Protein backbone angle prediction with machine learning. approaches
|
|
|
- Meghan Green
- 5 years ago
- Views:
Transcription
1 Bioinformatics Advance Access published February 26, 2004 Bioinfor matics Oxford University Press 2004; all rights reserved. Protein backbone angle prediction with machine learning approaches Rui Kuang 1, Christina S. Leslie 1,4, and An-Suei Yang 2,3,4 *, 1 Department of Computer Science, 2 Department of Pharmacology, 3 Columbia Genome Center, and 4 Center for Computational Biology and Bioinformatics Columbia University 630 West 168 th street, PH 7 W Room 318, New York, NY corresponding author: A.-S.Yang ay1@columbia.edu Department of Pharmacology, Columbia University, 630 West 168 th street, New York, NY (phone), (FAX) Key words: backbone torsion angle prediction, local structure prediction, machine learning, artificial neural network, support vector machine. 1
2 Abstract Motivation: Protein backbone torsion angle prediction provides useful local structural information that goes beyond conventional three-state (,, and coil) secondary structure predictions. Accurate prediction for protein backbone torsion angles will substantially improve modeling procedures for local structures of protein sequence segments, especially in modeling loop conformations that do not form regular structures as in - helices or -strands. Results: We have devised two novel automated methods in protein backbone conformational state prediction: one method is based on support vector machines (SVMs); the other method combines a standard feed-forward back-propagation artificial neural network (NN) with a local structure-based sequence profile database (LSBSP1). Extensive benchmark experiments demonstrate that both methods have improved the prediction accuracy rate over the previously published methods for conformation state prediction when using an alphabet of three or four states. Availability: LSBSP1 and neural network algorithm have been implemented in PrISM.1, which is available from: Supplementary data for the SVM method can be downloaded from the website: Contact: ay1@columbia.edu 2
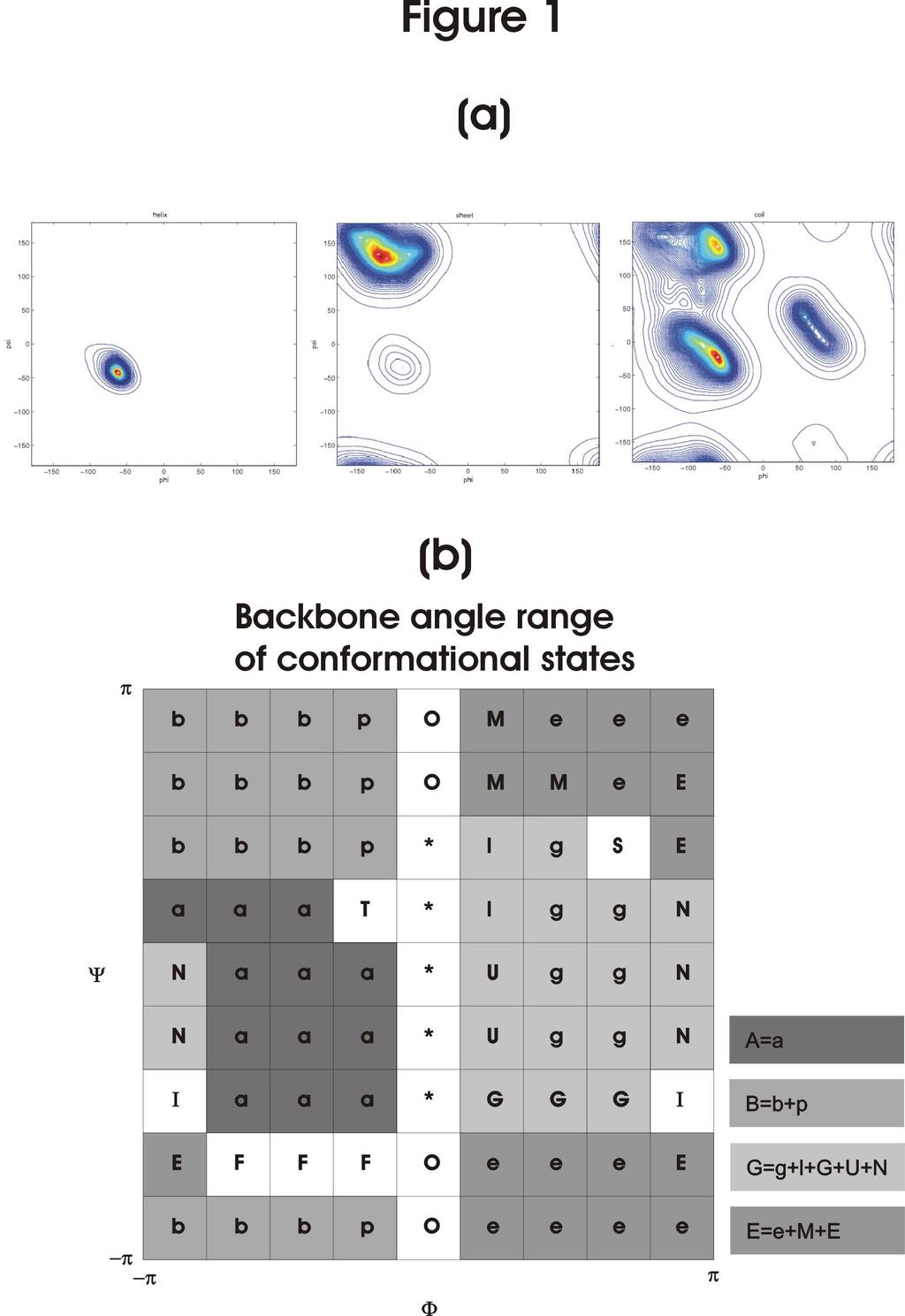
3 Introduction Protein backbone torsion (, ) angles are highly correlated to protein secondary structures. The distribution of the - angles in protein structures is mostly clustered around the alpha (centered at =-60, =-40 ), beta (centered at =-120, =120 ), and L-alpha (centered at =60, =0 ) regions of the Ramachandran plot. -helices and - sheets are comprised of residues with backbone torsion angles distributed mostly in the alpha and beta - angle regions respectively. Backbone structures in the loop regions are not as regular as in -helices and -sheets and can have - angles in any regions of the Ramachandran plot. Figure 1a summarizes the relationship between backbone torsion angles and secondary structure by plotting the distributions of - angles in the alpha, beta and loop regions. Both -helices and -strands are relatively straight in structure; the turning points in protein chains are made up of residues in loop regions. The loop residues in a protein chain play important roles as structural determinants in connecting regular secondary structure elements, leading to a specific protein folding topology for the protein structure (Richardson, 1981). Moreover, many loop residues involve enzymatic activities and protein-protein interactions, such as in antibody-antigen interactions. Local structural information provided by predictive algorithms will significantly facilitate the analysis of protein sequence-structure-function relationships. Although many loop regions contain recurrent local structural motifs (see recent reviews (de Brevern et al., 2002; Wojcik et al., 1999)), the large conformational 3
4 variability makes the characterization and prediction of loop conformations one of the most challenging molecular modeling problems (see for example (de Bakker et al., 2003; Fiser et al., 2000; Galaktionov et al., 2001; Wojcik et al., 1999; Xiang et al., 2002)). Accurate predictions of protein backbone torsion angles will further improve the prediction capacities of loop modeling procedures. Three-state (,, and coil) secondary structure prediction methods have reached ~80% accuracy (Petersen et al., 2000; Pollastri et al., 2002; Rost, 2001). Although these methods are powerful tools in protein structure prediction from amino acid sequences, three-state secondary structure predictions do not distinguish one loop conformation from the other. Backbone torsion angle predictions, on the other hand, provide local structural information that is useful in defining local structures for highly variable loop regions in amino acid sequences. While three-state secondary structure prediction methods have been developed with increasing accuracy, the prediction procedure for protein backbone torsion angle has received relatively little attention. The hidden Markov model HMMSTR based on local sequence-structure correlations in proteins has been demonstrated to make backbone torsion angle prediction with significant accuracy (Bystroff et al., 2000). This was the first protein backbone torsion angle prediction method benchmarked with a large set of test proteins (Bystroff et al., 2000). HMMSTR uses an alphabet of 11 conformation states, 10 corresponding to - angle regions and one for cis peptide bonds. 4
5 Other authors have presented an extensive study of the predictability of different definitions and alphabet sizes of local structural states (de Brevern et al., 2000). One of the goal of using predicted local structure is to improve performance of profile HMMs for fold recognition (Karchin et al., 2003). The focus of this work, however, is on the fold recognition problem rather than optimizing conformation state prediction. Recently, a local structure prediction method based on a local structure-based sequence profile database (LSBSP1) has been devised and tested for prediction accuracy (Yang & Wang, 2003). Although the LSBSP1 local structure prediction procedure has been demonstrated to predict reasonably accurate local structures for sequence segments of consecutive 9 residues based on the RMSD (root mean square deviation) measure, the backbone torsion angle prediction accuracy of the LSBSP1 based procedure has made only marginal progress in comparison with HMMSTR (Yang & Wang, 2003) (see also Table II for the comparison of the two published prediction results). In this work, we report two novel protein backbone torsion angle prediction procedures. One extends the previous LSBSP1 prediction procedure by using an artificial neural network (NN) algorithm to process and summarize the prediction results. The other uses support vector machine (SVM) to make protein backbone torsion angle prediction based on protein sequence profile produced with PSI-BLAST (Altschul et al., 1997) and the three-state secondary structure prediction from PSI-PRED (Jones, 1999). The goal of the prediction procedures is to predict the backbone conformational state of each residue in protein chains. Instead of using fine conformational states as in, for 5
6 example HMMSTR, our prediction procedures focused on prediction accuracy based on four (A, B, G, and E, for definition see Figure 1b) or three (A, B, G/E) conformational states. This goal reflects the general observation that, as shown in Figure 1a, there are only three major backbone conformational states for residues in proteins. Bystroff and Baker (Bystroff & Baker, 1998) have demonstrated that the backbones of two eightresidue segments can be superimposed with RMSD less than 1.4 A if none of the backbone torsion angles in one segment deviate from the corresponding torsion angles in the other segment by more than 120 degrees. This indicates that accurate course-grained backbone conformational state prediction can be extremely useful in local structure prediction. This is particularly true for the highly variable local structures as in the coil regions, which comprise slightly less than half of the residues in proteins and the conventional three-state (,, and coil) secondary structure prediction methods have provided essentially no local structural information for these residues. The two prediction methods have been benchmarked against extensive testing cases. The results show that these two methods improve backbone torsion angle prediction accuracy over those from previous published results. Method Protein backbone torsion angle conformational states Following previous work, protein backbone torsion angles are mapped onto the - plot (Oliva et al., 1997; Yang & Wang, 2003). We divided the - map into four 6
7 major conformational states: A, B, G, E. Figure 1b shows the - angle ranges of these conformational states. Almost all the residues in our training/testing proteins (see below for more details on the training/testing sets) have backbone torsion angles distributed in one of the four major conformational states. Only 0.38% of the residues have backbone conformation outside the four major conformational states. In addition, 1.8% of the residues, most of which are the N- or C-terminal residues, are not assigned to any of the conformational states because of lacking well-defined backbone atoms to calculate the - angles. These residues were removed from our training/testing set. Local structure-based sequence profile database LSBSP1 We only briefly describe the construction of the LSBSP1 database; more details can be found in a recently published work (Yang & Wang, 2003). The procedure has also been summarized in a flow chart available from our ftp server (ftp://ps7ayang.cpmc.columbia.edu/pub/lsbsp1flow1.pdf). The LSBSP1 database contains a total of position specific score matrices (PSSM). Each PSSM has the dimension of 9 by 20. Each of the PSSM was calculated from a structure-based multiple alignment constructed with a seed 9-residue segment from a protein structure. The seed segments in LSBSP1 are 9 consecutive residue sequence segments from the nonredundant protein structures in PDB_SELECT_25 (PDB_SELECT_25 (Hobohm et al., 1992) version Feb/2001, with no pair-wise sequence identify greater than 25%). To construct a PSSM based on a seed segment, we first used the seed segment as a probe to search through the non-redundant proteins. Sequence segments from the non-redundant protein set that are identical in backbone conformational states (see Figure 1) and have 7
