Corso di Laurea Magistrale in Chimica e Tecnologia Farmaceutiche E25. Fabbricazione Industriale dei Medicinali 4 CFU Prof.
|
|
|
- Trevor Lane
- 6 years ago
- Views:
Transcription



1 Sezione di Tecnologia e Legislazione Farmaceutiche Maria Edvige Sangalli Corso di Laurea Magistrale in Chimica e Tecnologia Farmaceutiche E25 Fabbricazione Industriale dei Medicinali 4 CFU Prof. Andrea Gazzaniga Stability program in the development of a medicinal product Dott. Carlo Vecchio

2 Stability program in the development of a medicinal product Carlo Vecchio 2

3 Introduction A medicinal product is designed to possess certain desirable properties of which the stability is of major importance. When the product is administered by the specified route, the active substance should achieve the required rate of absorption and extent of bioavailability. The product itself should be efficacious, safe, and acceptable to the patient; it should be convenient in use and stable. 3

4 Introduction The stability of a product relates to its resistance to the various chemical, physical, and microbiological reactions that may change the original properties of the preparation during transport, storage, and use. Other criteria of stability are the effects of such changes on the fitness of the product for use as a medicine. 4
5 Criteria for acceptable level of stability Type of stability Chemical Physical Microbiological Therapeutic Toxicological Conditions maintain thought the shelf-life of the drug product Each active ingredient retains its chemical integrity and labeled potency, within the specific limits. The original physical properties, including appearance, palatability, uniformity, dissolution, and suspendability are retained. Sterility or resistance to microbial growth requirements. Antimicrobial agents that are present retain effectiveness within the specific limits. The therapeutic effect remains unchanged. No significant increase in toxicity occurs. 5

6 What is the goal of stability program? The goal of a stability program is not uniquely defined, but depends on the stage of the development of the product in question. 6
7 Drug development Drug development is the process of bringing a new pharmaceutical drug to the market once a lead compound has been identified through the process of drug discovery. It includes pre-clinical research on microorganisms and animals, clinical trials on humans, and may include the step of obtaining regulatory approval to market the drug. 7
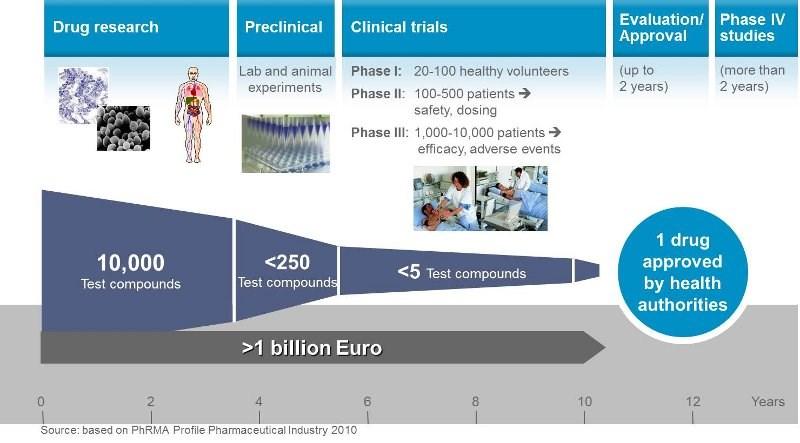
8 Drug development 8
and physical ( polymorphs, particle size/surface area, other attributes as resuspendibility and aggregation) - Essential for developing analytical methods - Essential")
9 The Role of Stability in Drug Development - Stability studies play a central role in drug development - Permit understanding of the molecule Mechanism of instability: chemical (labile centers, external factors, degradation)and physical ( polymorphs, particle size/surface area, other attributes as resuspendibility and aggregation) - Essential for developing analytical methods - Essential for selecting packaging for drug substance and drug product Linked to mechanisms of instability (protect from light and moisture), potential drug absorption and adsorption to container closure), container orientation and potential for leachables (container, closure, glue and ink) - Essential for choosing storage conditions for drug substance and drug product 9

10 Definitions Absorption Assimilation of molecular species throughout the bulk of the solid or liquid is termed as absorption. Adsorption Accumulation of the molecular species at the surface rather than in the bulk of the solid or liquid is termed as adsorption. Extractables Compounds that can be extracted from the container closure system when in the presence of a solvent. Leachables Compounds that leach into the drug product formulation from the container closure as a result of direct contact with the formulation. 10
11 Stability Quality by Design Pharmaceutical product stability is a function of: Drug Substance Excipients Understanding Product Manufacturing Process Container Closure 11

12 Goals of the stability in preformulation At the very onset of development it is desired to know: a. what the inherent stability of the drug substance is and b. what interaction with excipients can be expected. 12
 or equivalent. This also applies to the toxicology formulations as a recommendations for stability of the drug substance in the vehicle used in the animal trials.")
13 On the analytical side in preformulation The stability program is usually supported by an analytical research group, which is responsible for the stability indicating assay that goes into the NDA (New Drug Application) or equivalent. This also applies to the toxicology formulations as a recommendations for stability of the drug substance in the vehicle used in the animal trials. 13
 has become more clearly mandated.")
14 Stability-indicating assay The stability-indicating assay is a method that is employed for the analysis of stability samples in pharmaceutical industry. With the advent of ICH guidelines, the requirement of establishment of stability-indicating assay method (SIAM) has become more clearly mandated. The guidelines explicitly require conduct of forced decomposition studies under a variety of conditions, like ph, light, oxidation, dry heat, etc. and separation of drug from degradation products. The method is expected to allow analysis of individual degradation products. 14
15 Goals of the stability in preclinical formulation In the preclinical formulation phase the choice of formula is the primary goal. This is an important aspect, since this will be the temporary formula framework which is entered into IND (Investigational New Drug Application) or equivalent. 15
16 Goals of the stability in clinical formulations In the new product stability phase, clinical batches are involved and the goal of the stability program is: a) to ascertain that the batches tested in the clinic are stable, and b) they serve to support for the final NDA. Analytical support comes from analytical research up until this point, and at NDA time from routine QC (quality control). 16

17 Goals of the stability in product monitoring In the product stability monitoring, the goal is: - to fulfill the commitment part of the NDA and - to adhere to GMP requirements that a program exist to ascertain stability of marketed product. This is spelled out in the guidelines that the first three market batches and at least one batch per year be tested on stability. 17

18 Goals of the stability in post-nda changes Finally, it is obvious that a marketed product could be undergone to substantial changes in: - formulation, supplier, container-closure - new manufacturing facilities, and the goal is to show that the change does not adversely affect stability. Post-NDA changes are usually handled by a production support group and QC on the analytical side. 18

19 Stability functions Hence, stability in itself does not define a single function. There are functionally five types of stability functions: 1. Preformulation 2. Preclinical formulation 3. Clinical and NDA formulation 4. Commitment and product monitoring 5. Post-NDA change of formulation. 19
20 Stability studies during drug product Lifecycle File IND File NDA Approval Drug Discovery Preclinical Phase I Phase II Phase III Final Labelling Discussion Phase IV PLCM (Product Life Cycle Management) Rx to OTC Stability studies to support formulation development and clinical studies Stability studies to support marketing application Stability studies to monitor product quality and support post approval changes 20
 sensitivity to moisture, heat, light and oxygen, b) interaction probabilities (compatibility with excipients), c) optimum ph (when in")
21 Preformulation The early phase of development of new drug includes the conveyance of information relating to the formulation design to the formulators. The most salient aspects are: a) sensitivity to moisture, heat, light and oxygen, b) interaction probabilities (compatibility with excipients), c) optimum ph (when in solution or suspension). 21
 drug substance as is 2) drug substance with moisture added as water (approximately 5%) 3) drug substance equilibrated at high relative humidity (75%")
22 Heat-stability program Accurately weighed samples of drug substance are placed into appropriate containers. The following are typical variations studied: 1) drug substance as is 2) drug substance with moisture added as water (approximately 5%) 3) drug substance equilibrated at high relative humidity (75% RH or greater) 4) overlaying the headspace of the container with nitrogen versus air prior to sealing. 22
 1 2 4 8 Refrigeration (x) (x) (x) (x) Room temperature (x) (x) (x) (x) 55 C x x x x 75 C x x x 95 C x x a Parentheses indicate samples pulled but not assay")
23 Typical short-term stress stability program The containers are sealed and placed at several temperature conditions and testing according to a typical scheme shown in the table. Temperature Duration a (weeks) Refrigeration (x) (x) (x) (x) Room temperature (x) (x) (x) (x) 55 C x x x x 75 C x x x 95 C x x a Parentheses indicate samples pulled but not assay (as controls) 23
.")
24 Light stability program The standard conditions for photostability testing are described in ICH Q1B guidelines. Exposure samples with a validated chemical actinometric system to both the cool white fluorescent and near ultra violet lamps for appropriate duration of time (Option 2). Exposure of the drug substance in a open petri disches to 900 footcandles (fc) of illumination for 4 weeks is adequate to provide some idea whether light protection is required. 24
, placed in a oven at a constant temperature. These data are useful in determining if the material should be protected and stored in controlled low-humidity environment.")
25 Humidity stability program The dependence on the ambient moisture is known by exposing the drug substance to different relative humidity conditions using laboratory desiccators (containing saturated Solutions of various salts), placed in a oven at a constant temperature. These data are useful in determining if the material should be protected and stored in controlled low-humidity environment. 25
 is a gravimetric technique that measures how quickly and how much of a solvent is absorbed by a sample: such as a dry powder absorbing water.")
26 Humidity stability program Dynamic. vapor sorption (DVS) is a gravimetric technique that measures how quickly and how much of a solvent is absorbed by a sample: such as a dry powder absorbing water. It does this by varying the vapor concentration surrounding the sample and measuring the change in mass which this produces. 26

