel Ensure they receive continuous training, support and consistent supervision 38
39 Investigator Oversight Tips and Techniques If other entities perform trial-related duties and/or functions You must ensure they are qualified to do so and that their performance of the services is compliant Collect/review information to qualify and document review of performance on a regular basis May be at the institutional level ( If the investigator/institution retains the services ) Examples: Central IRB or a central clinical trial services group 39


40 Section 4 - INVESTIGATOR 40 This Photo by Unknown Author is licensed under CC BY-NC-ND
41 Section 5 - SPONSOR Section with the most significant revisions Not imposing new requirements but considerably clarifying sponsor responsibilities for Ensuring quality and compliance in every aspect of the clinical trial Using a risk based approach (which ICH, EMA and FDA have been advocating for many years - at least since 2013) 41
42 Section 5 - SPONSOR As a clinical investigator/study site, you may experience changes in sponsor approach and requirements Should be familiar with the revisions to Section 5 The addition of Section 5.0, Quality Management ADDENDUM on risk-based monitoring 42
43 Section 5 - SPONSOR If you are a sponsor-investigator, you are obligated to carry out both sponsor and investigator responsibilities A multi-institution trial where you are acting as Program Director, Lead PI or Coordinating Investigator confers the sponsor responsibilities on that role 43

44 Section 5 - SPONSOR 44
45 Section 5 - SPONSOR 45
46 Section 5 SPONSOR Tips and Techniques You have an excellent resource for assistance: qualitymanagementplan.html Another one that may be helpful: 46
47 47
48 48

49 Risk Assessment / Risk Management We all perform risk assessment and risk management on a daily basis This Photo by Unknown Author is licensed under CC BY-SA 49
50 Risk Assessment Tips and Techniques At the most basic level, risk assessment is: A. Defining the possible unintended occurrences, consequences, outcomes B. Determining how likely or unlikely it is that these may occur C. Evaluating what the impact of each could be particularly on study subject rights, safety and welfare and on data integrity D. Understanding how easy or hard it would be to detect the issues identified Then ranking these risks from high (critical) to low (minimal) Tip: There is no such thing as no risk! 50
51 Risk Management Tips and Techniques At the most basic level, risk management is: Developing and executing actions to maximally control for the impact and occurrence the risks identified in the assessment process Risk Management Plan Focus greatest effort on the high risks but don t ignore the others, particularly any with impact on study subject safety, rights, and welfare or data integrity Possible actions? Processes and procedures, standardized work, job aids, quality control, monitoring, quality assurance oversight, etc. 51

52 Section 5 - SPONSOR 52

53 Section 5 - SPONSOR 53

54 Section 5 - SPONSOR 54
55 Section 5 SPONSOR Tips and Techniques Not New! Guidance for Industry: Oversight of Clinical Investigations A Risk-Based Approach to Monitoring, U.S. Department of Health and Human Services, Food and Drug Administration, August
56 FDA Guidance 56
57 Section 5 - SPONSOR 57
58 Section 5 SPONSOR Tips and Techniques As an auditor, I see underutilization of regular use of data review techniques Intra-Subject Logic Inter-Site Logic Quality of Documentation Repetitive Errors Much of this can be done centrally 58
59 Section 5 - SPONSOR 59

60 Section 5 SPONSOR Tips and Techniques perform a root cause analysis and implement appropriate corrective and preventive actions. 60 This Photo by Unknown Author is licensed under CC BY-NC-ND
61 Root Cause Analysis and CAPA Boils down to this: Determine the true reason(s) why an unintended consequence occurred Evaluate what action(s) directly related to the cause(s) and take the action(s) Develop and execute activities to prevent future / similar occurrence 61

62 Root Cause Analysis and CAPA Basic Example: Enrolled a subject who did not meet all the eligibility criteria Initial root cause: Human error! 62 This Photo by Unknown Author is licensed under CC BY-NC-ND
63 Root Cause Analysis and CAPA Why did the human make the error? Used an Eligibility Checklist that was not consistent with the latest protocol amendment. Why? Person who received and processed the protocol amendment did not review it against the Checklist. Why? Procedure for processing protocol amendments does not include a step to Review amendment to determine if inclusion/exclusion criteria changed and compare to/revise the Eligibility Checklist. 63
64 Root Cause Analysis and CAPA What actions can mitigate the ineligible subject s enrollment? What can correct the cause of the problem going forward? What similar issues might occur in the future and what can be done? 64
65 Root Cause Analysis and CAPA Don t find fault, find a remedy. - Henry Ford This is a good reminder that effective root cause analysis needs to identify the reason(s) an issue occurred, not place blame or just default to 'human error The word "remedy" is a great metaphor for developing and implementing effective corrective and preventive actions that 'cure,' not just bandage, those true root cause(s) 65

66 Section 8 ESSENTIAL DOCUMENTS 66
67 Section 8 ESSENTIAL DOCUMENTS This passage seems to be causing Sponsors a lot of stress ICH did previously indicate that the Investigator should have a copy of the CRF Impact being seen specifically with EDC studies not paper-based CRFs Tip: As an investigator, make sure you have access to a readable copy of the complete CRFs at the conclusion of the study 67


68 ICH GCP E6(R2): What to Remember Clarifications and additional information that is helpful to facilitate compliance No new requirements per se Repeated emphasis on protecting research subject s rights, safety and welfare Directly equates quality to those objectives Importance of oversight, risk management, and record-keeping in more contemporary ways 68

69 Questions? 69
![References ICH E6_R2, INTEGRATED ADDENDUM TO ICH E6(R1): GUIDELINE FOR GOOD CLINICAL PRACTICE, Current Step 4 version dated 9 November 2016 EMA/269011/2013 [18 November 2013]: Reflection paper on](/docs-images/73/69523124/images/70-1.jpg "risk based quality management in clinical trials Guidance for Industry: Oversight of Clinical Investigations A Risk-Based Approach to Monitoring, U.S.")