8 the amino acid replacement scores above a threshold in comparison with the seed sequence were aligned to construct a preliminary local structure-based sequence profile for the seed segment. The sequence similarity was calculated with the structure-specific amino acid substitution matrices that we have developed to align distantly related protein pairs (Yang, 2002). This preliminary local structure-based sequence profile was then converted to a position specific score matrix (pre-pssm) in half-bit units with the Bayesian prediction pseudo-count method (Tatusov et al., 1994) : qji W Ji = 2log2( ) (1) p i where p i is the background probability (Tatusov et al., 1994) for amino acid type i, and q Ji = C Ji + ( B + M M + B 20 k = C Jk ) p i 1 (2) C Ji is the number of amino acid type i that appears in the column J of the sequence profile. M is the number of rows in the sequence profile. The term (B+M- k=1,20 C Jk ) in the numerator is the pseudo-count, where B=M 0.5 is considered adequate (Tatusov et al., 1994). This pre-pssm ([W Ji ], a 20 x 9 matrix) was further refined by removing in the preliminary structure-based multiple alignment the sequence segments that did not score higher than a threshold (>15) with the pre-pssm. The remaining set of segments form a refined local structure-based sequence profile and the PSSM was recalculated and saved along with the sequence and structural information of the seed 9-residue segment in the LSBSP1 database. The procedure described above was applied to all the 9-residue sequence segments in the non-redundant protein structures to construct the LSBSP1 database in the PrISM.1 system. 8
9 Protein backbone torsion angle prediction with LSBSP1 database and artificial neural network algorithm: the LSBSP1+NN method The goal of the LSBSP1+NN prediction procedure is to predict the backbone conformational state of the central residue in a 9-residue segment from a query protein sequence. A standard feed-forward back-propagation artificial neural network (Rumelhart et al., 1986) with single hidden layer is used in the torsion angle prediction procedure. The input layer has 216 input units, representing a window of 9 consecutive residues in a protein chain. The hidden layer has 50 units. Architectures with more hidden layer units did not improve the performance of the prediction capacities. The output layer has 3 units, representing A, B, or G/E backbone conformational state of the central residue in the input 9-residue segment. We group the G and E states into one class in the prediction output because the E conformational state has only 1.7% of the total training cases. The scarcity of the training cases made the prediction for E conformational state by itself extremely difficult with the artificial neural network and the SVM algorithm (see for example the results shown in Table II). By grouping together the G and E training cases (6.4% of the training cases), we were able to train the neural network algorithm to predict residues in the G/E state with reasonable accuracy. The 216 input units are divided into 9 groups, representing a window of 9 consecutive residues. We use a 9-residue window for prediction input because the LSBSP1 database was constructed with 9-residue segments (see above). Orthogonal representation of an amino acid type requires 21 input units. The input unit that specifies the amino acid type is set to 1 while all other 19 input units are set to zero. The 21 th input 9
10 unit is set to 1 for residue positions in the 9-residue window outside N- or C-terminus of the protein chain. The last three (22 th -24 th ) input units in each group are encoded with values summarized from the backbone torsion angle predictions with the LSBSP1 database. To make the LSBSP1-based backbone torsion angle prediction, we use each window of 9-residue segments in the query protein sequence as a probe sequence segment to match for 9-residue structure-based sequence profiles in the LSBSP1 database. All the LSBSP1 profiles for which the matching scores are more than a threshold of 20 and for which the secondary structure assignments are consistent with the PSI-PRED secondary structure prediction of the query sequence segment by more than 50% (Yang & Wang, 2003) are aligned to the query sequence to form a multiple alignment. Positions in the multiple alignment represent the predictions of the backbone conformational states, which can be A, B, or G/E, for the corresponding residue in the query sequence. As the 9-residue window slides through the query protein chain one residue at a time, the backbone conformational state predictions accumulate in the multiple alignment. Each column in the multiple alignment shows all the backbone conformational state predictions for the corresponding residue in the query protein. After the predictions for all 9-residue windows in the query protein, the multiple alignment for all the predicted backbone conformational states is then converted to a position specific scoring matrix with Equation (1) to calculate W Ji, where i can be A, B or G/E. The W Ji is a log-odds ratio in half-bit unit for the backbone conformational state predictions versus random predictions based on background probabilities of the conformational states at position J in the query sequence; a large positive W Ji value indicates that the residue position J is consistently predicted to be i conformational state, and a negative W Ji value 10
11 indicates that the J position is consistently predicted to be non-i conformational state. Three values within the range between 0 and 1 are calculated from W Ji for each position J in the query protein sequence using the standard logistic function (Jones, 1999): a Ji 1 = W 1 + e Ji (3), where i can be A, B or G/E. These a Ji values are used in 22 th -24 th input units for each group to encode the information of predicted backbone conformational states. An on-line back-propagation training procedure was used to update weights connecting the nodes after each training pattern presentation (Rumelhart et al., 1986). Each training pattern was randomly selected from a pool of 9-residue structural segments from training proteins. The input units are encoded based on the sequence of the 9- residue segment and the a Ji values derived from Equation (3) (see above). The three output target values are set to 0 or 1 based on the backbone conformational state of the central residue of the 9-residue segment. The momentum value of 0.9 is used to prevent oscillation. The learning rate of was found to be adequate in all the training procedures. A total of residue segments derived from a non-redundant protein set were used as training and testing cases. These non-redundant proteins are from the PDB_SELECT_25 list (version Dec/2002) and are not related to any of the proteins in LSBSP1 (from PDB_SELECT_25 version Feb/2001) with p-value threshold of 10-6 (or average sequence ID < 18%). The proteins in the non-redundant protein set are not related to each other by more than 25% sequence identity. It is important to have the 11
12 training/testing proteins unrelated to the proteins in LSBSP1 because close homologues to the proteins in LSBSP1 tend to have high accuracy in backbone torsion angle prediction with the LSBSP1-based method. The training and testing processes were carried out with 10-fold jackknife cross validation: 10% of the segments were used as testing cases while the rest 90% segments were used in training; the processes repeat 10 times, each with different 10% of the 9-residue segments. For each training-testing process, the training iteration was terminated when the prediction capacities of the network started to degrade on the 10% testing cases. The prediction accuracy was calculated by averaging the prediction accuracy rate for the testing cases over the 10-fold cross validation processes. Prediction accuracies were calculated by comparing the true backbone conformational state with the predicted conformational state. The predicted conformational state was indicated by the output node with the largest output value. Finally, the neural network was again trained with all the residue segments and the trained network was tested with a set of recently released proteins that are not related to any of the training proteins and the proteins in LSBSP1. Same p-value threshold described in the previous paragraph was used to identify the test proteins in a recent released PDB_SELECT_25 list (version Apr/2003). The prediction accuracy for the new test proteins was then compared with the average accuracy from the 10-fold cross validation. The comparison is to ensure that the LSBSP1+NN method has not been over-trained and the benchmarked accuracy is generally applicable to protein sequences of unknown structure. 12
13 The NN input nodes combine two types of information: amino acid sequence of the query sequence segment (1 st 21 st input unit) and the a Ji values, the information of predicted backbone conformational states (22 th -24 th input units). The combination of these two types of information gave the optimum overall prediction accuracy of 78.2% in the 10-fold cross validation results (see Table IV for details). Two tests have been performed to isolate the prediction effect of the two types of information: First, we used only the sequence information and removed 22 nd -24 th input units from the input nodes and re-run the 10-fold cross validation (see above). The results showed an overall prediction accuracy rate of 61.5%. Second, we used only the a Ji values, the information of predicted backbone conformational states (22 th -24 th input units) and removed 1 st -21 st input units from the input nodes and re-run the 10-fold cross validation. The results showed an overall prediction accuracy rate of 67.8%. These tests indicate that the latter information contributed more to the prediction accuracy and the artificial neural network algorithm does indeed combine these two types of information to make optimum prediction. Protein backbone torsion angle prediction with support vector machine and PSI-PRED We also developed a support vector machine (SVM) method to predict the backbone conformation of the middle amino acid in a 9-residue sequence segment. Here again, we classify either four types of conformational states (A, B, E and G), or we combine the two smallest states into a single class (E/G) for three state classification. Our main effort is to design the feature representation of 9-residue amino acid segments. The three kinds of information we use for features are amino acid sequences, PSI-BLAST 13
14 (Altschul et al., 1997) profiles and secondary structures, which are known for training data and predicted by PSI-PRED (Jones, 1999) for test data. Support Vector Machines. Support Vector Machines (SVMs) are a family of algorithms for classification problems (Vapnik, 1998). Given a training data set with m labeled training samples ( x i, y i ) (1 i m, xi n and yi {1,-1}), the goal is to learn a large margin linear classifier f to discriminate between the two classes. Here, a linear classifier can be represented as a function f(x)=<w, x>+b (w n, x n, b ) (4) The decision boundary is a hyperplane <w, x>+b =0, and the margin for a training example x is the value y f x ) (>0 if the example is correctly classified). A test i i ( i example will be classified as positive if f(x) > 0, negative otherwise. The linear classification function can be learned with a soft margin SVM (Cristianini & Shawe- Talor, 2000), which incorporates a trade-off between maximizing the geometric margin and minimizing margin violations on the training set. An important property of the SVM optimization problem is that we can replace the inner product x, x > by a kernel < i j function K ( x, y ) ; here, the kernel implicitly represents the inner product between feature vectors for pairs of input examples K ( x, y ) = < ( x ), ( y ) > for some feature mapping from the original input vector space to a feature space N (or a Hilbert space). d Typical kernels include polynomial kernels, K ( x, y ) = ( < x, y > + 1 ), or radial basis x y 2 kernels, K ( x, y ) = exp( )