27 Effect of oxygen The stability investigation of the sensitivity of drug substance to atmospheric oxygen can often be combined with elevated temperature studies comparing samples under inert gas and air. Samples are placed at C for a week and analyzed. 27
.")
28 ph-stability profile A ph stability profile experiment is performed with solution samples between ph 2 and 12 at a selected elevated temperature. Analytically prepared solutions, close to the desired concentration, are prepared used buffer solutions within selected ph range and filled in ampuls (air and nitrogen headspace). The samples are maintained at a specific temperature between 55 and 95 C for two weeks. 28

29 Autoclaving studies Since autoclaving is a preferred means of achieving the sterility of the solutions, an early determination of stability to autoclaving should be made. Ampuls containing solutions at the optimum ph range previously established are exposed to autoclaving conditions of 121 C at 2 bar for 20, 30, 45, and 90 minutes. 29
 are prepared and subjected to: - thermal analysis (DSC) and/or - stress studies (e.g., 55 C for 2 weeks) before analysis by chromatographic method.")
30 Drug excipient compatibility One of the objective of a preformulation study is to identify compatible, potentially useful excipient so that a stable formulation can be developed quickly. Generally, several binary mixtures of a drug and various excipients (generally 1:1 ratio, but lubricants 10:1) are prepared and subjected to: - thermal analysis (DSC) and/or - stress studies (e.g., 55 C for 2 weeks) before analysis by chromatographic method. Also the presence and absence of oxygen, light, humidity can be investigated. From an experience of verification of compatibility with excipients of twenty active principles showed that 10 days at 55 C corresponded to 24 months of storage at 25 C for most of the tested substances. 30
31 Drug excipient compatibility Scheme to identify chemically compatible excipients using DSC with confirmatory HPLC No Interaction Drug Excipient 50% w/w mixture DSC (Differential Scanning Calorometry) Interaction Recommen d excipient HPLC Alternative excipient Yes Significant breakdown No 31

32 What is important in this phase? A good communication between preformulator and formulator as well as both with analyst is essential. The development of a drug dosage form is a team effort, and the responsible of the project development should ascertain this. The best way of communication, as long as there is harmony among people. 32

33 Preclinical formulation Formulation is thought by many to be an art, and there are certain aspects of it that are. Each company has the old timer type of formulator who has a feel for what done with a formula. Such persons are worth their weight in silver, and if coupled with a Knowledgeable formulation scientist become twice their weight s worth in gold. Again, teamwork and good communication are the key words. 33

34 Preclinical formulation The challenge of the formulator is to develop the initial and final dosage forms (selecting the most stable) to the highest quality in the shortest time. It is essential that the formulator understand and effectively utilize the preformulation information on stability. If the drug substance is, e.g., incompatible with magnesium stearate, he cannot take the stand that it is impossible to formulate the product without magnesium stearate. There simply has to be an attempt. He must also stay in touch with the preformulation group, if he thinks that other raw material may be of advantage in the formula. 34
.")
35 Clinical formulation After a product has passed the phase I (safety and appropriate dosage), clinical batches limited to small and medium size are made by clinical manufacturing group, that establishes what the formulation characteristics (specifications). The required stability aspects of clinical are simply to ascertain that each clinical batch is within specifications for the duration of investigation. 35
.")
36 Clinical formulation Many companies place every clinical batch on stability according ICH Q1A guidelines. Others use skip-lot sampling (only a fraction of the submitted lots are inspected to demonstrate that the quality of the submitted product is very good). They place the first clinical batch on accelerated and room temperature stability storage, and, if no adverse stability results are gathered, they simply apply a skiplot principle and place every nth batch on stability and keep stability room temperature retention on remaining lots. 36
 would be advisable.")
37 Clinical formulation Skip-lot sampling is somewhat dangerous practice, because in the early phases there may be substantial batch-to-batch variation. So at least some accelerated studies on more batches (e.g. two weeks at 55 C) would be advisable. Also, clinical batches may provide very valuable backup stability data that will help in the argument the FDA applies against obtaining expiration periods by extrapolation of the data of the final formula. 37
38 Late clinical and first pilot batches In the late phase III clinical, larger batches are made of the product with the formulation which is become firmer and few changes are anticipated. Close to a NDA submission, a scale-up pilot batch (usually half-size) will be made in production equipment and formulation changes must be expected. Stability data on the final batches are analyzed statistically to set a suggested expiration period for the NDA. 38

39 Marketed product stability At the time the NDA is filed, the large clinical and the scale-up batches are still fairly young (usually data from these batches not in excess of 18 months). The FDA does not allow extrapolation, but an extrapolated expiration date is usually calculated and then requested. The FDA often grants two years. There is then the commitment clause, that the follow-up stability be carried out. 39

40 An advise for marketed product stability If smaller problems exist with the formula, it is usually wise to ask for approval of NDA with the formula in hand, and after approval submit an amendment. This would have had to be submitted to equivalence testing relative to both stability and biopharmaceutics. 40

41 Routine monitoring In the marketed product stability the first three to five production batches (as well as one batch per year) will be monitoring by the analytical or quality control department. The stability now becomes the formal stability testing of the marketed product. The first (yearly) report will reassess the temporary expiration date. 41

42 Changes in formula or procedure of existing products When the procedure or formula has been changed for pressing reasons, the change must be filed in a amendment and stability data must be included. Usually, accelerated data demonstrating compatibility with the previous approved drug product plus the standard commitment to continue the stability study, will suffice. Since reaction orders are known at this point, extrapolation can be used with more confidence. 42

43 Conclusions Stability testing of drug substances and drug products begins as part of drug discovery and synthesis or development-preformulation and ends only with the demise of the compound or commercial product. 43
44 A glance to ICH (International Conference Harmonization) guidelines 44