70 References ICH E6_R2, INTEGRATED ADDENDUM TO ICH E6(R1): GUIDELINE FOR GOOD CLINICAL PRACTICE, Current Step 4 version dated 9 November 2016 EMA/269011/2013 [18 November 2013]: Reflection paper on risk based quality management in clinical trials Guidance for Industry: Oversight of Clinical Investigations A Risk-Based Approach to Monitoring, U.S. Department of Health and Human Services, Food and Drug Administration, August
71 Visit our website: Follow us on LinkedIn! Contact us at: 71
TOP 10 INVESTIGATOR RESPONSIBILITIES WHEN CONDUCTING HUMAN SUBJECTS RESEARCH
 Investigators Responsibility #1: Design and Implement Ethical Research Consistent with the Three Ethical Principles Delineated in The Belmont Report. The Belmont Report: Three Basic Ethical Principles:
Investigators Responsibility #1: Design and Implement Ethical Research Consistent with the Three Ethical Principles Delineated in The Belmont Report. The Belmont Report: Three Basic Ethical Principles:
ELEMENTS OF A DATA MONITORING PLAN
 ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
It s All About The RISK
 Session Q 2: esource Document Verification A Case for CRF Mapping Glenda Guest It s All About The RISK What are some of the realized risks that auditors are observing? Multiple sources, how to ensure consistency
Session Q 2: esource Document Verification A Case for CRF Mapping Glenda Guest It s All About The RISK What are some of the realized risks that auditors are observing? Multiple sources, how to ensure consistency
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
Conducted Under an IND to Support a
 Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Sponsor-Investigator Responsibilities In Clinical Trials
 In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
Research Professionals Network Workshop Series
 Research Professionals Network Workshop Series SO YOU THINK YOU KNOW GCP? Mary-Tara Roth Director, CRRO mtroth@bu.edu Gina Daniels Human Research Quality Manager gdaniels@bu.edu What is [ICH] GCP? an
Research Professionals Network Workshop Series SO YOU THINK YOU KNOW GCP? Mary-Tara Roth Director, CRRO mtroth@bu.edu Gina Daniels Human Research Quality Manager gdaniels@bu.edu What is [ICH] GCP? an
How did it evolve? o Public disasters, serious fraud and abuse of human rights. o Trials of War criminals-nuremberg code 1949
 ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
ICH GCP Overview What is ICH? ICH is a joint initiative involving both the regulators and the industry as equal partners in the scientific and technical discussions of the testing procedures which are
GCP Refresher and GCP/GCDMP Trends. in the CTN. Denise King, MS, RD, CCRA & Lauren Yesko, BS. Presented by:
 GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
GCP Refresher and GCP/GCDMP Trends Presented by: in the CTN Denise King, MS, RD, CCRA & Lauren Yesko, BS CTN WEB SEMINAR SERIES: A FORUM TO EXCHANGE RESEARCH KNOWLEDGE Produced by: CTN Training This training
GCP Basics - refresher
 p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
Demystifying Audits. Audits and Audit Preparation 5/23/2016. What is an Audit?
 Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
Risk Management in Clinical Trials in Today s ICH-GCP(R2) Framework
 Framework") Clinical Research in Resource Limited Settings: Mission Impossible or Role Model for Future Drug Development? Risk Management in Clinical Trials in Today s ICH-GCP(R2) Framework Ingrid Klingmann, MD, PhD,
Clinical Research in Resource Limited Settings: Mission Impossible or Role Model for Future Drug Development? Risk Management in Clinical Trials in Today s ICH-GCP(R2) Framework Ingrid Klingmann, MD, PhD,
Outline of Discussion
 This final rule is intended to better protect human subjects involved in research, while facilitating valuable research and reducing burden, delay, and ambiguity for investigators. These revisions are
This final rule is intended to better protect human subjects involved in research, while facilitating valuable research and reducing burden, delay, and ambiguity for investigators. These revisions are
1 The Clinical Research Coordinator (CRC)... 1
... 1") TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
TABLE OF CONTENTS Dedication... iii Introduction... xi 1 The Clinical Research Coordinator (CRC)... 1 Role and Responsibilities of the CRC...1 Personality and Skills... 3 Where Do CRCs Work?... 3 CRC Responsibilities...