15 One-vs-all classification is used to make multiclass predictions from trained binary SVM classifiers. We simply choose our prediction to be the class that gives the maximum margin for each test example x : y = arg max ( f j ( x)), where f j is the SVM classifier for the th j class. We use the publicly available SVM light package to learn the binary classifiers (Joachims, 1999) in our experiments. Binary Encoding Feature Map. A simple way of representing an amino acid sequence is through binary encoding. Here, each amino acid is represented as a 21-dimensional vector, where each dimension corresponds to one type of amino acid or to a special null character which is used to fill in the blank positions in window segments containing entries before the beginning or after the end of a protein sequence. The introduction of a null character helps to make predictions for boundary positions without affecting the overall accuracy. To encode an amino acid or blank at a particular position in the sequence, we put a positive constant in the corresponding entry in the feature vector and 0 in all other entries. A length 9-segment S is mapped to a 189 (9x21)-dimension vector by the feature map binary. PSI-BLAST Profile Feature Map. Instead of using a binary encoding of the amino acid sequence, we can represent the sequence segment with its PSI-BLAST log-odds score profile. These scores are calculated as [ln( Q i / Pi )]/ i, where Q i is the estimated probability for residue i to be found in that column, P i is the background frequency of i and i is a scale parameter. The way we construct the profile kernel is as follows: the 15
16 position specific score matrix (PSSM) is constructed for all protein sequences with PSI- BLAST program running under the standard setup of PSI-PRED, and then these PSSMs are cut along with the sequence into 9x20 matrices (each of the 9 positions encode a probability distribution over 20 amino acids). For the blank positions at the beginning and the end of a protein sequence, 20 zeros are filled in to represent the background distribution. The feature map 9 20-dimension vector in the PSSM. profile assigns to a length-9 segment S the concatenation of Predicted Secondary Structure Feature Mapping. In addition to the protein sequence, another useful source of information we can use is the secondary structure. The secondary structure of training sequences is derived from DSSP program (Kabsch & Sander, 1983). For testing sequences, secondary structures are predicted with the PSI- PRED program (Jones, 1999). Similar to the binary encoding for amino acid sequence, we can represent a length 9 segment as a binary encoding of 9 positions, each of which has a 4-dimensional vector (three kinds of secondary structures plus the blank position). We denote the secondary structure feature map as. Different feature representations can be combined by concatenation of feature sec vectors (direct product of vectors); we write, for example, binary x sec for direct product of binary and secondary structure feature maps. Two datasets are used for the evaluation of the SVM method: PDB_SELECT_25 and a modified version of the Dunbrack-culled PDB (Karchin et al., 2003). This version 16
17 of Dunbrack-culled PDB has a sequence identity cutoff of 20%, resolution cutoff of 3.0 Å, R-factor cutoff of 1.0 with fragments shorter that 20 residues removed. The Dunbrack-culled PDB dataset provides a more accurate non-redundant benchmark and allows us to compare SVM performance with other results from the literature. For the PDB_SELECT_25 dataset, we performed two sets of benchmark experiments for evaluation of the SVM classifiers. First we used proteins in the LSBSP1 database (PDB_SELECT_25 version Feb/2001) as the training set, and we tested on PDB_SELECT_25 (version Dec/2002) proteins that are not related to any proteins in the training set. For the second set of experiments, we performed 10-fold cross-validation on PDB_SELECT_25 as described in the neural net approach above. The results produced from the two training/testing procedures were essentially identical, and we report only the 10-fold cross-validation experiments below. Finally, we performed 3-fold crossvalidation experiments on the dunbrack-in-scop dataset, where the number of folds was chosen for consistency with previous published results (Karchin et al., 2003) in order allow for comparison. Results and Discussion Feature Representations and Kernel Selection for SVM (Table I): We first report results on the PDB_SELECT_25 dataset. Table I compares the prediction accuracy between four different types of feature maps for SVM classification. The profile feature mapping outperforms the binary mappingby around 6% and is outperformed by the 17
18 secondary structure feature mapping by 3%. A breakdown of results by conformation state suggests that both the profile and secondary structure feature maps have good results in alpha and beta regions but are less helpful in loop regions; in particular, the binary encoding is more successful than profile or secondary structure for prediction of the E/G state, which almost always occurs in loops. These results are understandable given the strong correlation between secondary structure and local conformation for alpha helices and beta strands, and given that profiles help predict these regular secondary structures; for loop regions, secondary structure information is complementary to local conformation and not directly useful for prediction. By integrating the secondary structure with the profile or binary mapping, both are improved significantly to approximately the same accuracy. The similarity in performance could be explained by the fact that the predicted secondary structures used for testing segments are also derived from profiles in PSI-PRED. Finally, in the last two columns of table I, we use true secondary structure and predicted secondary structure for both training and testing. We find that using true secondary structure dramatically improves results for conformation state A but slightly degrades performance for E/G, again showing that secondary structure information is not predictive of conformation states in loop regions. All the results shown in the table are produced with linear kernels. We also performed experiments using polynomial kernels and RBF kernels on the combined profile feature map and secondary structure feature map and obtained a slight improvement of about 1% in each case (see supplementary website for results). 18
19 Comparison with other methods (Table II and Table III): Table II compares the SVM results of linear kernel with profile and secondary structure prediction with results reproduced from our previous work (LSBSP1+consensus prediction) (Yang & Wang, 2003) and the published HMMSTR prediction accuracy (Bystroff et al., 2000). Since HMMSTR uses a larger alphabet of 11 conformation states, we facilitate the comparison by grouping the 10 states that correspond to - angle ranges into four A, B, G and E states. (The final state corresponds to cis peptide bonds rather than backbone angle state, and we omit this small set of residues in the comparison.) One might consider this comparison unfair to HMMSTR, which is trying to perform a more difficult multi-class prediction problem; however, if the predictions, when grouped into this coarser four-state setting, are less accurate than four-state prediction methods, one could argue that a smaller alphabet is better justified. We present the SVM prediction accuracy in Table II by making explicit G and E state predictions. The separation of the two states slightly decreases the overall prediction accuracy (see Table I and Table II). Even so, the comparisons are not straightforward because the three methods were benchmarked with different sets of test cases. One interesting negative result is that the SVM method performs poorly on the smallest class (E) compared to both LSBSP1+consensus and HMMSTR, indicating perhaps that the simple feature representation is not expressive enough to detect this class. However, overall, the SVM prediction clearly outperforms the previous methods by a few percent. More importantly, the SVM prediction were benchmarked with a much larger set of test cases: the SVM test set is three-fold larger than the test set used in benchmarking the LSBSP1+consensus method and is five-fold larger than the test set used in benchmarking the HMMSTR method. Based on the large 19
20 benchmark, we expect that the SVM prediction accuracy will generalize to backbone torsion angle predictions for protein sequences of unknown structure. Table III further shows the SVM prediction (Table II) details in number of predicted residues. Prediction errors are shown as the off-diagonal numbers. The SVM prediction has been validated with the same procedure and parameters but with a different protein set: dunbrack-in-scop dataset. The results are compared side-by-side in Table III. The differences are comparable with less than 1% (see also Table IV and Table V), indicating that the benchmark results shown in this work is relatively insensitive to the choice of the test and/or the training datasets. Prediction Accuracy in Loop Region (Table IV and V): The trained LSBSP1+NN was tested with proteins in the most recent PDB_SELECT_25 list (Apr, 2003). All new non-redundant proteins that are not related to the training proteins for the LSBSP1+NN method are used to test the prediction method. The results are summarized as follows: residues are in A backbone conformational state, and 81.5% are correctly predicted; residues are in B backbone conformational state, and 76.6% are correctly predicted; 2065 residues are in G/E backbone conformational state, and 45.6% are correctly predicted. Overall, 77.0% of the residues are correctly predicted. The test results are similar to the prediction accuracy derived from the 10-fold cross validation (see Table IV), indicating that the prediction capacities of LSBSP1+NN method shown in Table IV have not been over-trained. 20
21 Table IV compares the prediction performance of the SVM method and the LSBSP1+NN method. The comparison shows that the SVM prediction is slightly better than the LSBSP1+NN method but by less than 1%. To further compare the two methods on more leveled ground, we trained the SVM methods with the proteins used in constructing the LSBSP1 database and then tested on the test cases. The prediction accuracies are almost identical to the results shown in Table I and II. We conclude that the SVM method is more accurate than the LSBSP1+NN by a small margin. Finally, to verify our results, one additional SVM experiment is done on the dunbrack-in-scop dataset with the profile and secondary structure feature map using 3- fold cross-validation. The results shown in Table V are slightly better than those on PDB_SELECT_25, probably due to cleaner structural data in the second dataset. (We can compare the overall accuracy of 79.5% on this dataset for 3-state prediction and 78.4% for 4-state prediction with previous results using neural nets (Karchin et al., 2003), which found accuracy of 58.8% on a 10-state conformation alphabet and 64.9% on a 4-state alphabet.) The usefulness of the backbone torsion angle prediction resides in the prediction of the local structures in protein sequences of unknown structure. Our prediction assessments have shown that the backbone torsion angle predictions for -helix and - strand residues are highly accurate (89% on average and 59% baseline; baseline is evaluated with random prediction based on the conformational state population in the training proteins). However, local structures in -helices and -strands are relatively 21