45 ICH Stability Guidelines The ICH Stability Guideline Q1A(R2) defines the stability data package for a new drug substance or drug product that is sufficient for a registration application within the three regions of the EC, Japan, and the United States. Also adopted by some non-ich countries including Canada, Australia, Switzerland. 45
46 General Principles The purpose of stability testing is: - to provide evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as temperature, humidity, and light, and - to establish a re-test period for the drug substance or a shelf life for the drug product and recommended storage conditions. The choice of test conditions defined in this guideline is based on an analysis of the effects of climatic conditions in the three regions of the EC, Japan and the United States (climatic zones I and II). 46
 testing conditions hot/higher humidity World view of climatic zones")
47 ICH Stability Zones Zone Zone I Zone II Zone III Zone IV Zone IVb Type of Climate Temperate zone Mediterranean/subtropical zone Hot dry zone Hot humid/tropical zone ASEAN (Association of Southeast Asian Nations) testing conditions hot/higher humidity World view of climatic zones 47
48 Long Term Testing Conditions Climatic Zone Temperature Humidity Minimum Duration Zone I 21ºC ± 2ºC 45% RH ± 5% RH 12 Months Zone II 25ºC ± 2ºC 60% RH ± 5% RH 12 Months Zone III 30ºC ± 2ºC 35% RH ± 5% RH 12 Months Zone IV 30ºC ± 2ºC 65% RH ± 5% RH 12 Months Zone IVb 30ºC ± 2ºC 75% RH ± 5% RH 12 Months Refrigerated 5ºC ± 3ºC No Humidity 12 Months Frozen -15ºC ± 5ºC No Humidity 12 Months 48
49 Accelerated and Intermediate Testing Conditions Climatic Zone Temperature Humidity Minimum Duration Accelerated Ambient 40ºC ± 2ºC 75% RH ± 5% RH 6 Months Accelerated Refrigerated 25ºC ± 2ºC 60% RH ± 5% RH 6 Months Accelerated Frozen 5ºC ± 3ºC No Humidity 6 Months Intermediate 30ºC ± 2ºC 65% RH ± 5% RH 6 Months 49
50 Drug Product Stability General The design of the formal stability studies for the drug product should be based on knowledge of the behavior and properties of the drug substance and from stability studies on the drug substance and on experience gained from clinical formulation studies. Photostability Testing It should be conducted on at least one primary batch of the drug product (ICH Q1B). Selection of Batches Data from stability studies should be provided on at least three primary batches of the drug product. The primary batches should be of the same formulation and packaged in the same container closure system as proposed for marketing. Container Closure System Stability testing should be conducted on the dosage form packaged in the container closure system proposed for marketing. 50
51 Drug Product Stability Specification Specification is a list of tests, reference to analytical procedures, and proposed acceptance criteria, including the concept of different acceptance criteria for release and shelf life specifications (ICH Q6A and Q6B); in addition, specification for degradation products in a drug product (Q3B R2). Stability studies should include testing of those attributes of the drug product that are susceptible to change during storage and are likely to influence quality, safety, and/or efficacy. The testing should cover the physical, chemical, biological, and microbiological attributes, preservative content (e.g., antioxidant, antimicrobial preservative), and functionality tests (e.g., for a dose delivery system). Analytical procedures should be fully validated and stability indicating. 51
52 Drug Product Stability Testing Frequency For long term studies, frequency of testing should be sufficient to establish the stability profile of the drug product. For products with a proposed shelf life of at least 12 months, the frequency of testing at the long term storage condition should normally be every 3 months over the first year, every 6 months over the second year, and annually thereafter through the proposed shelf life. At the accelerated storage condition, a minimum of three time points, including the initial and final time points (e.g., 0, 3, and 6 months), from a 6-month study is recommended 52
53 Drug Product Stability Storage Conditions In general, a drug product should be evaluated under storage conditions that test its thermal stability and its sensitivity to moisture or potential for solvent loss. The storage conditions and the lengths of studies chosen should be sufficient to cover storage, shipment, and subsequent use. The long term testing should cover a minimum of 12 months duration on at least three primary batches at the time of submission and should be continued for a period of time sufficient to cover the proposed shelf-life. Data from the accelerated storage condition and, if appropriate, from the intermediate storage condition can be used to evaluate the effect of short term excursions outside the label storage conditions (such as might occur during shipping). 53
54 General case Drug Product Stability Study Storage condition Minimum time period covered by data at submission Long term* 25 C ± 2 C/60% RH ± 5% RH or 30 C ± 2 C/65% RH ± 5% RH 12 months Intermediate** 30 C ± 2 C/65% RH ± 5% RH 6 months Accelerated 40 C ± 2 C/75% RH ± 5% RH 6 months *It is up to the applicant to decide whether long term stability studies are performed at 25 ± 2 C/60% RH ± 5% RH or 30 C ± 2 C/65% RH ± 5% RH. **If 30 C ± 2 C/65% RH ± 5% RH is the long-term condition, there is no intermediate condition. 54
55 Drug Product Stability If long-term studies are conducted at 25 C ± 2 C/60% RH ± 5% RH and significant change occurs at any time during 6 months testing at the accelerated storage condition, additional testing at the intermediate storage condition should be conducted and evaluated against significant change criteria. The initial application should include a minimum of 6 months data from a 12-month study at the intermediate storage condition. If the long-term data show variability, verification of the proposed retest period or shelf life by statistical analysis can be appropriate. 55
56 Drug Product Stability In general, significant change for a drug product is defined as: 1. A 5% change in assay from its initial value; or failure to meet the acceptance criteria for potency when using biological or immunological procedures; 2. Any degradation product s exceeding its acceptance criterion; 3. Failure to meet the acceptance criteria for appearance, physical attributes, and functionality test (e.g., color, phase separation, resuspendibility, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g., softening of suppositories, melting of creams) may be expected under accelerated conditions ; and, as appropriate for the dosage form: 4. Failure to meet the acceptance criterion for ph; or 5. Failure to meet the acceptance criteria for dissolution for 12 dosage units. 56
57 Drug Product Stability Stability Commitment When available long term stability data on primary batches do not cover the proposed shelf life granted at the time of approval, a commitment should be made to continue the stability studies post approval in order to firmly establish the shelf life. If the submission includes data from stability studies on at least three production batches, a commitment should be made to continue the long term studies through the proposed shelf life and the accelerated studies for 6 months. 57
58 Drug Product Stability Evaluation A systematic approach should be adopted in the presentation and evaluation of the stability information, which should include, results from the physical, chemical, biological, and microbiological tests, including particular attributes of the dosage form (for example, dissolution rate for solid oral dosage forms). Where the data show so little degradation and so little variability, it is normally unnecessary to go through the formal statistical analysis. An approach for analyzing data of a quantitative attribute that is expected to change with time is to determine the time at which the 95 one-sided confidence limit for the mean curve intersects the acceptance criterion. 58
59 Drug Product Stability Statements/Labeling A storage statement should be based on the stability evaluation of the drug product. Where applicable, specific instruction should be provided, particularly for drug products that cannot tolerate freezing. Terms such as ambient conditions or room temperature should be avoided. There should be a direct link between the label storage statement and the demonstrated stability of the drug product. An expiration date should be displayed on the container label. 59
60 Degradation Products in New Drug Products The summary of the degradation products observed during manufacture and/or stability studies should be based on sound scientific appraisal of potential degradation pathways in the new drug product and impurities arising from the interaction with excipients and/or the immediate container closure system. In addition Any degradation product observed in stability studies conducted at the recommended storage condition should be identified when present at a level greater than (>) the identification thresholds. Degradation products present at a level of not more than ( ) the identification threshold generally would not need to be identified. However, analytical procedures should be developed for those degradation products that are suspected to be unusually potent, producing toxic or significant pharmacological effects at levels not more than ( ) the identification threshold. 60
61 Thresholds for Degradation Products in New Drug Products Reporting Thresholds Maximum Daily Dose 1 Threshold 2,3 1 g 0.1% > 1 g 0.05% Identification Thresholds Maximum Daily Dose 1 Threshold 2, 3 < 1 mg 1.0% or 5 μg TDI, whichever is lower 1 mg - 10 mg 0.5% or 20 μg TDI, whichever is lower >10 mg - 2 g 0.2% or 2 mg TDI, whichever is lower > 2 g 0.10% Qualification Thresholds Maximum Daily Dose 1 Threshold 2,3 < 10 mg 1.0% or 50 μg TDI, whichever is lower 10 mg mg 0.5% or 200 μg TDI, whichever is lower >100 mg - 2 g 0.2% or 3 mg TDI, whichever is lower >2 g 0.15% 1 The amount of drug substance administered per day 2 Thresholds for degradation products are expressed either as a percentage of the drug substance or as total daily intake (TDI) of the degradation product. Lower thresholds can be appropriate if the degradation product is unusually toxic. 3 Higher thresholds should be scientifically justified. 61
62 Specifications: New Chemical Drug Substances and Products A specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a new drug substance or new drug product should conform to be considered acceptable for its intended use. "Conformance to specifications" means that the drug substance and / or drug product, when tested according to the listed analytical procedures, will meet the listed acceptance criteria. 62