ICH E6 Guideline R 2 優良臨床試驗指引修改及增編附錄
 ICH E6 Guideline R 2 優良臨床試驗指引修改及增編附錄 Mandy Liu 劉文婷 COQM/ Boehringer Ingelheim Taiwan Ltd Former Team leader GCP inspection team/ CDE Member of Expert Working Group of ICH E6 2016.11.23 Statement of the
ICH E6 Guideline R 2 優良臨床試驗指引修改及增編附錄 Mandy Liu 劉文婷 COQM/ Boehringer Ingelheim Taiwan Ltd Former Team leader GCP inspection team/ CDE Member of Expert Working Group of ICH E6 2016.11.23 Statement of the
Human Subject Protection; Acceptance of Data From Clinical Investigations for Medical
 This document is scheduled to be published in the Federal Register on 02/21/2018 and available online at https://federalregister.gov/d/2018-03244, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 02/21/2018 and available online at https://federalregister.gov/d/2018-03244, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
WCG ACADEMY COURSE OVERVIEW
 WCG ACADEMY COURSE OVERVIEW Investigator Course Title Documentation of Informed Consent Duration Ethical Principles Underlying Consent Documentation Long Form Short Form Consent Records Special Considerations
WCG ACADEMY COURSE OVERVIEW Investigator Course Title Documentation of Informed Consent Duration Ethical Principles Underlying Consent Documentation Long Form Short Form Consent Records Special Considerations
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd.
 Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Human Research Protection Program Policy
 Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices
 Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
Trends in Oversight of Human Research Protections?
 Trends in Oversight of Human Research Protections? Greg Koski, PhD, MD Associate Professor, Harvard Medical School Senior Scientist, James Mongan Institute for Health Policy Former Director, Office for
Trends in Oversight of Human Research Protections? Greg Koski, PhD, MD Associate Professor, Harvard Medical School Senior Scientist, James Mongan Institute for Health Policy Former Director, Office for
Human Research Protection Program Policy
 Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
HUMAN RESEARCH PROTECTION PROGRAM PLAN
 HUMAN RESEARCH PROTECTION PROGRAM PLAN HRP 101 June 2017 706-542-3199 irb@uga.edu https://research.uga.edu/hso/ Table of Contents Scope... 2 Purpose... 2 Definitions... 2 Agent... 2 Clinical Trial... 2
HUMAN RESEARCH PROTECTION PROGRAM PLAN HRP 101 June 2017 706-542-3199 irb@uga.edu https://research.uga.edu/hso/ Table of Contents Scope... 2 Purpose... 2 Definitions... 2 Agent... 2 Clinical Trial... 2
Human Research Protection Program. Plan
 Human Research Protection Program Plan Revised September 6, 2017 HRP-101 9/6/17 2 of 13 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Human Research Protection Program Plan Revised September 6, 2017 HRP-101 9/6/17 2 of 13 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Implementing Good Clinical Practice at an Academic Research Institution
 Implementing Good Clinical Practice at an Academic Research Institution Maintaining Essential Documents Partners Human Research Quality Improvement (QI) Program Stephen W. Hayes Outline Significance of
Implementing Good Clinical Practice at an Academic Research Institution Maintaining Essential Documents Partners Human Research Quality Improvement (QI) Program Stephen W. Hayes Outline Significance of
Guidelines for Setting Up a Regulatory Binder
 Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Advocate Health Care Network. Human Research Protection Program. Plan
 Advocate Health Care Network Human Research Protection Program Plan Revised October 19, 2014 Page 1 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Clinical Trial... 3 Engaged in Human Research...
Advocate Health Care Network Human Research Protection Program Plan Revised October 19, 2014 Page 1 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Clinical Trial... 3 Engaged in Human Research...