22 regular, and hence torsion angle predictions are not particularly informative in providing additional structural information in comparison with three-state secondary structure predictions. In contrast, local structures in loop regions are highly variable. Table IV and V show that it is more difficult to make accurate backbone torsion angle prediction for loop residues. Still, the prediction accuracies with both the prediction methods are far greater than the 39.0% baseline calculated with random assignment of backbone conformational states to residues in the test set with background probabilities. Figure 2 analyzes the backbone torsion angle prediction capacities on loop residues using LSBSP1+NN. The solid line in Figure 2(a) shows that 30% of the loops (1394 out of 4673 loops) with 2~10 residues can be predicted with high accuracy (more than 80% residues in the loop are predicted correctly). In contrast, the dotted line in Figure 2(a) indicates that random prediction can only produce high accuracy prediction for 4% of the loops. Figure 2(b) shows that the average backbone torsion angle predictions are relatively insensitive of the loop length. Together, Figure 2(a) and (b) indicate that some of the local conformation for residues in protein loop regions are recognizable from their local amino acid sequences, and these local structural motifs are not restricted to short segments of residues connecting two secondary structural elements. Conclusion Optimally combining available information is one of the difficult challenges in knowledge-based protein structure prediction procedures. The SVM and the LSBSP1+NN methods are fundamentally different in using structural and sequence 22
23 information derived from the database of known protein structures. Similar prediction accuracy rate suggests that we are approaching the prediction limit for the prediction methods with current knowledge in protein structural database. The results show that backbone torsion angles in regular secondary structure elements can be predicted with high accuracy, and backbone torsion angles in loop residues are more difficult to predict. However, the current prediction accuracy for loop backbone torsion angles suggests that some of the structural motifs in loop regions can be recognized with high accuracy from local sequence information alone. The predictions will provide useful information for modeling protein structures from protein sequences. With the upcoming expansion of protein structural databases, we expect to further improve our prediction capacities in recognizing protein local conformations from local sequence segments. Acknowledgement: This work was partially supported by the NIH (PAR ). Planning Grant: National Programs of Excellence in Biomedical Computing for Computational Biology and Bioinformatics. CL was also supported by an Award in Informatics from PhRMA Foundation. 23
24 Figure legend Figure 1. (a) Density plot of the joint distribution of - angles in alpha-helix, betasheet and loop (coil) region from left to right. The density at a point in the plot is estimated by the area of the disk that is centered at the point and contains exactly 100 observations. (b) Protein backbone conformational states. The backbone torsion angle ranges of the backbone conformational states (A,B,G, and E) are defined in the righthand-side of the Ramachandran plot. The definitions of the conformational states shown in the Ramachandran plot were obtained from Oliva et al (Oliva et al., 1997). Figure 2. (a) Distribution of protein loops against backbone torsion angle prediction accuracy rate. A total of 4673 loops with 2 to 10 residues were used as test loops, which have not been used in the training of the LSBSP1+NN prediction methods. Prediction accuracy for each loop was calculated as the ratio of the correctly predicted residues over the residues in the loop. The distribution of the loops against the prediction accuracy rate is shown in the solid line. The dotted line shows the distribution of the same set of loops against a random prediction accuracy rate. The random prediction accuracy rate for each loop was calculated with random predictions for the loop residues based on the background probabilities for the conformational states. (b) Average prediction accuracy rate plotted against loop length. The data set shown in this plot is the same as the data set shown in Table III. In this figure, the data set was subdivided into groups based on the loop length, and the average accuracy rate for each subgroup was calculated by averaging over residues in the subgroup. 24
25 Table I Prediction results using 10-fold cross-validation by SVM classification with various feature maps (PDB_SELECT_25 dataset). The combination of included since binary and profile is not profile is a richer representation of binary. The definition of these feature Test Case mappings are described in the Methods section. binary profile sec binary x sec profile x sec profile x true _ sec A % 77.30% 71.30% 80.50% 82.00% 83.41% 68.31% B % 65.40% 87.22% 78.80% 79.00% 86.78% 89.51% G/E % 42.60% 7.76% 48.50% 51.10% 52.68% 54.28% total % 70.10% 73.70% 77.70% 78.70% 82.78% 76.15% * profile x sec : uses true secondary structure for training and predicted secondary structure for testing * profile x true _ sec : uses true secondary structure for both training and testing x _ sec : uses predicted secondary structure for both training and testing * profile pred profile x pred _ sec 25
26 Table II Comparison of the prediction accuracies from SVM method against the two previously published results. The training and testing of the SVM method are described in the Method section. The LSBSP1+consensus data are reproduced from Table I of Yang and Wang (Yang & Wang, 2003). The HMMSTR data are reproduced from Table 5 of Bystroff et al (Bystroff et al., 2000). The definitions of the conformational states in HMMSTR predictions are not completely identical to the definitions of the A, B, G, E conformational states shown in Figure 1. For comparison, A =H+G, B =B+E+d+b+e, G =L+l, and E =x; the backbone conformational states on the right-hand side of the equations were defined by Bystroff et al (Bystroff et al., 2000). The A, B, G and E states are approximately equivalent to the A, B, G, and E states defined in Figure 1. The test cases listed under the HMMSTR predictions are the residues in the A, B, G, and E backbone conformational state respectively. SVM profile x sec LSBSP1+consensus prediction (consensus level=1) HMMSTR test cases accuracy test cases accuracy test cases accuracy A % % A = % B % % B = % G % % G = % E % % E = % total % % % 26
27 Table III Position-wise predicted conformation states are tabulated according to true values. dunbrack-in-scop PDB_SELECT_25 A pred B pred G/E pred Total A pred B pred G/E pred Total A obs A obs B obs B obs G/E obs G/E obs Total Total
28 Table IV Comparison of the prediction results from SVM with linear kernel and LSBSP1+NN methods. In here, the loop residues are the residues in the coil regions that connect two flanking regular secondary structure elements in the test proteins. Coil residues in the N- and C-termini are not included. Regular secondary structure elements were defined by the DSSP program: -helices are regions with at least four consecutive H residues characterized by the DSSP program, and -strands are regions with at least two consecutive E residues characterized by the DSSP program. The test cases are obtained from PDB_SELECT_25 dataset. All residues Loop residues only Test SVM LSBSP1 Test SVM LSBSP1 cases +NN cases +NN A % 81.9% % 61.3% B % 78.0% % 70.1% G/E % 50.1% % 47.1% total % 78.2% % 63.5% 28
29 Table V SVM predictions for test cases from the dunbrack-in-scop dataset. All residues Loop residues only Test cases SVM Test cases SVM A % % B % % G/E % % total % % 29
30 References: Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17), Bystroff, C., Thorsson, V. & Baker, D. (2000). HMMSTR: a hidden markov model for local sequence-structure correlation in proteins. J Mol Biol 301(1), Bystroff, C. and Baker, D. (1998). Prediction of local structure in proteins using a library of sequence-structure motif. J Mol Biol 281(3), Cristianini, N. & Shawe-Talor, J. (2000). A introduction to support vector machines, Cambridge. de Bakker, P. I., DePristo, M. A., Burke, D. F. & Blundell, T. L. (2003). Ab initio construction of polypeptide fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Born solvation model. Proteins. 51(1), de Brevern, A. G., Valadie, H., Hazout, S. & Etchebest, C. (2002). Extension of a local backbone description using a structural alphabet: a new approach to the sequencestructure relationship. Protein Science. 11(12), de Brevern, A. G., Etchebest, C. Hazout, S. (2000) Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins. 41(3), Fiser, A., Do, R. K. & Sali, A. (2000). Modeling of loops in protein structures. Protein Science. 9(9),
31 Galaktionov, S., Nikiforovich, G. V. & Marshall, G. R. (2001). Ab initio modeling of small, medium, and large loops in proteins. Biopolymers. 60(2), Hobohm, U., Scharf, M., Schneider, R. & Sander, C. (1992). Selection of representative protein data sets. Protein Sci 1(3), Joachims, T. (1999). Making large-scale SVM Learning Practical. Advances in Kernel Methods - Support Vector Learning (Schölkopf, B., Burges, C. & Smola, A., Eds.), MIT. Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrix. J Mol Biol 292, Kabsch, W. & Sander, C. (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12), Karchin, R., Cline, M., Mandel-Gutfreund, Y., and Karplus, K. (2003). Hidden Markov Models That Use Predicted Local Structure for Fold Recognition: Alphabets of Backbone Geometry. Proteins: Structure, Function, and Genetics 51: Oliva, B., Bates, P. A., Querol, E., Aviles, F. X. & Sternberg, M. J. (1997). An automated classification of the structure of protein loops. J Mol Biol 266(4), Petersen, T. N., Lundegaard, C., Nielsen, M., Bohr, H., Bohr, J., Brunak, S., Gippert, G. P. & Lund, O. (2000). Prediction of protein secondary structure at 80% accuracy. Proteins. 41(1), Pollastri, G., Przybylski, D., Rost, B. & Baldi, P. (2002). Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins. 47(2),