63 Specifications: New Chemical Drug Substances Universal Test / Criteria The following tests and acceptance criteria are considered generally applicable to all new drug substances. a) Description b) Identification. c) Assay d) Impurities (organic impurities and residual solvents) Specific Tests / Criteria In addition to the universal tests listed above, the following tests may be considered on a case by case basis for drug substances a) Physicochemical properties (ph of an aqueous solution, melting point, and refractive index) b) Particle size c) Polymorphic forms (including pseudopolymorphs, and amorphous forms. Solid state IR, X-ray powder diffraction, thermal analysis procedures like DSC, TGA and DTA, Raman spectroscopy, optical microscopy, and solid state NMR should be applied). d) Tests for chiral new drug substances (Where a new drug substance is predominantly one enantiomer, the opposite enantiomer is excluded from the qualification and identification thresholds) e) Water content f) Inorganic impurities 63 g) Microbial limits
64 Specifications: New Chemical Drug Products The following tests and acceptance criteria are considered generally applicable to all new drug products: a) Description b) Identification c) Assay d) Impurities (organic and inorganic impurities, as degradation products, and residual solvents) Specific Tests / Criteria Additional tests and acceptance criteria generally should be included for particular new drug products (solid oral drug products, liquid oral drug products, and parenterals). Solid oral drug products The following tests are applicable to tablets (coated and uncoated) and hard capsules. One or more of these tests may also be applicable to soft capsules and granules. a) Dissolution b) Disintegration c) Hardness/friability d) Uniformity of dosage units e) Water content f) Microbial limits 64
65 Specifications: New Chemical Drug Products The following tests and acceptance criteria are considered generally applicable to all new drug products: a) Description b) Identification c) Assay d) Impurities (organic and inorganic impurities and residual solvents) Specific Tests / Criteria Additional tests and acceptance criteria generally should be included for particular new drug products (solid oral drug products, liquid oral drug products, and parenterals). Solid oral drug products The following tests are applicable to tablets (coated and uncoated) and hard capsules. One or more of these tests may also be applicable to soft capsules and granules. a) Dissolution b) Disintegration c) Hardness/friability d) Uniformity of dosage units e) Water content f) Microbial limits 65
66 Specifications: New Chemical Drug Products Oral liquid drug products One or more of the following specific tests will normally be applicable to oral liquids and to powders intended for reconstitution as oral liquids. a) Uniformity of dosage units b) ph c) Microbial limits d) Antimicrobial preservative content (for oral liquids needing an antimicrobial preservative ) e) Antioxidant preservative content f) Extractables (where data demonstrate the need to test extractables from the container/closure system components, e.g., rubber stopper, cap liner, plastic bottle, etc.) g) Alcohol content h) Dissolution (for oral suspensions and dry powder products for resuspension) i) Particle size distribution (for suspensions) j) Redispersibility k) Rheological properties l) Reconstitution time m) Water content (for oral products requiring reconstitution) 66
67 Specifications: New Chemical Drug Products Parenteral Drug Products The following tests may be applicable to parenteral drug products. a) Uniformity of dosage units. b) ph c) Sterility d) Endotoxins e) Particulate matter f) Water content (for non-aqueous parenterals, and for products for reconstitution) g) Antimicrobial preservative content (for multidose parenteral products) h) Antioxidant preservative i) Extractables (for parenteral products packaged in non-glass systems or in glass containers with elastomeric closures) j) Functionality testing of delivery systems (parenteral formulations packaged in prefilled syringes, autoinjector cartridges) k) Osmolarity (when the tonicity of a product is declared in its labeling) l) Particle size distribution (for injectable suspensions) m) Redispersibility (for injectable suspensions which settle on storage, produce sediment) n) Reconstitution time (for all parenteral products which require reconstitution). 67
68 Validation of Analytical Procedures The objective of the analytical procedure should be clearly understood since this will govern the validation characteristics which need to be evaluated. Typical validation characteristics which should be considered are listed below: Accuracy Precision - Repeatability - Intermediate Precision Specificity Detection Limit Quantitation Limit Linearity Range. 68
69 Validation of Analytical Procedures ANALYTICAL PROCEDURE The analytical procedure refers to the way of performing the analysis and should describe in detail the steps necessary to perform each analytical test. This may include but is not limited to: the sample, the reference standard and the reagents preparations, use of the apparatus, generation of the calibration curve, use of the formulae for the calculation, etc. SPECIFICITY Specificity is the ability to assess unequivocally the analyte in the presence of components which may be expected to be present, typically including impurities, degradants, matrix, etc. This definition has the following implications: identification, purity, assay (content or potency). 69
70 Validation of Analytical Procedures ACCURACY The accuracy of an analytical procedure expresses the closeness of agreement between the value which is accepted either as a conventional true value or an accepted reference value and the value found. PRECISION The precision of an analytical procedure expresses the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample. Precision should be investigated using homogeneous, authentic samples. However, if it is not possible to obtain a homogeneous sample it may be investigated using artificially prepared samples or a sample solution. The precision of an analytical procedure is usually expressed as the variance, standard deviation or coefficient of variation of a series of measurements. 70
71 Validation of Analytical Procedures Precision may be considered at three levels: repeatability, intermediate precision and reproducibility. Repeatability Repeatability expresses the precision under the same operating conditions over a short interval of time. Repeatability is also termed intra-assay precision. Intermediate precision Intermediate precision expresses within-laboratories variations: different days, different analysts, different equipment, etc. Reproducibility Reproducibility expresses the precision between laboratories (collaborative studies, usually applied to standardization of methodology). 71
72 Validation of Analytical Procedures DETECTION LIMIT The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be detected but not necessarily quantitated as an exact value. QUANTITATION LIMIT The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy. The quantitation limit is a parameter of quantitative assays for low levels of compounds in sample matrices, and is used particularly for the determination of impurities and/or degradation products. 72
73 Validation of Analytical Procedures LINEARITY The linearity of an analytical procedure is its ability (within a given range) to obtain test results which are directly proportional to the concentration (amount) of analyte in the sample. RANGE The range of an analytical procedure is the interval between the upper and lower concentration (amounts) of analyte in the sample for which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy and linearity. ROBUSTNESS The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage. 73
WHO GUIDELINE Stability testing of active pharmaceutical ingredients and finished pharmaceutical products
 WHO GUIDELINE Stability testing of active pharmaceutical ingredients and finished pharmaceutical products 1. Introduction 1.1 Objectives of these guidelines 1.2 Scope of these guidelines 1.3 General principles
WHO GUIDELINE Stability testing of active pharmaceutical ingredients and finished pharmaceutical products 1. Introduction 1.1 Objectives of these guidelines 1.2 Scope of these guidelines 1.3 General principles
STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS
FOOD AND DRUGS AUTHORITY
 G H A N A FOOD AND DRUGS AUTHORITY GUIDELINES FOR STABILITY TESTING OF ACTIVE PHARMACEUTICAL INGREDIENTS AND FINISHED PHARMACEUTICAL PRODUCTS Document No: FDA/DRI/DER/GL-STP/2013/07 Date of First Adoption:
G H A N A FOOD AND DRUGS AUTHORITY GUIDELINES FOR STABILITY TESTING OF ACTIVE PHARMACEUTICAL INGREDIENTS AND FINISHED PHARMACEUTICAL PRODUCTS Document No: FDA/DRI/DER/GL-STP/2013/07 Date of First Adoption:
The GCC Guidelines for Stability Testing of Drug Substances and Pharmaceutical Products EDITION TWO 1428 H 2007 G
 The GCC Guidelines for Stability Testing of Drug Substances and Pharmaceutical Products EDITION TWO 1428 H 2007 G (1) INTRODUCTION The following guideline defines the stability data package for a drug
The GCC Guidelines for Stability Testing of Drug Substances and Pharmaceutical Products EDITION TWO 1428 H 2007 G (1) INTRODUCTION The following guideline defines the stability data package for a drug
Draft regional guidelines on stability testing of active substances and pharmaceutical products
 Regional Committee for the EM/RC53/12 Eastern Mediterranean August 2006 Fifty-third Session Original: Arabic Agenda item 17 Draft regional guidelines on stability testing of active substances and pharmaceutical
Regional Committee for the EM/RC53/12 Eastern Mediterranean August 2006 Fifty-third Session Original: Arabic Agenda item 17 Draft regional guidelines on stability testing of active substances and pharmaceutical
STABILITY: STABILITY TESTING OF NEW VETERINARY DRUG SUBSTANCES (REVISION)
") VICH GL3(R) (QUALITY) January 2007 Revision at Step 9 For Implementation at Step 7 STABILITY: STABILITY TESTING OF NEW VETERINARY DRUG SUBSTANCES (REVISION) Recommended for Adoption at Step 7 of the VICH
VICH GL3(R) (QUALITY) January 2007 Revision at Step 9 For Implementation at Step 7 STABILITY: STABILITY TESTING OF NEW VETERINARY DRUG SUBSTANCES (REVISION) Recommended for Adoption at Step 7 of the VICH
COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP)