Regulatory Document Guidelines for DMID Clinical Studies. Version Oct-2005
 Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1
 Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
Volunteering for Clinical Trials
 Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Volunteering for Clinical Trials Volunteering for Clinical Trials When considering volunteering for a clinical trial, it is important to make an informed decision. Below are answers to frequently asked
Sven Trelle CTU Bern University of Bern
 International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E6(R2) Integrated Addendum to Good Clinical Practice (GCP) Guideline Sven Trelle CTU Bern University
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E6(R2) Integrated Addendum to Good Clinical Practice (GCP) Guideline Sven Trelle CTU Bern University
Clinical Trials & Regulatory
 Clinical Trials & Regulatory Infrastructures t MENA Raeda Mustafa, B.Sc.Pharm., MBA, CCRA, CCRC/ Molecule CRO Updates to ICH GCP E6 (R2) and BE Studies 3rd MENA Regulatory Conference on Bioequivalence,
Clinical Trials & Regulatory Infrastructures t MENA Raeda Mustafa, B.Sc.Pharm., MBA, CCRA, CCRC/ Molecule CRO Updates to ICH GCP E6 (R2) and BE Studies 3rd MENA Regulatory Conference on Bioequivalence,
Considerations for Using etools in Research: Part 11 and System Validation
 Considerations for Using etools in Research: Part 11 and System Validation Mitchell Parrish, JD, RAC, CIP, VP of Legal Affairs James Riddle, MCSE, CIP, CPIA, VP of Client Services 04/19/18 description
Considerations for Using etools in Research: Part 11 and System Validation Mitchell Parrish, JD, RAC, CIP, VP of Legal Affairs James Riddle, MCSE, CIP, CPIA, VP of Client Services 04/19/18 description
\\NAS1\George\Docs\SoCRA\CCRP communications\study guide management
 Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
Five Content Areas Percent of Scored Test Items (Range) in Each Area This table shows the percent of scored test questions that are included in each major content area. Five Content Areas Ethical Principles
Clinical Research: A Multifaceted Discipline
 Clinical Research: A Multifaceted Discipline Anuradha Kulkarni a, Arun Bhatt b* a Senior Associate - Medical & Regulatory Affairs, Clininvent Research Pvt Ltd, A-302, Everest Chambers, Marol Naka, Andheri
Clinical Research: A Multifaceted Discipline Anuradha Kulkarni a, Arun Bhatt b* a Senior Associate - Medical & Regulatory Affairs, Clininvent Research Pvt Ltd, A-302, Everest Chambers, Marol Naka, Andheri
1.4 Applicable Regulatory Requirement(s) Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.
 Any law(s) and regulation(s) addressing the conduct of clinical trials of investigational products.") 1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
1.1 Adverse Drug Reaction (ADR) In the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) may not be established: all noxious and unintended
GCP/Clinical Investigation in Japan
 GCP/Clinical Investigation in Japan 27-28 August, 2018 Shinwa Shibata Office of Non-clinical and Clinical Compliance Pharmaceuticals and Medical Devices Agency 1 Today s Agenda 1. Japanese-GCP (J-GCP)
GCP/Clinical Investigation in Japan 27-28 August, 2018 Shinwa Shibata Office of Non-clinical and Clinical Compliance Pharmaceuticals and Medical Devices Agency 1 Today s Agenda 1. Japanese-GCP (J-GCP)
Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework
 1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
1 Quality Assurance for the Research Team: Connecting Day-to-Day Operations to a Regulatory Framework Mandy Morneault Research Compliance Monitor University of Washington Institute of Translational Health
Human Subjects Protection Program Plan
 December 1, 2016 HRP-101 12/1/16 2 of 12 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research... 3 Human Research:... 4 Human Subject as Defined
December 1, 2016 HRP-101 12/1/16 2 of 12 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research... 3 Human Research:... 4 Human Subject as Defined
FDA Sponsor and Investigator Responsibility Checklist
 FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
3.1. Overall Principal Investigator (PI), who holds the IDE and/or is the Sponsor
, who holds the IDE and/or is the Sponsor") POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities
POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)
 SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)") POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
Quality Assurance in Clinical Trials
 Quality Assurance in Clinical Trials Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 5th, 2011 1 Introduction: quality in clinical research in human subjects Regulatory requirements:
Quality Assurance in Clinical Trials Doctor Catherine CORNU, Lyon clinical Investigation Centre EUDIPHARM December 5th, 2011 1 Introduction: quality in clinical research in human subjects Regulatory requirements:
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval
 Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
Data Quality and Integrity: From Clinical Monitoring to Marketing Approval Nancy Detich, Ph.D., C.C.R.P. Senior Scientist, Clinical Strategy 18 November 2010 1 Objectives Identify the importance of accuracy,
1. POLICY STATEMENT: 2. BACKGROUND:
 POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
Good Clinical Practice. Martin Rose, MD, JD February 8, 2018 ASQ
 Good Clinical Practice Martin Rose, MD, JD February 8, 2018 ASQ Disclaimer The views expressed in this presentation are those of the presenter and do not necessarily represent the official position of