32 Richardson, J. S. (1981). The anatomy and taxonomy of protein structure. Advances in Protein Chemistry. 34, Rost, B. (2001). Review: protein secondary structure prediction continues to rise. Journal of Structural Biology. 134(2-3), Rumelhart, D. E., Hinton, G. E. & Williams, R. J. (1986). Learning representations by back-propagating errors. Nature 323, Tatusov, R. L., Altschul, S. F. & Koonin, E. V. (1994). Detection of conserved segments in proteins: iterative scanning of sequence databases with alignment blocks. Proc Natl Acad Sci USA 91(25), Vapnik, V. N. (1998). Statistical Learning Theory, Springer. Wojcik, J., Mornon, J. P. & Chomilier, J. (1999). New efficient statistical sequencedependent structure prediction of short to medium-sized protein loops based on an exhaustive loop classification. J Mol Biol 289(5), Xiang, Z., Soto, C. S. & Honig, B. (2002). Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proceedings of the National Academy of Sciences of the United States of America. 99(11), Yang, A. S. (2002). Structure-dependent sequence alignment for remotely related proteins. Bioinformatics 18(12), Yang, A. S. & Wang, L. (2003). Local structure prediction with local structure-based sequence profiles. Bioinformatics 19(10),
33 33
34 34
Sequence Analysis '17 -- lecture Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction
 Sequence Analysis '17 -- lecture 16 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction Alpha helix Right-handed helix. H-bond is from the oxygen at i to the nitrogen
Sequence Analysis '17 -- lecture 16 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction Alpha helix Right-handed helix. H-bond is from the oxygen at i to the nitrogen
Local structure prediction with local structure-based sequence profiles
 BIOINFORMATICS Vol. 19 no. 10 2003, pages 1267 1274 DOI: 10.1093/bioinformatics/btg151 Local structure prediction with local structure-based sequence profiles An-Suei Yang and Lu-yong Wang Department of
BIOINFORMATICS Vol. 19 no. 10 2003, pages 1267 1274 DOI: 10.1093/bioinformatics/btg151 Local structure prediction with local structure-based sequence profiles An-Suei Yang and Lu-yong Wang Department of
Finding Regularity in Protein Secondary Structures using a Cluster-based Genetic Algorithm
 Finding Regularity in Protein Secondary Structures using a Cluster-based Genetic Algorithm Yen-Wei Chu 1,3, Chuen-Tsai Sun 3, Chung-Yuan Huang 2,3 1) Department of Information Management 2) Department
Finding Regularity in Protein Secondary Structures using a Cluster-based Genetic Algorithm Yen-Wei Chu 1,3, Chuen-Tsai Sun 3, Chung-Yuan Huang 2,3 1) Department of Information Management 2) Department
Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS*
 COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 9(1-2) 93-100 (2003/2004) Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS* DARIUSZ PLEWCZYNSKI AND LESZEK RYCHLEWSKI BiolnfoBank
COMPUTATIONAL METHODS IN SCIENCE AND TECHNOLOGY 9(1-2) 93-100 (2003/2004) Ab Initio SERVER PROTOTYPE FOR PREDICTION OF PHOSPHORYLATION SITES IN PROTEINS* DARIUSZ PLEWCZYNSKI AND LESZEK RYCHLEWSKI BiolnfoBank
Bayesian Inference using Neural Net Likelihood Models for Protein Secondary Structure Prediction
 Bayesian Inference using Neural Net Likelihood Models for Protein Secondary Structure Prediction Seong-gon KIM Dept. of Computer & Information Science & Engineering, University of Florida Gainesville,
Bayesian Inference using Neural Net Likelihood Models for Protein Secondary Structure Prediction Seong-gon KIM Dept. of Computer & Information Science & Engineering, University of Florida Gainesville,
Bioinformatics & Protein Structural Analysis. Bioinformatics & Protein Structural Analysis. Learning Objective. Proteomics
 The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
The molecular structures of proteins are complex and can be defined at various levels. These structures can also be predicted from their amino-acid sequences. Protein structure prediction is one of the
Assessing a novel approach for predicting local 3D protein structures from sequence
 Assessing a novel approach for predicting local 3D protein structures from sequence Cristina Benros*, Alexandre G. de Brevern, Catherine Etchebest and Serge Hazout Equipe de Bioinformatique Génomique et
Assessing a novel approach for predicting local 3D protein structures from sequence Cristina Benros*, Alexandre G. de Brevern, Catherine Etchebest and Serge Hazout Equipe de Bioinformatique Génomique et
A Protein Secondary Structure Prediction Method Based on BP Neural Network Ru-xi YIN, Li-zhen LIU*, Wei SONG, Xin-lei ZHAO and Chao DU
 2017 2nd International Conference on Artificial Intelligence: Techniques and Applications (AITA 2017 ISBN: 978-1-60595-491-2 A Protein Secondary Structure Prediction Method Based on BP Neural Network Ru-xi
2017 2nd International Conference on Artificial Intelligence: Techniques and Applications (AITA 2017 ISBN: 978-1-60595-491-2 A Protein Secondary Structure Prediction Method Based on BP Neural Network Ru-xi
Combining PSSM and physicochemical feature for protein structure prediction with support vector machine
 See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/316994523 Combining PSSM and physicochemical feature for protein structure prediction with
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/316994523 Combining PSSM and physicochemical feature for protein structure prediction with
Molecular Modeling Lecture 8. Local structure Database search Multiple alignment Automated homology modeling
 Molecular Modeling 2018 -- Lecture 8 Local structure Database search Multiple alignment Automated homology modeling An exception to the no-insertions-in-helix rule Actual structures (myosin)! prolines
Molecular Modeling 2018 -- Lecture 8 Local structure Database search Multiple alignment Automated homology modeling An exception to the no-insertions-in-helix rule Actual structures (myosin)! prolines
Statistical Machine Learning Methods for Bioinformatics VI. Support Vector Machine Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics VI. Support Vector Machine Applications in Bioinformatics Jianlin Cheng, PhD Computer Science Department and Informatics Institute University of
Statistical Machine Learning Methods for Bioinformatics VI. Support Vector Machine Applications in Bioinformatics Jianlin Cheng, PhD Computer Science Department and Informatics Institute University of
Database Searching and BLAST Dannie Durand
 Computational Genomics and Molecular Biology, Fall 2013 1 Database Searching and BLAST Dannie Durand Tuesday, October 8th Review: Karlin-Altschul Statistics Recall that a Maximal Segment Pair (MSP) is
Computational Genomics and Molecular Biology, Fall 2013 1 Database Searching and BLAST Dannie Durand Tuesday, October 8th Review: Karlin-Altschul Statistics Recall that a Maximal Segment Pair (MSP) is
Molecular Modeling 9. Protein structure prediction, part 2: Homology modeling, fold recognition & threading
 Molecular Modeling 9 Protein structure prediction, part 2: Homology modeling, fold recognition & threading The project... Remember: You are smarter than the program. Inspecting the model: Are amino acids
Molecular Modeling 9 Protein structure prediction, part 2: Homology modeling, fold recognition & threading The project... Remember: You are smarter than the program. Inspecting the model: Are amino acids
Discovering Sequence-Structure Motifs from Protein Segments and Two Applications. T. Tang, J. Xu, and M. Li
 Discovering Sequence-Structure Motifs from Protein Segments and Two Applications T. Tang, J. Xu, and M. Li Pacific Symposium on Biocomputing 10:370-381(2005) DISCOVERING SEQUENCE-STRUCTURE MOTIFS FROM
Discovering Sequence-Structure Motifs from Protein Segments and Two Applications T. Tang, J. Xu, and M. Li Pacific Symposium on Biocomputing 10:370-381(2005) DISCOVERING SEQUENCE-STRUCTURE MOTIFS FROM
An Overview of Protein Structure Prediction: From Homology to Ab Initio
 An Overview of Protein Structure Prediction: From Homology to Ab Initio Final Project For Bioc218, Computational Molecular Biology Zhiyong Zhang Abstract The current status of the protein prediction methods,
An Overview of Protein Structure Prediction: From Homology to Ab Initio Final Project For Bioc218, Computational Molecular Biology Zhiyong Zhang Abstract The current status of the protein prediction methods,
CS273: Algorithms for Structure Handout # 5 and Motion in Biology Stanford University Tuesday, 13 April 2004
 CS273: Algorithms for Structure Handout # 5 and Motion in Biology Stanford University Tuesday, 13 April 2004 Lecture #5: 13 April 2004 Topics: Sequence motif identification Scribe: Samantha Chui 1 Introduction
CS273: Algorithms for Structure Handout # 5 and Motion in Biology Stanford University Tuesday, 13 April 2004 Lecture #5: 13 April 2004 Topics: Sequence motif identification Scribe: Samantha Chui 1 Introduction
Representation in Supervised Machine Learning Application to Biological Problems
 Representation in Supervised Machine Learning Application to Biological Problems Frank Lab Howard Hughes Medical Institute & Columbia University 2010 Robert Howard Langlois Hughes Medical Institute What
Representation in Supervised Machine Learning Application to Biological Problems Frank Lab Howard Hughes Medical Institute & Columbia University 2010 Robert Howard Langlois Hughes Medical Institute What
I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure
 W306 W310 Nucleic Acids Research, 2005, Vol. 33, Web Server issue doi:10.1093/nar/gki375 I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure Emidio Capriotti,
W306 W310 Nucleic Acids Research, 2005, Vol. 33, Web Server issue doi:10.1093/nar/gki375 I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure Emidio Capriotti,
NNvPDB: Neural Network based Protein Secondary Structure Prediction with PDB Validation
 www.bioinformation.net Web server Volume 11(8) NNvPDB: Neural Network based Protein Secondary Structure Prediction with PDB Validation Seethalakshmi Sakthivel, Habeeb S.K.M* Department of Bioinformatics,
www.bioinformation.net Web server Volume 11(8) NNvPDB: Neural Network based Protein Secondary Structure Prediction with PDB Validation Seethalakshmi Sakthivel, Habeeb S.K.M* Department of Bioinformatics,
Structural Bioinformatics (C3210) Conformational Analysis Protein Folding Protein Structure Prediction
 Conformational Analysis Protein Folding Protein Structure Prediction") Structural Bioinformatics (C3210) Conformational Analysis Protein Folding Protein Structure Prediction Conformational Analysis 2 Conformational Analysis Properties of molecules depend on their three-dimensional
Structural Bioinformatics (C3210) Conformational Analysis Protein Folding Protein Structure Prediction Conformational Analysis 2 Conformational Analysis Properties of molecules depend on their three-dimensional
Supplement for the manuscript entitled
 Supplement for the manuscript entitled Prediction and Analysis of Nucleotide Binding Residues Using Sequence and Sequence-derived Structural Descriptors by Ke Chen, Marcin Mizianty and Lukasz Kurgan FEATURE-BASED
Supplement for the manuscript entitled Prediction and Analysis of Nucleotide Binding Residues Using Sequence and Sequence-derived Structural Descriptors by Ke Chen, Marcin Mizianty and Lukasz Kurgan FEATURE-BASED
Ranking Beta Sheet Topologies of Proteins
 , October 20-22, 2010, San Francisco, USA Ranking Beta Sheet Topologies of Proteins Rasmus Fonseca, Glennie Helles and Pawel Winter Abstract One of the challenges of protein structure prediction is to
, October 20-22, 2010, San Francisco, USA Ranking Beta Sheet Topologies of Proteins Rasmus Fonseca, Glennie Helles and Pawel Winter Abstract One of the challenges of protein structure prediction is to