") The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 17 December 2003 CPMP/QWP/122/02, rev 1 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) GUIDELINE
The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 17 December 2003 CPMP/QWP/122/02, rev 1 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) GUIDELINE
NOTICE Our file number:
 September 25, 2003 NOTICE Our file number: 03-118437-914 Adoption of ICH 1 Guidance: - This guidance document is a revised version of the ICH Q1A(R) guidance document and defines the stability data package
September 25, 2003 NOTICE Our file number: 03-118437-914 Adoption of ICH 1 Guidance: - This guidance document is a revised version of the ICH Q1A(R) guidance document and defines the stability data package
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE
GUIDELINE FOR THE STABILITY TESTING
 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 GUIDELINE FOR THE STABILITY TESTING OF NON-PRESCRIPTION (OTC)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 GUIDELINE FOR THE STABILITY TESTING OF NON-PRESCRIPTION (OTC)
Annex 10. Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. Introduction and background
 Annex 10 Stability testing of active pharmaceutical ingredients and finished pharmaceutical products Introduction and background The guidance on Stability testing of active pharmaceutical ingredients and
Annex 10 Stability testing of active pharmaceutical ingredients and finished pharmaceutical products Introduction and background The guidance on Stability testing of active pharmaceutical ingredients and
VICH Topic GL3. Step 7 (after revision at step 9) GUIDELINE ON STABILITY: STABILITY TESTING OF NEW VETERINARY DRUG SUBSTANCES AND MEDICINAL PRODUCTS
 GUIDELINE ON STABILITY: STABILITY TESTING OF NEW VETERINARY DRUG SUBSTANCES AND MEDICINAL PRODUCTS") European Medicines Agency Veterinary Medicines and Inspections London, 19 February 2007 Doc. Ref.EMEA/CVMP/VICH/899/99-Rev.1 VICH Topic GL3 Step 7 (after revision at step 9) GUIDELINE ON STABILITY: STABILITY
European Medicines Agency Veterinary Medicines and Inspections London, 19 February 2007 Doc. Ref.EMEA/CVMP/VICH/899/99-Rev.1 VICH Topic GL3 Step 7 (after revision at step 9) GUIDELINE ON STABILITY: STABILITY
FDA S GUIDANCE FOR INDUSTRY ANDAS: STABILITY TESTING OF DRUG SUBSTANCES AND PRODUCTS
 FDA S GUIDANCE FOR INDUSTRY ANDAS: STABILITY TESTING OF DRUG SUBSTANCES AND PRODUCTS 02-December-2014 San Diego, CA Kim Huynh-Ba Executive Director PHARMALYTIK Kim.huynhba@pharmalytik.com Overview Stability
FDA S GUIDANCE FOR INDUSTRY ANDAS: STABILITY TESTING OF DRUG SUBSTANCES AND PRODUCTS 02-December-2014 San Diego, CA Kim Huynh-Ba Executive Director PHARMALYTIK Kim.huynhba@pharmalytik.com Overview Stability
DEVELOPMENT PHARMACEUTICS AND PROCESS VALIDATION
 DEVELOPMENT PHARMACEUTICS AND PROCESS VALIDATION Guideline Title Development Pharmaceutics and Process Validation Legislative basis Directive 75/318/EEC as amended Date of first adoption April 1988 Date
DEVELOPMENT PHARMACEUTICS AND PROCESS VALIDATION Guideline Title Development Pharmaceutics and Process Validation Legislative basis Directive 75/318/EEC as amended Date of first adoption April 1988 Date
GUIDELINES ON THE STABILITY DATA REQUIRED FOR REGISTRATION OF STOCK REMEDIES IN SOUTH AFRICA
 GUIDELINES ON THE STABILITY DATA REQUIRED FOR REGISTRATION OF STOCK REMEDIES IN SOUTH AFRICA Issued by the Registrar: Act No. 36 of 1947, Private Bag X343, Pretoria 0001 Republic of South Africa Tel. (**27
GUIDELINES ON THE STABILITY DATA REQUIRED FOR REGISTRATION OF STOCK REMEDIES IN SOUTH AFRICA Issued by the Registrar: Act No. 36 of 1947, Private Bag X343, Pretoria 0001 Republic of South Africa Tel. (**27
á1225ñ VALIDATION OF COMPENDIAL PROCEDURES
 1640 á1224ñ Transfer of Analytical Procedures / General Information USP 39 THE ANALYTICAL PROCEDURE The procedure should be written with sufficient detail and explicit instructions, so that a trained analyst
1640 á1224ñ Transfer of Analytical Procedures / General Information USP 39 THE ANALYTICAL PROCEDURE The procedure should be written with sufficient detail and explicit instructions, so that a trained analyst
DECISION TREE #1: ESTABLISHING ACCEPTANCE CRITERION FOR A SPECIFIED IMPURITY IN A NEW DRUG SUBSTANCE
 DECISION TREE #1: ESTABLISHING ACCEPTANCE CRITERION FOR A SPECIFIED IMPURITY IN A NEW DRUG SUBSTANCE Determine impurity level in relevant batches 1 Determine mean + upper confidence limit for the impurity
DECISION TREE #1: ESTABLISHING ACCEPTANCE CRITERION FOR A SPECIFIED IMPURITY IN A NEW DRUG SUBSTANCE Determine impurity level in relevant batches 1 Determine mean + upper confidence limit for the impurity
DEPARTMENT OF AGRICULTURE, FORESTRY & FISHERIES STABILITY
 DEPARTMENT OF AGRICULTURE, FORESTRY & FISHERIES STABILITY This guideline is intended to provide recommendations to applicants wishing to submit applications for the registration of Stock Remedies. It represents
DEPARTMENT OF AGRICULTURE, FORESTRY & FISHERIES STABILITY This guideline is intended to provide recommendations to applicants wishing to submit applications for the registration of Stock Remedies. It represents
TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR NEW VETERINARY DRUG SUBSTANCES AND NEW MEDICINAL PRODUCTS: CHEMICAL SUBSTANCES
 VICH GL39 (QUALITY) November 2005 For implementation at Step 7 TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR NEW VETERINARY DRUG SUBSTANCES AND NEW MEDICINAL PRODUCTS: CHEMICAL SUBSTANCES Recommended for
VICH GL39 (QUALITY) November 2005 For implementation at Step 7 TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR NEW VETERINARY DRUG SUBSTANCES AND NEW MEDICINAL PRODUCTS: CHEMICAL SUBSTANCES Recommended for
Q8 Pharmaceutical Development
 Q8 Pharmaceutical Development For questions regarding this draft document contact (CDER) Ajaz Hussain at 301-594-2847 or (CBER) Christopher Joneckis at 301-435-5681. This draft guidance, when finalized,
Q8 Pharmaceutical Development For questions regarding this draft document contact (CDER) Ajaz Hussain at 301-594-2847 or (CBER) Christopher Joneckis at 301-435-5681. This draft guidance, when finalized,
ANNEX V ASEAN GUIDELINES ON STABILITY STUDY AND SHELF-LIFE OF HEALTH SUPPLEMENTS
 Association of South East Asian Nations (ASEAN) ANNEX V ASEAN GUIDELINES ON STABILITY STUDY AND SHELF-LIFE OF HEALTH SUPPLEMENTS Disclaimer: This document is provided for information purpose only and subject
Association of South East Asian Nations (ASEAN) ANNEX V ASEAN GUIDELINES ON STABILITY STUDY AND SHELF-LIFE OF HEALTH SUPPLEMENTS Disclaimer: This document is provided for information purpose only and subject
Setting Specifications
 536 LCGC NORTH AMERICA VOLUME 22 NUMBER 6 JUNE 2004 www.chromatographyonline.com Setting Specifications Validation Viewpoint Specifications establish the criteria to which a drug substance or product should
536 LCGC NORTH AMERICA VOLUME 22 NUMBER 6 JUNE 2004 www.chromatographyonline.com Setting Specifications Validation Viewpoint Specifications establish the criteria to which a drug substance or product should
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT. Version 4.0. Update revision : May Document Control
 ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT Version 4.0 Update revision : May 2012 Document Control Version Date 1.0 July 2004 (8 th ACSQ PPWG Meeting; Bangkok) 2.0 February 2005 (9 th ACSQ PPWG
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT Version 4.0 Update revision : May 2012 Document Control Version Date 1.0 July 2004 (8 th ACSQ PPWG Meeting; Bangkok) 2.0 February 2005 (9 th ACSQ PPWG
Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review
 Review") Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review Presented by Erika E. Englund, Ph.D. Slides courtesy of Dorota Matecka, Ph.D. Office of Pharmaceutical
Quality Issues for Clinical Trial Materials: The Chemistry, Manufacturing and Controls (CMC) Review Presented by Erika E. Englund, Ph.D. Slides courtesy of Dorota Matecka, Ph.D. Office of Pharmaceutical
BRIEFING. . Over time, 466 may be used less frequently and may be withdrawn.
 Page 1 of 13 BRIEFING 1086 USP 37 page 828. As part of an ongoing monograph modernization initiative, the United States Pharmacopeial Convention (USP) is updating this general chapter, 1086 Impurities
Page 1 of 13 BRIEFING 1086 USP 37 page 828. As part of an ongoing monograph modernization initiative, the United States Pharmacopeial Convention (USP) is updating this general chapter, 1086 Impurities
Guidance for Industry Q3B(R2) Impurities in New Drug Products
 Impurities in New Drug Products") Guidance for Industry Q3B(R2) Impurities in New Drug Products U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics
Guidance for Industry Q3B(R2) Impurities in New Drug Products U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics
Guidelines for Pharmaceutical Equivalence Requirements
 Guidelines for Pharmaceutical Equivalence Requirements Version 1.1 1 September 2010 Page 1 of 9 Guidelines for Pharmaceutical Equivalence Requirements Version 1.1 Drug Sector Saudi Food & Drug Authority
Guidelines for Pharmaceutical Equivalence Requirements Version 1.1 1 September 2010 Page 1 of 9 Guidelines for Pharmaceutical Equivalence Requirements Version 1.1 Drug Sector Saudi Food & Drug Authority
EVALUATION FOR STABILITY DATA
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE EVALUATION FOR STABILITY DATA Q1E Recommended for
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE EVALUATION FOR STABILITY DATA Q1E Recommended for
International Journal of Generic Drugs
 Photostability STABILITY TESTING in New Drug Products evaluating photostability is foremost for new chemical entities only - not in generic drugs, provided the container-closure protection is the same
Photostability STABILITY TESTING in New Drug Products evaluating photostability is foremost for new chemical entities only - not in generic drugs, provided the container-closure protection is the same
How to Avoid Common Deficiencies in INDs and NDAs. Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER
 How to Avoid Common Deficiencies in INDs and NDAs Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER 1 Structure of FDA Office of Commissioner Chief Scientist FOODS Medical Products & Tobacco
How to Avoid Common Deficiencies in INDs and NDAs Ramesh Raghavachari, Ph.D. Branch Chief, Branch IX ONDQA/OPS/CDER 1 Structure of FDA Office of Commissioner Chief Scientist FOODS Medical Products & Tobacco
Approval Review of Generic Drugs. Office of Generic/OTC Drugs, PMDA Kazuyuki SAITO, Ph.D.
 Approval Review of Generic Drugs Office of Generic/OTC Drugs, PMDA Kazuyuki SAITO, Ph.D. Outline of Presentation Introduction Approval Review of Generic Drugs Equivalency review Conformity audit Conclusion