Good Clinical Practice Martin Rose, MD, JD February 8, 2018 ASQ Disclaimer The views expressed in this presentation are those of the presenter and do not necessarily represent the official position of
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES
 IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals
 ORIENTATION FOR NEW CLINICAL RESEARCH COORDINATORS Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals Overall Agenda for Orientation Module 1: Introduction
ORIENTATION FOR NEW CLINICAL RESEARCH COORDINATORS Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals Overall Agenda for Orientation Module 1: Introduction
European Academy of Hospital Pharmacy Biotechnology Educational Summit. Clinical trials David Gerrett (Acknowledgment Mark Howells)
") European Academy of Hospital Pharmacy Biotechnology Educational Summit Clinical trials David Gerrett (Acknowledgment Mark Howells) Objectives Have an appreciation and overview of clinical trials Understand
European Academy of Hospital Pharmacy Biotechnology Educational Summit Clinical trials David Gerrett (Acknowledgment Mark Howells) Objectives Have an appreciation and overview of clinical trials Understand
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
ClinicalTrials.gov Registration Guide
 ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
Ethics Committees/IRBs Today: Challenges for Efficiency and Quality
 Marjorie A. Speers, Ph.D. President and CEO Ethics Committees/IRBs Today: Challenges for Efficiency and Quality Copyright 2013 AAHRPP All rights reserved Goal Clinical Research Globally Access to patients
Marjorie A. Speers, Ph.D. President and CEO Ethics Committees/IRBs Today: Challenges for Efficiency and Quality Copyright 2013 AAHRPP All rights reserved Goal Clinical Research Globally Access to patients
Inspections: an academic perspective
 Inspections: an academic perspective Patricia Henley Quality and Governance Manager Head, Research Governance & Integrity Office London School of Hygiene & Tropical Medicine Email: patricia.henley@lshtm.ac.uk
Inspections: an academic perspective Patricia Henley Quality and Governance Manager Head, Research Governance & Integrity Office London School of Hygiene & Tropical Medicine Email: patricia.henley@lshtm.ac.uk
Developing and Implementing Standard Operating Procedures
 Developing and Implementing Standard Operating Procedures L A U R A T U T T L E, M A, C C R P U N C T R I A L I N N O V A T I O N U N I T P R O G R A M M A N A G E R NC T R A C S I N S T I T U T E Objectives
Developing and Implementing Standard Operating Procedures L A U R A T U T T L E, M A, C C R P U N C T R I A L I N N O V A T I O N U N I T P R O G R A M M A N A G E R NC T R A C S I N S T I T U T E Objectives
Learning about Clinical Trials
 Learning about Clinical Trials A Guide for Individuals and Their Loved Ones INTRODUCTION Clinical trials help researchers answer important medical questions, providing information that may help with the
Learning about Clinical Trials A Guide for Individuals and Their Loved Ones INTRODUCTION Clinical trials help researchers answer important medical questions, providing information that may help with the
Applicability of US Regulations to Canadian Research
 Applicability of US Regulations to Canadian Research Mary Kate Needler Capital District Health Authority Halifax, Nova Scotia, Canada N2 Stakeholder Meeting August 18, 2014 MK s Conventions: 1. US Regulation
Applicability of US Regulations to Canadian Research Mary Kate Needler Capital District Health Authority Halifax, Nova Scotia, Canada N2 Stakeholder Meeting August 18, 2014 MK s Conventions: 1. US Regulation
ClinicalTrials.gov REGISTRATION REQUIREMENTS
 ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
ICH GCP Revision and EU Clinical Trial Regulation
 ICH GCP Revision and EU Clinical Trial Regulation Sinead Curran, GCP/PhV Inspection Manager Irish Research Nurses Network Annual National Conference Friday, 17 November 2017 Overview Guideline for Good
ICH GCP Revision and EU Clinical Trial Regulation Sinead Curran, GCP/PhV Inspection Manager Irish Research Nurses Network Annual National Conference Friday, 17 November 2017 Overview Guideline for Good
Human Research Protection Program. Investigator Manual
 Human Research Protection Program Revised July 7, 2014 HRP-910 7/7/2014 2 of 10 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection
Human Research Protection Program Revised July 7, 2014 HRP-910 7/7/2014 2 of 10 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection
FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up?
 FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up? The following framework of characteristics focuses on attributes that are within the control of investigators and their
FRAMEWORK OF CHARACTERISTICS OF A QUALIFIED SITE TEAM: How Does Yours Measure Up? The following framework of characteristics focuses on attributes that are within the control of investigators and their
GW Policy on Human Research Protection Program
 GW Policy on Human Research Protection Program 1. PURPOSE 1.1. This policy establishes the [Organization] s Human Research Protection Program (HRPP) and its commitment to protect the rights and welfare
GW Policy on Human Research Protection Program 1. PURPOSE 1.1. This policy establishes the [Organization] s Human Research Protection Program (HRPP) and its commitment to protect the rights and welfare
Device research sponsors, whether companies or investigators, are held responsible for meeting the same regulations.
 POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: A DF/HCC Investigator who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities that
POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: A DF/HCC Investigator who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities that
Conducting and reporting a GCP Inspection. Gunnar Danielsson Medical Products Agency
 Conducting and reporting a GCP Inspection Gunnar Danielsson Medical Products Agency Preparation for an inspection Inspection plan Create worksheets for the inspection general project specific study specific
Conducting and reporting a GCP Inspection Gunnar Danielsson Medical Products Agency Preparation for an inspection Inspection plan Create worksheets for the inspection general project specific study specific
Ethical Issues in Clinical Trial & Development of Regulation Policy on Clinical Research
 Ethical Issues in Clinical Trial & Development of Regulation Policy on Clinical Research Melody Lin, Ph.D. Deputy Director, Office for Human Research Protections Director, International Activities Department
Ethical Issues in Clinical Trial & Development of Regulation Policy on Clinical Research Melody Lin, Ph.D. Deputy Director, Office for Human Research Protections Director, International Activities Department
TITLE: DF/HCC Investigator-Sponsored Multi-Center Research POLICY #: MULTI-100 Page: 1 of 4 Effective Date: 1/31/19
 POLICY #: MULTI-100 Page: 1 of 4 1. POLICY STATEMENT: The DF/HCC Sponsor-Investigator, Coordinating Center and External Sites collaborating in DF/HCC Investigator-Sponsored Multi-Center research must comply
POLICY #: MULTI-100 Page: 1 of 4 1. POLICY STATEMENT: The DF/HCC Sponsor-Investigator, Coordinating Center and External Sites collaborating in DF/HCC Investigator-Sponsored Multi-Center research must comply
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
Regulatory Binder Guidance
 Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
ClinicalTrials.gov Registration and Results Reporting: Updates and Recent Activity
 Vol. 7, No. 2, February 2011 Can You Handle the Truth? ClinicalTrials.gov Registration and Results Reporting: Updates and Recent Activity By Matthew Lester and Barbara Godlew The Food and Drug Administration
Vol. 7, No. 2, February 2011 Can You Handle the Truth? ClinicalTrials.gov Registration and Results Reporting: Updates and Recent Activity By Matthew Lester and Barbara Godlew The Food and Drug Administration
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
Human Research Protection Program. Plan
 Human Research Protection Program Plan Dated: September 4, 2014 HRP-101 9/4/2014 2 of 14 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Human Research Protection Program Plan Dated: September 4, 2014 HRP-101 9/4/2014 2 of 14 Table of Contents Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Standard Operating Procedures (SOPs)
") Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Organizational Strategies for Clinical Trials Standard Operating Procedures (SOPs) SUSAN JACKSON, MPA UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL CENTER FOR GASTROINTESTINAL BIOLOGY AND DISEASE UNC CENTER
Human Research Participant Protection Program Institutional Review Board (IRB)
") Human Research Participant Protection Program Institutional Review Board (IRB) Procedure 1: Determining whether a research activity needs IRB review and approval 1. Subject All research activities that
Human Research Participant Protection Program Institutional Review Board (IRB) Procedure 1: Determining whether a research activity needs IRB review and approval 1. Subject All research activities that
Vanessa Laroche, CCRC, CCRA, CIP, CQA Quality Assurance Specialist Technical Resources International (TRI), Inc.
, Inc.") December 8, 2016 C2012 Web Seminar Series Identifying and Reporting Protocol Deviations Vanessa Laroche, CCRC, CCRA, CIP, CQA Quality Assurance Specialist Technical Resources International (TRI), Inc.
December 8, 2016 C2012 Web Seminar Series Identifying and Reporting Protocol Deviations Vanessa Laroche, CCRC, CCRA, CIP, CQA Quality Assurance Specialist Technical Resources International (TRI), Inc.
Human Research Protection Program Plan
 Revised January 19, 2018 Table of Contents Human Research Protection Program Plan HRP-101 1/19/2018 2 of 13 Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Revised January 19, 2018 Table of Contents Human Research Protection Program Plan HRP-101 1/19/2018 2 of 13 Scope... 3 Purpose... 3 Definitions... 3 Agent... 3 Clinical Trial... 3 Engaged in Human Research...
Outline 18/11/14. Implementation of the DoH in the Americas: Challenges and successes. ! Background and context. ! United States. !
 Implementation of the DoH in the Americas: Challenges and successes Dr. Jeff Blackmer Director of Ethics Canadian Medical Association 1 Outline! Background and context! United States! Canada! South America
Implementation of the DoH in the Americas: Challenges and successes Dr. Jeff Blackmer Director of Ethics Canadian Medical Association 1 Outline! Background and context! United States! Canada! South America
Establishing our Trajectory: An Overview & Assessment of the Impact of ICH E6 R2 on Sites and Sponsors
 Establishing our Trajectory: An Overview & Assessment of the Impact of ICH E6 R2 on Sites and Sponsors Module 1 of a 4 Part Series Nicole Stansbury Executive Director, Adaptive & Intelligent Monitoring,
Establishing our Trajectory: An Overview & Assessment of the Impact of ICH E6 R2 on Sites and Sponsors Module 1 of a 4 Part Series Nicole Stansbury Executive Director, Adaptive & Intelligent Monitoring,
Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective
 1 Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective Kate-Louise Gottfried, JD, MSPH, Senior Director Stephanie C. Guzik, RN, BSN, MBA, Assistant Director Research
1 Effectively Preparing for and Responding to an FDA Audit: The Research Team s Perspective Kate-Louise Gottfried, JD, MSPH, Senior Director Stephanie C. Guzik, RN, BSN, MBA, Assistant Director Research
TITLE: DF/HCC Investigator-Sponsored Multi-Center TrialsResearch POLICY #: MULTI-100 Page: 11 of 54 Effective Date: 8/31/17 1.