Protein Structure Prediction
 Homology Modeling Protein Structure Prediction Ingo Ruczinski M T S K G G G Y F F Y D E L Y G V V V V L I V L S D E S Department of Biostatistics, Johns Hopkins University Fold Recognition b Initio Structure
Homology Modeling Protein Structure Prediction Ingo Ruczinski M T S K G G G Y F F Y D E L Y G V V V V L I V L S D E S Department of Biostatistics, Johns Hopkins University Fold Recognition b Initio Structure
Designing and benchmarking the MULTICOM protein structure prediction system
 Li et al. BMC Structural Biology 2013, 13:2 RESEARCH ARTICLE Open Access Designing and benchmarking the MULTICOM protein structure prediction system Jilong Li 1, Xin Deng 1, Jesse Eickholt 1 and Jianlin
Li et al. BMC Structural Biology 2013, 13:2 RESEARCH ARTICLE Open Access Designing and benchmarking the MULTICOM protein structure prediction system Jilong Li 1, Xin Deng 1, Jesse Eickholt 1 and Jianlin
BIOINFORMATICS ORIGINAL PAPER
 BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 10 2005, pages 2370 2374 doi:10.1093/bioinformatics/bti358 Structural bioinformatics Improved method for predicting β-turn using support vector machine Qidong
BIOINFORMATICS ORIGINAL PAPER Vol. 21 no. 10 2005, pages 2370 2374 doi:10.1093/bioinformatics/bti358 Structural bioinformatics Improved method for predicting β-turn using support vector machine Qidong
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Secondary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Secondary Structure Prediction Secondary Structure Annotation Given a macromolecular structure Identify the regions of secondary structure
CMPS 6630: Introduction to Computational Biology and Bioinformatics Secondary Structure Prediction Secondary Structure Annotation Given a macromolecular structure Identify the regions of secondary structure
BIOINFORMATICS THE MACHINE LEARNING APPROACH
 88 Proceedings of the 4 th International Conference on Informatics and Information Technology BIOINFORMATICS THE MACHINE LEARNING APPROACH A. Madevska-Bogdanova Inst, Informatics, Fac. Natural Sc. and
88 Proceedings of the 4 th International Conference on Informatics and Information Technology BIOINFORMATICS THE MACHINE LEARNING APPROACH A. Madevska-Bogdanova Inst, Informatics, Fac. Natural Sc. and
Protein Domain Boundary Prediction from Residue Sequence Alone using Bayesian Neural Networks
 Protein Domain Boundary Prediction from Residue Sequence Alone using Bayesian s DAVID SACHEZ SPIROS H. COURELLIS Department of Computer Science Department of Computer Science California State University
Protein Domain Boundary Prediction from Residue Sequence Alone using Bayesian s DAVID SACHEZ SPIROS H. COURELLIS Department of Computer Science Department of Computer Science California State University
Textbook Reading Guidelines
 Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: May 1, 2009 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
PCA and SOM based Dimension Reduction Techniques for Quaternary Protein Structure Prediction
 PCA and SOM based Dimension Reduction Techniques for Quaternary Protein Structure Prediction Sanyukta Chetia Department of Electronics and Communication Engineering, Gauhati University-781014, Guwahati,
PCA and SOM based Dimension Reduction Techniques for Quaternary Protein Structure Prediction Sanyukta Chetia Department of Electronics and Communication Engineering, Gauhati University-781014, Guwahati,
VALLIAMMAI ENGINEERING COLLEGE
 VALLIAMMAI ENGINEERING COLLEGE SRM Nagar, Kattankulathur 603 203 DEPARTMENT OF COMPUTER SCIENCE AND ENGINEERING QUESTION BANK VII SEMESTER BM6005 BIO INFORMATICS Regulation 2013 Academic Year 2018-19 Prepared
VALLIAMMAI ENGINEERING COLLEGE SRM Nagar, Kattankulathur 603 203 DEPARTMENT OF COMPUTER SCIENCE AND ENGINEERING QUESTION BANK VII SEMESTER BM6005 BIO INFORMATICS Regulation 2013 Academic Year 2018-19 Prepared
Methods and tools for exploring functional genomics data
 Methods and tools for exploring functional genomics data William Stafford Noble Department of Genome Sciences Department of Computer Science and Engineering University of Washington Outline Searching for
Methods and tools for exploring functional genomics data William Stafford Noble Department of Genome Sciences Department of Computer Science and Engineering University of Washington Outline Searching for
COMBINING SEQUENCE AND STRUCTURAL PROFILES FOR PROTEIN SOLVENT ACCESSIBILITY PREDICTION
 195 COMBINING SEQUENCE AND STRUCTURAL PROFILES FOR PROTEIN SOLVENT ACCESSIBILITY PREDICTION Rajkumar Bondugula Digital Biology Laboratory, 110 C.S. Bond Life Sciences Center, University of Missouri Columbia,
195 COMBINING SEQUENCE AND STRUCTURAL PROFILES FOR PROTEIN SOLVENT ACCESSIBILITY PREDICTION Rajkumar Bondugula Digital Biology Laboratory, 110 C.S. Bond Life Sciences Center, University of Missouri Columbia,
Title: A topological and conformational stability alphabet for multi-pass membrane proteins
 Supplementary Information Title: A topological and conformational stability alphabet for multi-pass membrane proteins Authors: Feng, X. 1 & Barth, P. 1,2,3 Correspondences should be addressed to: P.B.
Supplementary Information Title: A topological and conformational stability alphabet for multi-pass membrane proteins Authors: Feng, X. 1 & Barth, P. 1,2,3 Correspondences should be addressed to: P.B.
A Hidden Markov Model for Identification of Helix-Turn-Helix Motifs
 A Hidden Markov Model for Identification of Helix-Turn-Helix Motifs CHANGHUI YAN and JING HU Department of Computer Science Utah State University Logan, UT 84341 USA cyan@cc.usu.edu http://www.cs.usu.edu/~cyan
A Hidden Markov Model for Identification of Helix-Turn-Helix Motifs CHANGHUI YAN and JING HU Department of Computer Science Utah State University Logan, UT 84341 USA cyan@cc.usu.edu http://www.cs.usu.edu/~cyan
Simple jury predicts protein secondary structure best
 Simple jury predicts protein secondary structure best Burkhard Rost 1,*, Pierre Baldi 2, Geoff Barton 3, James Cuff 4, Volker Eyrich 5,1, David Jones 6, Kevin Karplus 7, Ross King 8, Gianluca Pollastri
Simple jury predicts protein secondary structure best Burkhard Rost 1,*, Pierre Baldi 2, Geoff Barton 3, James Cuff 4, Volker Eyrich 5,1, David Jones 6, Kevin Karplus 7, Ross King 8, Gianluca Pollastri
Prediction of DNA-Binding Proteins from Structural Features
 Prediction of DNA-Binding Proteins from Structural Features Andrea Szabóová 1, Ondřej Kuželka 1, Filip Železný1, and Jakub Tolar 2 1 Czech Technical University, Prague, Czech Republic 2 University of Minnesota,
Prediction of DNA-Binding Proteins from Structural Features Andrea Szabóová 1, Ondřej Kuželka 1, Filip Železný1, and Jakub Tolar 2 1 Czech Technical University, Prague, Czech Republic 2 University of Minnesota,
UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics
 UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, 2010 SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics DURATION: 3 HOURS TOTAL MARKS: 125 Internal Examiner: Dr. Ché Pillay External Examiner: Prof. Nicola
UNIVERSITY OF KWAZULU-NATAL EXAMINATIONS: MAIN, 2010 SUBJECT, COURSE AND CODE: GENE 320: Bioinformatics DURATION: 3 HOURS TOTAL MARKS: 125 Internal Examiner: Dr. Ché Pillay External Examiner: Prof. Nicola
RosettainCASP4:ProgressinAbInitioProteinStructure Prediction
 PROTEINS: Structure, Function, and Genetics Suppl 5:119 126 (2001) DOI 10.1002/prot.1170 RosettainCASP4:ProgressinAbInitioProteinStructure Prediction RichardBonneau, 1 JerryTsai, 1 IngoRuczinski, 1 DylanChivian,
PROTEINS: Structure, Function, and Genetics Suppl 5:119 126 (2001) DOI 10.1002/prot.1170 RosettainCASP4:ProgressinAbInitioProteinStructure Prediction RichardBonneau, 1 JerryTsai, 1 IngoRuczinski, 1 DylanChivian,
Protein Structure Prediction. christian studer , EPFL
 Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
Protein Structure Prediction christian studer 17.11.2004, EPFL Content Definition of the problem Possible approaches DSSP / PSI-BLAST Generalization Results Definition of the problem Massive amounts of
Prot-SSP: A Tool for Amino Acid Pairing Pattern Analysis in Secondary Structures
 Mol2Net, 2015, 1(Section F), pages 1-6, Proceedings 1 SciForum Mol2Net Prot-SSP: A Tool for Amino Acid Pairing Pattern Analysis in Secondary Structures Miguel de Sousa 1, *, Cristian R. Munteanu 2 and
Mol2Net, 2015, 1(Section F), pages 1-6, Proceedings 1 SciForum Mol2Net Prot-SSP: A Tool for Amino Acid Pairing Pattern Analysis in Secondary Structures Miguel de Sousa 1, *, Cristian R. Munteanu 2 and
Local backbone structure prediction of proteins.
 Local backbone structure prediction of proteins. Alexandre De Brevern, Cristina Benros, Romain Gautier, Hélène Valadié, Serge Hazout, Catherine Etchebest To cite this version: Alexandre De Brevern, Cristina
Local backbone structure prediction of proteins. Alexandre De Brevern, Cristina Benros, Romain Gautier, Hélène Valadié, Serge Hazout, Catherine Etchebest To cite this version: Alexandre De Brevern, Cristina
3D Structure Prediction with Fold Recognition/Threading. Michael Tress CNB-CSIC, Madrid
 3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
3D Structure Prediction with Fold Recognition/Threading Michael Tress CNB-CSIC, Madrid MREYKLVVLGSGGVGKSALTVQFVQGIFVDEYDPTIEDSY RKQVEVDCQQCMLEILDTAGTEQFTAMRDLYMKNGQGFAL VYSITAQSTFNDLQDLREQILRVKDTEDVPMILVGNKCDL
Structure Prediction. Modelado de proteínas por homología GPSRYIV. Master Dianas Terapéuticas en Señalización Celular: Investigación y Desarrollo
 Master Dianas Terapéuticas en Señalización Celular: Investigación y Desarrollo Modelado de proteínas por homología Federico Gago (federico.gago@uah.es) Departamento de Farmacología Structure Prediction
Master Dianas Terapéuticas en Señalización Celular: Investigación y Desarrollo Modelado de proteínas por homología Federico Gago (federico.gago@uah.es) Departamento de Farmacología Structure Prediction
Protein Sequence Analysis. BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl)
") Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Protein Sequence Analysis BME 110: CompBio Tools Todd Lowe April 19, 2007 (Slide Presentation: Carol Rohl) Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical
Protein structure. Wednesday, October 4, 2006
 Protein structure Wednesday, October 4, 2006 Introduction to Bioinformatics Johns Hopkins School of Public Health 260.602.01 J. Pevsner pevsner@jhmi.edu Copyright notice Many of the images in this powerpoint
Protein structure Wednesday, October 4, 2006 Introduction to Bioinformatics Johns Hopkins School of Public Health 260.602.01 J. Pevsner pevsner@jhmi.edu Copyright notice Many of the images in this powerpoint
Mining Residue Contacts in Proteins Using Local Structure Predictions
 IEEE TRANSACTIONS ON SYSTEMS, MAN, AND CYBERNETICS PART B: CYBERNETICS, VOL. 33, NO. 5, OCTOBER 2003 789 Mining Residue Contacts in Proteins Using Local Structure Predictions Mohammed J. Zaki, Associate
IEEE TRANSACTIONS ON SYSTEMS, MAN, AND CYBERNETICS PART B: CYBERNETICS, VOL. 33, NO. 5, OCTOBER 2003 789 Mining Residue Contacts in Proteins Using Local Structure Predictions Mohammed J. Zaki, Associate
proteins Prediction of RNA binding sites in a protein using SVM and PSSM profile Manish Kumar, 1 M. Michael Gromiha, 2 and G. P. S.