Approval Review of Generic Drugs Office of Generic/OTC Drugs, PMDA Kazuyuki SAITO, Ph.D. Outline of Presentation Introduction Approval Review of Generic Drugs Equivalency review Conformity audit Conclusion
International Journal of Pharma and Bio Sciences DEVELOPMENT OF ACCELERATED STABILITY PROTOCOL FOR SILDENAFIL TABLETS A EUROPEAN PERSPECTIVE REVIEW
 International Journal of Pharma and Bio Sciences DEVELOPMENT OF ACCELERATED STABILITY PROTOCOL FOR SILDENAFIL TABLETS A EUROPEAN PERSPECTIVE REVIEW SUKHDEV SINGH *1 AND JASBIR SINGH 2 1 Rayat Institute
International Journal of Pharma and Bio Sciences DEVELOPMENT OF ACCELERATED STABILITY PROTOCOL FOR SILDENAFIL TABLETS A EUROPEAN PERSPECTIVE REVIEW SUKHDEV SINGH *1 AND JASBIR SINGH 2 1 Rayat Institute
PHARMACEUTICAL TESTING
 WHITEHOUSE, NJ PHARMACEUTICAL TESTING Pharmaceutical Expertise for GMP & CMC Testing Our Pharmaceutical Expertise With more than 20 years of experience in a variety of industries, our Whitehouse, New Jersey
WHITEHOUSE, NJ PHARMACEUTICAL TESTING Pharmaceutical Expertise for GMP & CMC Testing Our Pharmaceutical Expertise With more than 20 years of experience in a variety of industries, our Whitehouse, New Jersey
1-6 Specifications. Andrew Chemwolo, Technical Officer, WHO Prequalification Team Medicines Assessment
 1-6 Specifications Andrew Chemwolo, Technical Officer, WHO Prequalification Team Medicines Assessment Outline Definition Why are specifications important? Setting appropriate specifications PQT-medicines
1-6 Specifications Andrew Chemwolo, Technical Officer, WHO Prequalification Team Medicines Assessment Outline Definition Why are specifications important? Setting appropriate specifications PQT-medicines
IMPURITIES IN NEW DRUG PRODUCTS
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
Manual 055 Commercial Stability Testing For Formulated Products. This procedure applies to all drug products. The procedure covers:
 1 Purpose Manual 055 Commercial Stability Testing For Formulated Products The intent of this procedure is to provide to manufacturing and primary packaging sites the principles of a stability program.
1 Purpose Manual 055 Commercial Stability Testing For Formulated Products The intent of this procedure is to provide to manufacturing and primary packaging sites the principles of a stability program.
GUIDANCE NOTES ON ANALYTICAL METHOD VALIDATION
 ON ANALYTICAL METHOD VALIDATION HSA September 2004 Reproduction prohibited for commercial purposes. Reproduction for internal use is authorised, provided the source is acknowledged. MQA Dir: DISK1\GUIDE-MQA-012A-004.doc
ON ANALYTICAL METHOD VALIDATION HSA September 2004 Reproduction prohibited for commercial purposes. Reproduction for internal use is authorised, provided the source is acknowledged. MQA Dir: DISK1\GUIDE-MQA-012A-004.doc
Validation of Analytical Methods used for the Characterization, Physicochemical and Functional Analysis and of Biopharmaceuticals.
 Validation of Analytical Methods used for the Characterization, Physicochemical and Functional Analysis and of Biopharmaceuticals. 1 Analytical Method Validation: 1..1 Philosophy: Method validation is
Validation of Analytical Methods used for the Characterization, Physicochemical and Functional Analysis and of Biopharmaceuticals. 1 Analytical Method Validation: 1..1 Philosophy: Method validation is
VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY *)
") VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY *) Guideline Title Validation of Analytical Procedures: Methodology Legislative basis Directive 75/318/EEC as amended Date of first adoption December 1996
VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY *) Guideline Title Validation of Analytical Procedures: Methodology Legislative basis Directive 75/318/EEC as amended Date of first adoption December 1996
A STANDARD PROTOCOL for DERIVING and ASSESSMENT of STABILITY. Part 3 Oral Liquid Medicines (Solutions, Emulsions, Suspensions and Syrups)
") A STANDARD PROTOCOL for DERIVING and ASSESSMENT of STABILITY Part 3 Oral Liquid Medicines (Solutions, Emulsions, Suspensions and Syrups) EDITION 1 August 2014 NHS Pharmaceutical Quality Assurance Committee
A STANDARD PROTOCOL for DERIVING and ASSESSMENT of STABILITY Part 3 Oral Liquid Medicines (Solutions, Emulsions, Suspensions and Syrups) EDITION 1 August 2014 NHS Pharmaceutical Quality Assurance Committee
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT
 ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT Update revision : 22 February 2005. 9 th ACCSQ-PPWG Meeting, Philippines, 21-24 Feb 2005 LIST OF CONTENTS 1. INTRODUCTION 1 2. OBJECTIVES 1 3. SCOPES
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT Update revision : 22 February 2005. 9 th ACCSQ-PPWG Meeting, Philippines, 21-24 Feb 2005 LIST OF CONTENTS 1. INTRODUCTION 1 2. OBJECTIVES 1 3. SCOPES
Impurities from degradation of Drug Substances
 Impurities from degradation of Drug Substances REFERENCE 1. ICH - IMPURITIES IN NEW DRUG SUBSTANCES - Q3A(R2) 2. ICH - IMPURITIES IN NEW DRUG PRODUCTS - Q3B(R2) 3. ICH - VALIDATION OF ANALYTICAL PROCEDURES:
Impurities from degradation of Drug Substances REFERENCE 1. ICH - IMPURITIES IN NEW DRUG SUBSTANCES - Q3A(R2) 2. ICH - IMPURITIES IN NEW DRUG PRODUCTS - Q3B(R2) 3. ICH - VALIDATION OF ANALYTICAL PROCEDURES:
MINISTRY OF HEALTH AND SOCIAL SERVICES
 MINISTRY OF HEALTH AND SOCIAL SERVICES NAMIBIA MEDICINES REGULATORY COUNCIL POST REGISTRATION AMENDMENT GUIDELINES These guidelines are meant to provide assistance to industry and health care professionals
MINISTRY OF HEALTH AND SOCIAL SERVICES NAMIBIA MEDICINES REGULATORY COUNCIL POST REGISTRATION AMENDMENT GUIDELINES These guidelines are meant to provide assistance to industry and health care professionals
Application of Quality by Design (QbD) in product development. James E. Polli September 16, 2015
 in product development. James E. Polli September 16, 2015") Application of Quality by Design (QbD) in product development James E. Polli jpolli@rx.umaryland.edu September 16, 2015 Pharmaceutical Equivalence Same active ingredient(s) Same dosage form Same route
Application of Quality by Design (QbD) in product development James E. Polli jpolli@rx.umaryland.edu September 16, 2015 Pharmaceutical Equivalence Same active ingredient(s) Same dosage form Same route
Analytical and formulation attributes
 Peer reviewed article Analytical and formulation attributes in developing generic sterile injectable liquid and lyophilized drugs (part 1) Arindam Roy ARINDAM ROY 1,2 *, GURMUKH CHANANA 1 *Corresponding
Peer reviewed article Analytical and formulation attributes in developing generic sterile injectable liquid and lyophilized drugs (part 1) Arindam Roy ARINDAM ROY 1,2 *, GURMUKH CHANANA 1 *Corresponding
STABILITY TESTING: PHOTOSTABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE STABILITY TESTING: PHOTOSTABILITY TESTING OF NEW
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE STABILITY TESTING: PHOTOSTABILITY TESTING OF NEW
COMMERCIAL PRODUCT STABILITY
 COMMERCIAL PRODUCT STABILITY Being Responsible for your Tweener, Senior Citizen and Hospice Stage Products Melissa Lambert Global Head Stability in Quality & Compliance Management, R&D Director Quality
COMMERCIAL PRODUCT STABILITY Being Responsible for your Tweener, Senior Citizen and Hospice Stage Products Melissa Lambert Global Head Stability in Quality & Compliance Management, R&D Director Quality
ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
 European Medicines Agency August 2003 CPMP/ICH/4680/02 ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
European Medicines Agency August 2003 CPMP/ICH/4680/02 ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
VICH Topic GL2 (Validation: Methodology) GUIDELINE ON VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY
 GUIDELINE ON VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY") The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit CVMP/VICH/591/98-FINAL London, 10 December 1998 VICH Topic GL2 (Validation: Methodology) Step 7 Consensus
The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit CVMP/VICH/591/98-FINAL London, 10 December 1998 VICH Topic GL2 (Validation: Methodology) Step 7 Consensus
NEWSMAGAZINE FEB 13 REGULATORY SCIENCES. AAPS ELEARNING Advanced Computer Modeling Seen as Way to Dissect Drugs Complex Effects
 AAPS NEWSMAGAZINE FEB 13 REGULATORY SCIENCES AAPS ELEARNING Advanced Computer Modeling Seen as Way to Dissect Drugs Complex Effects PHARMACEUTICAL STABILITY: Doing the Right Thing versus Doing Things the
AAPS NEWSMAGAZINE FEB 13 REGULATORY SCIENCES AAPS ELEARNING Advanced Computer Modeling Seen as Way to Dissect Drugs Complex Effects PHARMACEUTICAL STABILITY: Doing the Right Thing versus Doing Things the
Contents. Contents (13) 1 Production (23)
 1 Production (23)") 1 Production (23) 1.A Sanitation (27) 1.A.1 Organisational prerequisites (27) 1.A.2 Sources of contamination (28) 1.A.3 Responsibilities and implementation (29) 1.B Personnel hygiene (31) 1.B.1 Clothing
1 Production (23) 1.A Sanitation (27) 1.A.1 Organisational prerequisites (27) 1.A.2 Sources of contamination (28) 1.A.3 Responsibilities and implementation (29) 1.B Personnel hygiene (31) 1.B.1 Clothing
Process Drift: When Do We Detect it? Richard L. Friedman Director, DMPQ CDER/Office of Compliance PQRI Process Drift Workshop December 1, 2010
 Process Drift: When Do We Detect it? Richard L. Friedman Director, DMPQ CDER/Office of Compliance PQRI Process Drift Workshop December 1, 2010 Overview Goal of Manufacturing Central Question: Why is process
Process Drift: When Do We Detect it? Richard L. Friedman Director, DMPQ CDER/Office of Compliance PQRI Process Drift Workshop December 1, 2010 Overview Goal of Manufacturing Central Question: Why is process
An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications
 An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications On behalf of Specifications Team Kirby Wong-Moon, Ph.D. Amgen Inc. Best Practices and Application of GMPs for
An Overview of IQ s Position Paper: Early Development GMPs for Small-Molecule Specifications On behalf of Specifications Team Kirby Wong-Moon, Ph.D. Amgen Inc. Best Practices and Application of GMPs for
Extractables and leachables: An Introduction
 Extractables and leachables: An Introduction Tim Hulme Smithers Rapra THulme@smithers.com 44(0)1939 252 418 1 Extractables and leachables: An Introduction Tim Hulme Smithers Rapra thulme@smithers.com 2
Extractables and leachables: An Introduction Tim Hulme Smithers Rapra THulme@smithers.com 44(0)1939 252 418 1 Extractables and leachables: An Introduction Tim Hulme Smithers Rapra thulme@smithers.com 2