 POLICY #: MULTI-100 Page: 11 of 54 1. POLICY STATEMENT: The DF/HCC Sponsor-Investigator, Coordinating Center and External Sites collaborating in DF/HCC Investigator-Sponsored Multi-Center Trialsresearch
POLICY #: MULTI-100 Page: 11 of 54 1. POLICY STATEMENT: The DF/HCC Sponsor-Investigator, Coordinating Center and External Sites collaborating in DF/HCC Investigator-Sponsored Multi-Center Trialsresearch
Good Clinical Practice
 Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Dublin Academic Medical Centre Good Clinical Practice Patrick Murray, MD, FASN, FRCPI Professor, University College Dublin, Mater Misericordiae University Hospital, Dublin, Ireland patrick.murray@ucd.ie
Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes
 Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes Martha E. Payne, PhD, MPH Office of Research Development, Duke University School of Medicine Objectives for Today What has
Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes Martha E. Payne, PhD, MPH Office of Research Development, Duke University School of Medicine Objectives for Today What has
Good Clinical Practice Compliance
 Good Clinical Practice Compliance Pharmaceutical Regulatory & Compliance Congress Greg Levine LLP Agenda? GCP compliance rules What is the law? What other (non-binding) standards apply? What are the unwritten
Good Clinical Practice Compliance Pharmaceutical Regulatory & Compliance Congress Greg Levine LLP Agenda? GCP compliance rules What is the law? What other (non-binding) standards apply? What are the unwritten
REVISION OF THE CLINICAL TRIALS DIRECTIVE 2001/20/EC UK RESPONSE TO CONCEPT PAPER
 REVISION OF THE CLINICAL TRIALS DIRECTIVE 2001/20/EC UK RESPONSE TO CONCEPT PAPER Introduction This letter provides the UK Government s response to the Commission s public consultation on the proposed
REVISION OF THE CLINICAL TRIALS DIRECTIVE 2001/20/EC UK RESPONSE TO CONCEPT PAPER Introduction This letter provides the UK Government s response to the Commission s public consultation on the proposed
RBM Risk Based Monitoring GCP Training 12/SEP/2015. Gabor Kiss Synexus Hungary2015
 RBM Risk Based Monitoring GCP Training 12/SEP/2015 Gabor Kiss Synexus Hungary2015 Agenda Why traditional monitoring must change? The landscape Regulatory Industry IT What can we expect? Transition period
RBM Risk Based Monitoring GCP Training 12/SEP/2015 Gabor Kiss Synexus Hungary2015 Agenda Why traditional monitoring must change? The landscape Regulatory Industry IT What can we expect? Transition period
OFFICE FOR RESEACH PROCEDURE. Site Initiation and Close-out
 OFFICE FOR RESEACH PROCEDURE Site Initiation and Close-out 1. Purpose: To describe the procedures related to site initiation and close-out of a clinical trial. 2. Scope: Applicable to all phases of clinical
OFFICE FOR RESEACH PROCEDURE Site Initiation and Close-out 1. Purpose: To describe the procedures related to site initiation and close-out of a clinical trial. 2. Scope: Applicable to all phases of clinical
Compliance and Quality Monitoring: What, Why, When, and How
 Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
Sponsor/Investigator Responsibilities
 Sponsor/Investigator Responsibilities Marian Serge, RN Division of Bioresearch Monitoring Office of Compliance Center for Devices and Radiological Health Food and Drug Administration Marian.Serge@fda.hhs.gov
Sponsor/Investigator Responsibilities Marian Serge, RN Division of Bioresearch Monitoring Office of Compliance Center for Devices and Radiological Health Food and Drug Administration Marian.Serge@fda.hhs.gov
Good Clinical Practice
 Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
Clinical Site Monitoring DMID/ICSSC 10/7/08 1 Monitoring ICH E6 5.18.1 The purpose of monitoring is to ensure that: The rights and well-being of subjects are being protected The data are accurate, complete
Evolving Role of the Data Manager
 07 November 2016 Evolving Role of the Data Manager 2 Evolution of the Data Manager Role... 2014 2010 Mobility Early 90 s We all simply did it OUR way! No formal measure of data managers' knowledge and
07 November 2016 Evolving Role of the Data Manager 2 Evolution of the Data Manager Role... 2014 2010 Mobility Early 90 s We all simply did it OUR way! No formal measure of data managers' knowledge and