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Prediction of RNA binding sites in a protein using SVM and PSSM profile Manish Kumar, 1 M. Michael Gromiha, 2 and G. P. S. Raghava 1 * 1 Bioinformatics Centre,
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Prediction of RNA binding sites in a protein using SVM and PSSM profile Manish Kumar, 1 M. Michael Gromiha, 2 and G. P. S. Raghava 1 * 1 Bioinformatics Centre,
CFSSP: Chou and Fasman Secondary Structure Prediction server
 Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Wide Spectrum, Vol. 1, No. 9, (2013) pp 15-19 CFSSP: Chou and Fasman Secondary Structure Prediction server T. Ashok Kumar Department of Bioinformatics, Noorul Islam College of Arts and Science, Kumaracoil
Artificial Intelligence in Prediction of Secondary Protein Structure Using CB513 Database
 Artificial Intelligence in Prediction of Secondary Protein Structure Using CB513 Database Zikrija Avdagic, Prof.Dr.Sci. Elvir Purisevic, Mr.Sci. Samir Omanovic, Mr.Sci. Zlatan Coralic, Pharma D. University
Artificial Intelligence in Prediction of Secondary Protein Structure Using CB513 Database Zikrija Avdagic, Prof.Dr.Sci. Elvir Purisevic, Mr.Sci. Samir Omanovic, Mr.Sci. Zlatan Coralic, Pharma D. University
CSE : Computational Issues in Molecular Biology. Lecture 19. Spring 2004
 CSE 397-497: Computational Issues in Molecular Biology Lecture 19 Spring 2004-1- Protein structure Primary structure of protein is determined by number and order of amino acids within polypeptide chain.
CSE 397-497: Computational Issues in Molecular Biology Lecture 19 Spring 2004-1- Protein structure Primary structure of protein is determined by number and order of amino acids within polypeptide chain.
A Combination of a Functional Motif Model and a Structural Motif Model for a Database Validation
 A Combination of a Functional Motif Model and a Structural Motif Model for a Database Validation Minoru Asogawa, Yukiko Fujiwara, Akihiko Konagaya Massively Parallel Systems NEC Laboratory, RWCP * 4-1-1,
A Combination of a Functional Motif Model and a Structural Motif Model for a Database Validation Minoru Asogawa, Yukiko Fujiwara, Akihiko Konagaya Massively Parallel Systems NEC Laboratory, RWCP * 4-1-1,
Multi-class protein Structure Prediction Using Machine Learning Techniques
 Multi-class protein Structure Prediction Using Machine Learning Techniques Mayuri Patel Information Technology Department A.D Patel College Institute oftechnology Vallabh Vidyanagar 388 120 mayuri.ldce@gmail.com
Multi-class protein Structure Prediction Using Machine Learning Techniques Mayuri Patel Information Technology Department A.D Patel College Institute oftechnology Vallabh Vidyanagar 388 120 mayuri.ldce@gmail.com
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 Programme 8.00-8.30 Last week s quiz results 8.30-9.00 Prediction of secondary structure & surface exposure 9.00-9.20 Protein disorder prediction 9.20-9.30 Break get computers upstairs 9.30-11.00 Ex.:
Programme 8.00-8.30 Last week s quiz results 8.30-9.00 Prediction of secondary structure & surface exposure 9.00-9.20 Protein disorder prediction 9.20-9.30 Break get computers upstairs 9.30-11.00 Ex.:
Dynamic Programming Algorithms
 Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Dynamic Programming Algorithms Sequence alignments, scores, and significance Lucy Skrabanek ICB, WMC February 7, 212 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
BIOINFORMATICS Sequence to Structure
 BIOINFORMATICS Sequence to Structure Mark Gerstein, Yale University gersteinlab.org/courses/452 (last edit in fall '06, includes in-class changes) 1 (c) M Gerstein, 2006, Yale, gersteinlab.org Secondary
BIOINFORMATICS Sequence to Structure Mark Gerstein, Yale University gersteinlab.org/courses/452 (last edit in fall '06, includes in-class changes) 1 (c) M Gerstein, 2006, Yale, gersteinlab.org Secondary
Textbook Reading Guidelines
 Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: January 16, 2013 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
Understanding Bioinformatics by Marketa Zvelebil and Jeremy Baum Last updated: January 16, 2013 Textbook Reading Guidelines Preface: Read the whole preface, and especially: For the students with Life Science
A Neural Network Method for Identification of RNA-Interacting Residues in Protein
 Genome Informatics 15(1): 105 116 (2004) 105 A Neural Network Method for Identification of RNA-Interacting Residues in Protein Euna Jeong I-Fang Chung Satoru Miyano eajeong@ims.u-tokyo.ac.jp cif@ims.u-tokyo.ac.jp
Genome Informatics 15(1): 105 116 (2004) 105 A Neural Network Method for Identification of RNA-Interacting Residues in Protein Euna Jeong I-Fang Chung Satoru Miyano eajeong@ims.u-tokyo.ac.jp cif@ims.u-tokyo.ac.jp
BMC Structural Biology
 BMC Structural Biology BioMed Central Methodology article A generic method for assignment of reliability scores applied to solvent accessibility predictions Bent Petersen 1, Thomas Nordahl Petersen 1,
BMC Structural Biology BioMed Central Methodology article A generic method for assignment of reliability scores applied to solvent accessibility predictions Bent Petersen 1, Thomas Nordahl Petersen 1,
Exploring Similarities of Conserved Domains/Motifs
 Exploring Similarities of Conserved Domains/Motifs Sotiria Palioura Abstract Traditionally, proteins are represented as amino acid sequences. There are, though, other (potentially more exciting) representations;
Exploring Similarities of Conserved Domains/Motifs Sotiria Palioura Abstract Traditionally, proteins are represented as amino acid sequences. There are, though, other (potentially more exciting) representations;
APPLYING FEATURE-BASED RESAMPLING TO PROTEIN STRUCTURE PREDICTION
 APPLYING FEATURE-BASED RESAMPLING TO PROTEIN STRUCTURE PREDICTION Trent Higgs 1, Bela Stantic 1, Md Tamjidul Hoque 2 and Abdul Sattar 13 1 Institute for Integrated and Intelligent Systems (IIIS), Grith
APPLYING FEATURE-BASED RESAMPLING TO PROTEIN STRUCTURE PREDICTION Trent Higgs 1, Bela Stantic 1, Md Tamjidul Hoque 2 and Abdul Sattar 13 1 Institute for Integrated and Intelligent Systems (IIIS), Grith
Bioinformatics Tools. Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine
 Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Bioinformatics Tools Stuart M. Brown, Ph.D Dept of Cell Biology NYU School of Medicine Overview This lecture will
BMB/Bi/Ch 170 Fall 2017 Problem Set 1: Proteins I
 BMB/Bi/Ch 170 Fall 2017 Problem Set 1: Proteins I Please use ray-tracing feature for all the images you are submitting. Use either the Ray button on the right side of the command window in PyMOL or variations
BMB/Bi/Ch 170 Fall 2017 Problem Set 1: Proteins I Please use ray-tracing feature for all the images you are submitting. Use either the Ray button on the right side of the command window in PyMOL or variations
ISSN: ISO 9001:2008 Certified International Journal of Engineering and Innovative Technology (IJEIT) Volume 7, Issue 11, May 2018
 Volume 7, Issue 11, May 2018") Application of Machine Learning to Immune Disease Prediction Kuan-Hui Lin, Yuh-Jyh Hu College of Computer Science, National Chiao Tung University, Hsinchu, Taiwan Abstract The intrusion of viruses, germs
Application of Machine Learning to Immune Disease Prediction Kuan-Hui Lin, Yuh-Jyh Hu College of Computer Science, National Chiao Tung University, Hsinchu, Taiwan Abstract The intrusion of viruses, germs
G4120: Introduction to Computational Biology
 ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
ICB Fall 2004 G4120: Computational Biology Oliver Jovanovic, Ph.D. Columbia University Department of Microbiology Copyright 2004 Oliver Jovanovic, All Rights Reserved. Analysis of Protein Sequences Coding
JPred and Jnet: Protein Secondary Structure Prediction.
 JPred and Jnet: Protein Secondary Structure Prediction www.compbio.dundee.ac.uk/jpred ...A I L E G D Y A S H M K... FUNCTION? Protein Sequence a-helix b-strand Secondary Structure Fold What is the difference
JPred and Jnet: Protein Secondary Structure Prediction www.compbio.dundee.ac.uk/jpred ...A I L E G D Y A S H M K... FUNCTION? Protein Sequence a-helix b-strand Secondary Structure Fold What is the difference
Progress Report: Predicting Which Recommended Content Users Click Stanley Jacob, Lingjie Kong
 Progress Report: Predicting Which Recommended Content Users Click Stanley Jacob, Lingjie Kong Machine learning models can be used to predict which recommended content users will click on a given website.
Progress Report: Predicting Which Recommended Content Users Click Stanley Jacob, Lingjie Kong Machine learning models can be used to predict which recommended content users will click on a given website.