Pharmaceutical Sciences
 SRI International Biosciences From Idea to IND Research on Disease Mechanisms Drug Discovery Drug Metabolism, Pharmacokinetics, & Toxicology Services Pharmaceutical Sciences Preclinical Development Planning
SRI International Biosciences From Idea to IND Research on Disease Mechanisms Drug Discovery Drug Metabolism, Pharmacokinetics, & Toxicology Services Pharmaceutical Sciences Preclinical Development Planning
New Drug Product Impurities
 Title Page Image - with courtesy of the FDA CDER original web site at www. cder.fda.gov/ New Drug Product Impurities IAGIM Scientific Committee Block JD; Holmann E ; West P NEW DRAFT GUIDANCE FDA Viewpoint
Title Page Image - with courtesy of the FDA CDER original web site at www. cder.fda.gov/ New Drug Product Impurities IAGIM Scientific Committee Block JD; Holmann E ; West P NEW DRAFT GUIDANCE FDA Viewpoint
API Stability Protocols and. Chris Byrne Tasmanian Alkaloids
 API Stability Protocols and Evaluations Chris Byrne Tasmanian Alkaloids API Stability Overview APIs = 100% pure Limited (if any) degradation No interactions with other agents in drug products Less likelihood
API Stability Protocols and Evaluations Chris Byrne Tasmanian Alkaloids API Stability Overview APIs = 100% pure Limited (if any) degradation No interactions with other agents in drug products Less likelihood
COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE:
 The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit - FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE: DEVELOPMENT PHARMACEUTICS FOR VETERINARY
The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit - FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE: DEVELOPMENT PHARMACEUTICS FOR VETERINARY
Future of Question-based Review and Regulatory Submissions
 Future of Question-based Review and Regulatory Submissions Robert Iser Associate Director for Policy Development (Acting) Office of Pharmaceutical Science / CDER / FDA FDA/PQRI Conference on Evolving Product
Future of Question-based Review and Regulatory Submissions Robert Iser Associate Director for Policy Development (Acting) Office of Pharmaceutical Science / CDER / FDA FDA/PQRI Conference on Evolving Product
Elemental impurities impact on APIs
 Regulatory Expectations on impurities in Drug Substances: Authority and Industry perspective Pavia, 2nd October 2015 Elemental impurities impact on APIs October 2 nd 2015 Annalisa Scali RegulatoryAffairsManager
Regulatory Expectations on impurities in Drug Substances: Authority and Industry perspective Pavia, 2nd October 2015 Elemental impurities impact on APIs October 2 nd 2015 Annalisa Scali RegulatoryAffairsManager
Statistical Evaluation Of Stability Data
 Reprinted from FDA s website by #219 GUIDANCE FOR INDUSTRY Statistical Evaluation Of Stability Data VICH GL51 DRAFT GUIDANCE This guidance document is being distributed for comment purposes only Submit
Reprinted from FDA s website by #219 GUIDANCE FOR INDUSTRY Statistical Evaluation Of Stability Data VICH GL51 DRAFT GUIDANCE This guidance document is being distributed for comment purposes only Submit
QbD in developing semisolid formulations
 QbD in developing semisolid formulations 4th Jerusalem Conference on Quality and Pharma Sciences Haim Barsimantov, COO, Sol-Gel 21.05.13 Outline Drug Product Specification - definition In Process Control
QbD in developing semisolid formulations 4th Jerusalem Conference on Quality and Pharma Sciences Haim Barsimantov, COO, Sol-Gel 21.05.13 Outline Drug Product Specification - definition In Process Control
LEGAL REQUIREMENTS FOR STABILITY
 BY DR. A.V.PRABHU LEGAL REQUIREMENTS FOR STABILITY 21 CFR 211.166- STABILITY TESTING GMP To assess stability characteristics to determine storage conditions and expiration dates. Written stability program
BY DR. A.V.PRABHU LEGAL REQUIREMENTS FOR STABILITY 21 CFR 211.166- STABILITY TESTING GMP To assess stability characteristics to determine storage conditions and expiration dates. Written stability program
Laboratory Services Alcami s comprehensive development and testing services are well established and ready to assist your pharmaceutical product
 Laboratory Services Alcami s comprehensive development and testing services are well established and ready to assist your pharmaceutical product needs. The Alcami Advantage Alcami specializes in all phases
Laboratory Services Alcami s comprehensive development and testing services are well established and ready to assist your pharmaceutical product needs. The Alcami Advantage Alcami specializes in all phases
ICH Q9 An Industry Perspective: Ensuring Quality to Patients in a Risk-Based Regulatory Environment
 ICH Q9 An Industry Perspective: Ensuring Quality to Patients in a Risk-Based Regulatory Environment Thomas Schultz, Ph.D. Director, Regulatory Sciences Johnson & Johnson September 12, 2007 Presentation
ICH Q9 An Industry Perspective: Ensuring Quality to Patients in a Risk-Based Regulatory Environment Thomas Schultz, Ph.D. Director, Regulatory Sciences Johnson & Johnson September 12, 2007 Presentation
GUIDANCE DOCUMENT Use of a Foreign-sourced Reference Product as a Canadian Reference Product
 GUIDANCE DOCUMENT Use of a Foreign-sourced Reference Product as a Canadian Reference Product Published by authority of the Minister of Health Date Adopted 2017/11/24 Effective Date 2017/11/24 Health Products
GUIDANCE DOCUMENT Use of a Foreign-sourced Reference Product as a Canadian Reference Product Published by authority of the Minister of Health Date Adopted 2017/11/24 Effective Date 2017/11/24 Health Products
Extractable and Leachable Challenges From a generic injectable drug development perspective
 Extractable and Leachable Challenges From a generic injectable drug development perspective Andrea Redd Director, US Regulatory Affairs Fresenius Kabi November 8, 2017 Disclaimer This presentation contains
Extractable and Leachable Challenges From a generic injectable drug development perspective Andrea Redd Director, US Regulatory Affairs Fresenius Kabi November 8, 2017 Disclaimer This presentation contains
Why Do I Test, What Do I Test & When Do I Test It? Ross Caputo, PhD Chief Technical Officer Eagle Analytical Services
 Why Do I Test, What Do I Test & When Do I Test It? Ross Caputo, PhD Chief Technical Officer Eagle Analytical Services Disclosures I, Ross Caputo, declare no conflicts of interest, real or apparent, and
Why Do I Test, What Do I Test & When Do I Test It? Ross Caputo, PhD Chief Technical Officer Eagle Analytical Services Disclosures I, Ross Caputo, declare no conflicts of interest, real or apparent, and
Stability of Biological Products
 Stability of Biological Products Dr Jurgen Lindner Principal, BioPharma Consulting & Executive Secretary, APIMAA Biological Products Functional Proteins or Polypeptides (mab s, enzymes & inhibitors, growth
Stability of Biological Products Dr Jurgen Lindner Principal, BioPharma Consulting & Executive Secretary, APIMAA Biological Products Functional Proteins or Polypeptides (mab s, enzymes & inhibitors, growth
PHOTOSTABILITY TESTING OF NEW ACTIVE SUBSTANCES AND MEDICINAL PRODUCTS *)
") PHOTOSTABILITY TESTING OF NEW ACTIVE SUBSTANCES AND MEDICINAL PRODUCTS *) Guideline Title Photostability Testing of New Active Substances and Medicinal Products *) Legislative basis Directive 75/318/EEC
PHOTOSTABILITY TESTING OF NEW ACTIVE SUBSTANCES AND MEDICINAL PRODUCTS *) Guideline Title Photostability Testing of New Active Substances and Medicinal Products *) Legislative basis Directive 75/318/EEC
On the Q&A to the Guideline for Common Technical Documents
 To: Public Health Bureau Prefectural Government Letter from PFSB/ELD 22 nd October 2001 From: Evaluation & Licensing Division, Pharmaceutical & Food Safety Bureau, The Ministry of Health, Labour and Welfare
To: Public Health Bureau Prefectural Government Letter from PFSB/ELD 22 nd October 2001 From: Evaluation & Licensing Division, Pharmaceutical & Food Safety Bureau, The Ministry of Health, Labour and Welfare
Chapter -1. Importance of Stability Indicating Analytical Methods for Analyzing Organic Impurities in Active Pharmaceutical Ingredients
 Chapter -1 Importance of Stability Indicating Analytical Methods for Analyzing Organic Impurities in Active Pharmaceutical Ingredients 1.1 Introduction and importance of stability indicating analytical
Chapter -1 Importance of Stability Indicating Analytical Methods for Analyzing Organic Impurities in Active Pharmaceutical Ingredients 1.1 Introduction and importance of stability indicating analytical
ICH Topic Q 3 B Impurities in New Medicinal Products
 The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit ICH Topic Q 3 B Impurities in New Medicinal Products Step 4, Consensus Guideline, 6 November 1996 TE FOR GUIDANCE
The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit ICH Topic Q 3 B Impurities in New Medicinal Products Step 4, Consensus Guideline, 6 November 1996 TE FOR GUIDANCE
Regulatory Assessment
 Implementation of ICH Q8, Q9, Q10 Regulatory Assessment International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Presentation Overview Goal
Implementation of ICH Q8, Q9, Q10 Regulatory Assessment International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Presentation Overview Goal
QUALITY OF PROLONGED RELEASE ORAL SOLID DOSAGE FORMS
 QUALITY OF PROLONGED RELEASE ORAL SOLID DOSAGE FORMS Guideline Title Quality of Prolonged Release Oral Solid Dosage Forms Legislative basis Directive 75/318/EEC as amended Date of first adoption October
QUALITY OF PROLONGED RELEASE ORAL SOLID DOSAGE FORMS Guideline Title Quality of Prolonged Release Oral Solid Dosage Forms Legislative basis Directive 75/318/EEC as amended Date of first adoption October
Perspectives on Method Validation: Importance of Adequate Method Validation
 Perspectives on Method Validation: Importance of Adequate Method Validation Heather Bridwell, Vikas Dhingra, Daniel Peckman, Jennifer Roark and Thomas Lehman The appropriate validation of analytical methods
Perspectives on Method Validation: Importance of Adequate Method Validation Heather Bridwell, Vikas Dhingra, Daniel Peckman, Jennifer Roark and Thomas Lehman The appropriate validation of analytical methods
ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances
 The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit CPMP/ICH/142/95 ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances Step 5 NOTE
The European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit CPMP/ICH/142/95 ICH Topic Q 3 A Impurities Testing Guideline: Impurities in New Drug Substances Step 5 NOTE
Design and Interpretation of Accelerated Stability Studies of Biologics