4.2. Investigator Regulatory Binder: Files, usually a binder, maintained by the investigator at the investigative site.
 POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
ROLE OF THE RESEARCH COORDINATOR Investigational New Drug Application-Sponsor Responsibilities 21CFR Part , subpart D
 Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
The Institutional Review Board (IRB) Manual
 Manual") The Institutional Review Board (IRB) Manual Investigational Devices Policy Research that involves the use of investigational devices must conform to Food and Drug Administration (FDA) and Department of
The Institutional Review Board (IRB) Manual Investigational Devices Policy Research that involves the use of investigational devices must conform to Food and Drug Administration (FDA) and Department of
Views of a Clinical Study Report
 Out-of-(CSR)-Body Experiences Tips on Assembling Appendices, Datasets, and CRFs Susan C Sisk, PhD, RAC 1 Views of a Clinical Study Report OR Photos courtesy of Leigh Vaughan and RAPS, 2008 2 Topics Process
Out-of-(CSR)-Body Experiences Tips on Assembling Appendices, Datasets, and CRFs Susan C Sisk, PhD, RAC 1 Views of a Clinical Study Report OR Photos courtesy of Leigh Vaughan and RAPS, 2008 2 Topics Process
Annual Research Administrators Symposium IRB Compliance. Thursday, July 31, 2014
 Annual Research Administrators Symposium IRB Compliance Thursday, July 31, 2014 Why IRB Compliance Is Required for Grant Submissions Topics to Cover Today What is an IRB? Why IRB review is required? Regulatory
Annual Research Administrators Symposium IRB Compliance Thursday, July 31, 2014 Why IRB Compliance Is Required for Grant Submissions Topics to Cover Today What is an IRB? Why IRB review is required? Regulatory
Guidance on Requirements of the Sponsor and the Investigator as a Sponsor
 Guidance on Requirements of the Sponsor and the Investigator as a Sponsor University of Colorado Denver (UCD) secures assurances from the sponsor or the investigator-sponsor* that the manufacture and formulation
Guidance on Requirements of the Sponsor and the Investigator as a Sponsor University of Colorado Denver (UCD) secures assurances from the sponsor or the investigator-sponsor* that the manufacture and formulation
RESEARCH AUDIT Standard Operating Procedure
 Reference Number: UHB 236 Version Number: 2 Date of Next Review: 17 th Oct 2020 Previous Trust/LHB Reference Number: N/A RESEARCH AUDIT Standard Operating Procedure Introduction and Aim As a legal Sponsor
Reference Number: UHB 236 Version Number: 2 Date of Next Review: 17 th Oct 2020 Previous Trust/LHB Reference Number: N/A RESEARCH AUDIT Standard Operating Procedure Introduction and Aim As a legal Sponsor
etmf A Regulators Perspective Paula Walker, GCP Operations Manager & Senior GCP Inspector
 etmf A Regulators Perspective Paula Walker, GCP Operations Manager & Senior GCP Inspector Agenda EU Legislation and Guidance News/Updates relating to TMF Structure and Content of e/tmf Inspecting etmfs
etmf A Regulators Perspective Paula Walker, GCP Operations Manager & Senior GCP Inspector Agenda EU Legislation and Guidance News/Updates relating to TMF Structure and Content of e/tmf Inspecting etmfs
Investigator Manual. Human Subjects Protection Program
 Human Subjects Protection Program HRP-103, Revised June 1, 2015 HRP-103 06/01/2015 2 of 33 Table of Contents Scope... 3 What is the purpose of this manual?... 3 What is Human Research?... 3 What is the
Human Subjects Protection Program HRP-103, Revised June 1, 2015 HRP-103 06/01/2015 2 of 33 Table of Contents Scope... 3 What is the purpose of this manual?... 3 What is Human Research?... 3 What is the
Ethics in CTs. The Role of the Regulator versus The Role of the REB
 gust 22, 2007 8:45 11:45 gust 22, 2007 8:45 11:45 ADVANCED WORKSHOP : REVIEW OF Helping the people of Canada maintain and improve their health Aider les Canadiens et les Canadiennes à maintenir et à améliorer
gust 22, 2007 8:45 11:45 gust 22, 2007 8:45 11:45 ADVANCED WORKSHOP : REVIEW OF Helping the people of Canada maintain and improve their health Aider les Canadiens et les Canadiennes à maintenir et à améliorer
Good Clinical Practice (GCP) & Clinical Trial Registries
 & Clinical Trial Registries") Good Clinical Practice (GCP) & Clinical Trial Registries The Fifth Annual Pharmaceutical Regulatory and Compliance Congress and Best Practice Forum November 14-17, 2004 Kate Maloney, RN, MS, CPHQ Manager,
Good Clinical Practice (GCP) & Clinical Trial Registries The Fifth Annual Pharmaceutical Regulatory and Compliance Congress and Best Practice Forum November 14-17, 2004 Kate Maloney, RN, MS, CPHQ Manager,
Investigator s Responsibility
 Investigator s Responsibility Introduction Investigator s Qualifications Clinical Trial Agreement Adequate Resources Medical Care of Trial Subjects Communication with IRB/IEC Study Initiation Patient Recruitment
Investigator s Responsibility Introduction Investigator s Qualifications Clinical Trial Agreement Adequate Resources Medical Care of Trial Subjects Communication with IRB/IEC Study Initiation Patient Recruitment