 Design and Interpretation of Accelerated Stability Studies of Biologics Sheila G. Magil, Ph.D. Cambridge Healthtech Institute s Fourth Annual The Bioprocessing Summit 2012 August 20-23, 2012 Boston, MA
Design and Interpretation of Accelerated Stability Studies of Biologics Sheila G. Magil, Ph.D. Cambridge Healthtech Institute s Fourth Annual The Bioprocessing Summit 2012 August 20-23, 2012 Boston, MA
Strategies for IND Filing Success: Chemistry, Manufacturing and Controls
 Strategies for IND Filing Success: Chemistry, Manufacturing and Controls October 21, 2016 Presented by: Sharon Ayd, Ph.D., MBA Chief Scientific Officer and SVP, Pharmaceuticals document contains proprietary
Strategies for IND Filing Success: Chemistry, Manufacturing and Controls October 21, 2016 Presented by: Sharon Ayd, Ph.D., MBA Chief Scientific Officer and SVP, Pharmaceuticals document contains proprietary
Good practices in quality control in pharmaceutical industry - Overview of regulatory guidelines
 Review Article Good practices in quality control in pharmaceutical industry - Overview of regulatory guidelines Chagi Venkatesh*, S. B. Puranik ABSTRACT Good practices in quality control (QC) department
Review Article Good practices in quality control in pharmaceutical industry - Overview of regulatory guidelines Chagi Venkatesh*, S. B. Puranik ABSTRACT Good practices in quality control (QC) department
MODULE 2.3 QUALITY OVERALL SUMMARY PRODUCT DOSSIER (QOS PD)
") MODULE 2.3 QUALITY OVERALL SUMMARY PRODUCT DOSSIER () INTRODUCTION The introduction should include proprietary name, non-proprietary name or common name of the drug substance, company name, dosage form(s),
MODULE 2.3 QUALITY OVERALL SUMMARY PRODUCT DOSSIER () INTRODUCTION The introduction should include proprietary name, non-proprietary name or common name of the drug substance, company name, dosage form(s),
Control Strategy. Implementation of ICH Q8, Q9, Q10
 Implementation of ICH Q8, Q9, Q10 Control Strategy International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Introduction Structure of this session
Implementation of ICH Q8, Q9, Q10 Control Strategy International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Introduction Structure of this session
Stability Report. Stability profile of. BIWG 98 SE tablets 40 mg SR of 133. This stability report comprises 133 pages.
 Stability Report Stability profile of BIWG 98 SE tablets 40 mg Number SR 2001-01-04-01 Date Page 00. 00. 0000 1 of 133 Responsible Company Successful Pharma KG Biberach This stability report comprises
Stability Report Stability profile of BIWG 98 SE tablets 40 mg Number SR 2001-01-04-01 Date Page 00. 00. 0000 1 of 133 Responsible Company Successful Pharma KG Biberach This stability report comprises
Analytical Methods Development and Validation
 Understanding and Implementing Efficient Analytical Methods Development and Validation Jay Breaux, Kevin Jones, and Pierre Boulas Analytical methods development and validation play important roles in the
Understanding and Implementing Efficient Analytical Methods Development and Validation Jay Breaux, Kevin Jones, and Pierre Boulas Analytical methods development and validation play important roles in the
PHARMACEUTICAL PRINCIPLES OF SOLID DOSAGE FORMS
 PHARMACEUTICAL PRINCIPLES OF SOLID DOSAGE FORMS J. T. Carstensen, Ph.D. Professor of Pharmacy University of Wisconsin Madison, Wisconsin TECHNOMIC ^PUBLISHING CO.. INC J 1.ANCASTER BASET, TABLE OF CONTENTS
PHARMACEUTICAL PRINCIPLES OF SOLID DOSAGE FORMS J. T. Carstensen, Ph.D. Professor of Pharmacy University of Wisconsin Madison, Wisconsin TECHNOMIC ^PUBLISHING CO.. INC J 1.ANCASTER BASET, TABLE OF CONTENTS
VICH GL51: Quality: statistical evaluation of stability data
 1 2 3 12 December 2011 EMA/CVMP/VICH/858875/2011 Committee for Medicinal Products for Veterinary Use (CVMP) 4 5 VICH GL51: Quality: statistical evaluation of stability data Draft Draft agreed by VICH Steering
1 2 3 12 December 2011 EMA/CVMP/VICH/858875/2011 Committee for Medicinal Products for Veterinary Use (CVMP) 4 5 VICH GL51: Quality: statistical evaluation of stability data Draft Draft agreed by VICH Steering
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C.
 Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Specifications Breakout Session 2 Pete Yehl and Mike Coutant, moderators
Best Practices and Application of GMPs for Small Molecule Drugs in Early Development IQ Workshop, Feb 4-5, 2014, Washington, D.C. Specifications Breakout Session 2 Pete Yehl and Mike Coutant, moderators
Guidance for Industry
 Guidance for Industry ANDAs: Impurities in Drug Products DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should
Guidance for Industry ANDAs: Impurities in Drug Products DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should
Review article Rapports De Pharmacie 2015;1(2):46-53 ISSN:
:46-53 ISSN:") Review article Rapports De Pharmacie 2015;1(2):46-53 ISSN: 2455-0507 ABSTRACT APPLICATION OF RISK ASSESSMENT IN PRODUCT QUALITY LIFECYCLE MANAGEMENT Subin Sankarankutty Regulatory Consultant, Health Sciences
Review article Rapports De Pharmacie 2015;1(2):46-53 ISSN: 2455-0507 ABSTRACT APPLICATION OF RISK ASSESSMENT IN PRODUCT QUALITY LIFECYCLE MANAGEMENT Subin Sankarankutty Regulatory Consultant, Health Sciences
ASMF/DMF Quality Assessment Report (QAR) IGDRP Quality Working Group
 IGDRP Quality Working Group") ASMF/DMF Quality Assessment Report (QAR) IGDRP Quality Working Group Version Description of Change Author Effective Date v 1.0 Original publication ASMF/DMF WG May 26, 2015 v 1.1 Watermark added ASMF/DMF
ASMF/DMF Quality Assessment Report (QAR) IGDRP Quality Working Group Version Description of Change Author Effective Date v 1.0 Original publication ASMF/DMF WG May 26, 2015 v 1.1 Watermark added ASMF/DMF
Derivation and Justification of Safety Thresholds
 Derivation and Justification of Safety Thresholds Douglas J. Ball, MS, DABT Chair, PQRI L&E Toxicology Subgroup Research Fellow, Safety Sciences - Pfizer, Inc. Agenda Basic Definitions Current Regulatory
Derivation and Justification of Safety Thresholds Douglas J. Ball, MS, DABT Chair, PQRI L&E Toxicology Subgroup Research Fellow, Safety Sciences - Pfizer, Inc. Agenda Basic Definitions Current Regulatory
Federal Register / Vol. 65, No. 251 / Friday, December 29, 2000 / Notices
 83041 evaluating the quality and reliability of information and data for use in developing the VADS system contents; (3) apply the principles of pharmacology in constructing therapeutic regimens for use
83041 evaluating the quality and reliability of information and data for use in developing the VADS system contents; (3) apply the principles of pharmacology in constructing therapeutic regimens for use
Reference Standards for Monoclonal Antibodies: Key Challenges Addressed
 CASSS WCBP 2012: 16th Symposium on the Interface of Regulatory and Analytical Sciences for Biotechnology Health Products January 23-25, 2012 Reference Standards for Monoclonal Antibodies: Key Challenges
CASSS WCBP 2012: 16th Symposium on the Interface of Regulatory and Analytical Sciences for Biotechnology Health Products January 23-25, 2012 Reference Standards for Monoclonal Antibodies: Key Challenges
USP Chapter 823 USP 32 (old) vs. USP 35 (new)
 vs. USP 35 (new)") USP Chapter 823 USP 32 (old) vs. USP 35 (new) Sally W. Schwarz, MS, BCNP Research Associate Professor of Radiology Washington University School of Medicine St. Louis, MO Why USP Chapter ? FDA has
USP Chapter 823 USP 32 (old) vs. USP 35 (new) Sally W. Schwarz, MS, BCNP Research Associate Professor of Radiology Washington University School of Medicine St. Louis, MO Why USP Chapter ? FDA has
Formulation Development & CTM Manufacturing Services
 Formulation Development & CTM Manufacturing Services VxP Pharma Purdue Research Park 5225 Exploration Drive Indianapolis, IN 46241 Tel: 317.759.2299 Fax: 317.713.2950 VxP Pharmaprovides an extensive range
Formulation Development & CTM Manufacturing Services VxP Pharma Purdue Research Park 5225 Exploration Drive Indianapolis, IN 46241 Tel: 317.759.2299 Fax: 317.713.2950 VxP Pharmaprovides an extensive range
1.1. Overview of Drug Stability Studies
 1.1. Overview of Drug Stability Studies Everything made by human hands is known to deteriorate and pharmaceutical product is no exception to it. Degradation is a phenomenon where certain measurements of
1.1. Overview of Drug Stability Studies Everything made by human hands is known to deteriorate and pharmaceutical product is no exception to it. Degradation is a phenomenon where certain measurements of
BIOEQUIVALENCE TRIAL INFORMATION FORM (Medicines and Allied Substances Act [No. 3] of 2013 Part V Section 39)
![BIOEQUIVALENCE TRIAL INFORMATION FORM (Medicines and Allied Substances Act [No. 3] of 2013 Part V Section 39)](/thumbs/90/102231627.jpg "BIOEQUIVALENCE TRIAL INFORMATION FORM (Medicines and Allied Substances Act [No. 3] of 2013 Part V Section 39)") ZAMRA BTIF BIOEQUIVALENCE TRIAL INFORMATION FORM (Medicines and Allied Substances Act [No. 3] of 2013 Part V Section 39) The Guidelines on Bioequivalence Studies to be consulted in completing this form.
ZAMRA BTIF BIOEQUIVALENCE TRIAL INFORMATION FORM (Medicines and Allied Substances Act [No. 3] of 2013 Part V Section 39) The Guidelines on Bioequivalence Studies to be consulted in completing this form.
GUIDELINES FOR INTRODUCING A LOCALLY MANUFACTURED NEW PHARMACEUTICAL PRODUCT ON THE UGANDA MARKET
 GUIDELINES FOR INTRODUCING A LOCALLY MANUFACTURED NEW PHARMACEUTICAL PRODUCT ON THE UGANDA MARKET National Drug Authority Head Office Rumee Towers Plot 19, Lumumba Avenue P. O. Box 23096 Kampala, Uganda
GUIDELINES FOR INTRODUCING A LOCALLY MANUFACTURED NEW PHARMACEUTICAL PRODUCT ON THE UGANDA MARKET National Drug Authority Head Office Rumee Towers Plot 19, Lumumba Avenue P. O. Box 23096 Kampala, Uganda
Inspection. Implementation of ICH Q8, Q9, Q10
 Implementation of ICH Q8, Q9, Q10 International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Outline Aim of - as a key part of the regulatory
Implementation of ICH Q8, Q9, Q10 International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Outline Aim of - as a key part of the regulatory
Workshop Summaries. Discussions of the Global Stability Workshop are summarized for the following topics:
 Workshop Summaries Discussions of the Global Stability Workshop are summarized for the following topics: QbD and Stability Program Dr. Alasandro Setting Specifications Dr. Gentry Challenges of Different
Workshop Summaries Discussions of the Global Stability Workshop are summarized for the following topics: QbD and Stability Program Dr. Alasandro Setting Specifications Dr. Gentry Challenges of Different