Correcting Sampling Bias in Structural Genomics through Iterative Selection of Underrepresented Targets
 Correcting Sampling Bias in Structural Genomics through Iterative Selection of Underrepresented Targets Kang Peng Slobodan Vucetic Zoran Obradovic Abstract In this study we proposed an iterative procedure
Correcting Sampling Bias in Structural Genomics through Iterative Selection of Underrepresented Targets Kang Peng Slobodan Vucetic Zoran Obradovic Abstract In this study we proposed an iterative procedure
Prediction of Protein Structure by Emphasizing Local Side- Chain/Backbone Interactions in Ensembles of Turn
 PROTEINS: Structure, Function, and Genetics 53:486 490 (2003) Prediction of Protein Structure by Emphasizing Local Side- Chain/Backbone Interactions in Ensembles of Turn Fragments Qiaojun Fang and David
PROTEINS: Structure, Function, and Genetics 53:486 490 (2003) Prediction of Protein Structure by Emphasizing Local Side- Chain/Backbone Interactions in Ensembles of Turn Fragments Qiaojun Fang and David
Predicting the Coupling Specif icity of G-protein Coupled Receptors to G-proteins by Support Vector Machines
 Article Predicting the Coupling Specif icity of G-protein Coupled Receptors to G-proteins by Support Vector Machines Cui-Ping Guan, Zhen-Ran Jiang, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular
Article Predicting the Coupling Specif icity of G-protein Coupled Receptors to G-proteins by Support Vector Machines Cui-Ping Guan, Zhen-Ran Jiang, and Yan-Hong Zhou* Hubei Bioinformatics and Molecular
Protein function prediction using sequence motifs: A research proposal
 Protein function prediction using sequence motifs: A research proposal Asa Ben-Hur Abstract Protein function prediction, i.e. classification of protein sequences according to their biological function
Protein function prediction using sequence motifs: A research proposal Asa Ben-Hur Abstract Protein function prediction, i.e. classification of protein sequences according to their biological function
Machine Learning and Graph Theory Approaches for Classification and Prediction of Protein Structure
 Georgia State University ScholarWorks @ Georgia State University Computer Science Dissertations Department of Computer Science 4-22-2008 Machine Learning and Graph Theory Approaches for Classification
Georgia State University ScholarWorks @ Georgia State University Computer Science Dissertations Department of Computer Science 4-22-2008 Machine Learning and Graph Theory Approaches for Classification
Molecular Biology and Pooling Design
 Molecular Biology and Pooling Design Weili Wu 1, Yingshu Li 2, Chih-hao Huang 2, and Ding-Zhu Du 2 1 Department of Computer Science, University of Texas at Dallas, Richardson, TX 75083, USA weiliwu@cs.utdallas.edu
Molecular Biology and Pooling Design Weili Wu 1, Yingshu Li 2, Chih-hao Huang 2, and Ding-Zhu Du 2 1 Department of Computer Science, University of Texas at Dallas, Richardson, TX 75083, USA weiliwu@cs.utdallas.edu
Protein 3D Structure Prediction
 Protein 3D Structure Prediction Michael Tress CNIO ?? MREYKLVVLGSGGVGKSALTVQFVQGIFVDE YDPTIEDSYRKQVEVDCQQCMLEILDTAGTE QFTAMRDLYMKNGQGFALVYSITAQSTFNDL QDLREQILRVKDTEDVPMILVGNKCDLEDER VVGKEQGQNLARQWCNCAFLESSAKSKINVN
Protein 3D Structure Prediction Michael Tress CNIO ?? MREYKLVVLGSGGVGKSALTVQFVQGIFVDE YDPTIEDSYRKQVEVDCQQCMLEILDTAGTE QFTAMRDLYMKNGQGFALVYSITAQSTFNDL QDLREQILRVKDTEDVPMILVGNKCDLEDER VVGKEQGQNLARQWCNCAFLESSAKSKINVN
BIOINFORMATICS Introduction
 BIOINFORMATICS Introduction Mark Gerstein, Yale University bioinfo.mbb.yale.edu/mbb452a 1 (c) Mark Gerstein, 1999, Yale, bioinfo.mbb.yale.edu What is Bioinformatics? (Molecular) Bio -informatics One idea
BIOINFORMATICS Introduction Mark Gerstein, Yale University bioinfo.mbb.yale.edu/mbb452a 1 (c) Mark Gerstein, 1999, Yale, bioinfo.mbb.yale.edu What is Bioinformatics? (Molecular) Bio -informatics One idea
Pacific Symposium on Biocomputing 4: (1999)
") U S I N G I N F O R M A T I O N T H E O R Y T O D I S C O V E R S I D E C H A I N R O T A M E R C L A S S E S : A N A L Y S I S O F T H E E F F E C T S O F L O C A L B A C K B O N E S T R U C T U R E J
U S I N G I N F O R M A T I O N T H E O R Y T O D I S C O V E R S I D E C H A I N R O T A M E R C L A S S E S : A N A L Y S I S O F T H E E F F E C T S O F L O C A L B A C K B O N E S T R U C T U R E J
NACSVM Pred : A MACHINE LEARNING APPROACH FOR PREDICTION OF NAC PROTEINS IN RICE USING SUPPORT VECTOR MACHINES
 NACSVM Pred : A MACHINE LEARNING APPROACH FOR PREDICTION OF NAC PROTEINS IN RICE USING SUPPORT VECTOR MACHINES Hemalatha N. 1,*, Rajesh M. K. 2 and Narayanan N. K. 3 1 AIMIT, St. Aloysius College, Mangalore,
NACSVM Pred : A MACHINE LEARNING APPROACH FOR PREDICTION OF NAC PROTEINS IN RICE USING SUPPORT VECTOR MACHINES Hemalatha N. 1,*, Rajesh M. K. 2 and Narayanan N. K. 3 1 AIMIT, St. Aloysius College, Mangalore,
Bioinformatics Advance Access published August 29, A New Representation for Protein Secondary Structure Prediction Based on Frequent Patterns
 Bioinformatics Advance Access published August 29, 2006 BIOINFORMATICS A New Representation for Protein Secondary Structure Prediction Based on Frequent Patterns Fabian Birzele a and Stefan Kramer b a
Bioinformatics Advance Access published August 29, 2006 BIOINFORMATICS A New Representation for Protein Secondary Structure Prediction Based on Frequent Patterns Fabian Birzele a and Stefan Kramer b a
Supersecondary Structure Motifs and De Novo Protein Structure Prediction
 Supersecondary Structure Motifs and De Novo Protein Structure Prediction Bryn Reinstadler Williams College Jennifer Van George Mason University 31 July 2012 Amarda Shehu George Mason University Abstract
Supersecondary Structure Motifs and De Novo Protein Structure Prediction Bryn Reinstadler Williams College Jennifer Van George Mason University 31 July 2012 Amarda Shehu George Mason University Abstract
Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices
 Article No. jmbi.1999.3091 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 292, 195±202 COMMUNICATION Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices
Article No. jmbi.1999.3091 available online at http://www.idealibrary.com on J. Mol. Biol. (1999) 292, 195±202 COMMUNICATION Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices
Bioinformation by Biomedical Informatics Publishing Group
 Algorithm to find distant repeats in a single protein sequence Nirjhar Banerjee 1, Rangarajan Sarani 1, Chellamuthu Vasuki Ranjani 1, Govindaraj Sowmiya 1, Daliah Michael 1, Narayanasamy Balakrishnan 2,
Algorithm to find distant repeats in a single protein sequence Nirjhar Banerjee 1, Rangarajan Sarani 1, Chellamuthu Vasuki Ranjani 1, Govindaraj Sowmiya 1, Daliah Michael 1, Narayanasamy Balakrishnan 2,
MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein-protein network mapping
 MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein-protein network mapping Chengxin Zhang, Wei Zheng, Peter L Freddolino, and Yang
MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein-protein network mapping Chengxin Zhang, Wei Zheng, Peter L Freddolino, and Yang
Extracting Database Properties for Sequence Alignment and Secondary Structure Prediction
 Available online at www.ijpab.com ISSN: 2320 7051 Int. J. Pure App. Biosci. 2 (1): 35-39 (2014) International Journal of Pure & Applied Bioscience Research Article Extracting Database Properties for Sequence
Available online at www.ijpab.com ISSN: 2320 7051 Int. J. Pure App. Biosci. 2 (1): 35-39 (2014) International Journal of Pure & Applied Bioscience Research Article Extracting Database Properties for Sequence
BIRKBECK COLLEGE (University of London)
") BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
BIRKBECK COLLEGE (University of London) SCHOOL OF BIOLOGICAL SCIENCES M.Sc. EXAMINATION FOR INTERNAL STUDENTS ON: Postgraduate Certificate in Principles of Protein Structure MSc Structural Molecular Biology
Bioinformatics. Microarrays: designing chips, clustering methods. Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute
 Bioinformatics Microarrays: designing chips, clustering methods Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Course Syllabus Jan 7 Jan 14 Jan 21 Jan 28 Feb 4 Feb 11 Feb 18 Feb 25 Sequence
Bioinformatics Microarrays: designing chips, clustering methods Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Course Syllabus Jan 7 Jan 14 Jan 21 Jan 28 Feb 4 Feb 11 Feb 18 Feb 25 Sequence
Motif Discovery from Large Number of Sequences: a Case Study with Disease Resistance Genes in Arabidopsis thaliana
 Motif Discovery from Large Number of Sequences: a Case Study with Disease Resistance Genes in Arabidopsis thaliana Irfan Gunduz, Sihui Zhao, Mehmet Dalkilic and Sun Kim Indiana University, School of Informatics
Motif Discovery from Large Number of Sequences: a Case Study with Disease Resistance Genes in Arabidopsis thaliana Irfan Gunduz, Sihui Zhao, Mehmet Dalkilic and Sun Kim Indiana University, School of Informatics
Predicting prokaryotic incubation times from genomic features Maeva Fincker - Final report
 Predicting prokaryotic incubation times from genomic features Maeva Fincker - mfincker@stanford.edu Final report Introduction We have barely scratched the surface when it comes to microbial diversity.
Predicting prokaryotic incubation times from genomic features Maeva Fincker - mfincker@stanford.edu Final report Introduction We have barely scratched the surface when it comes to microbial diversity.
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/19/07 CAP5510 1 HMM for Sequence Alignment 2/19/07 CAP5510 2
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/19/07 CAP5510 1 HMM for Sequence Alignment 2/19/07 CAP5510 2
Computational analysis and prediction of protein- RNA interactions
 Graduate Theses and Dissertations Graduate College 2008 Computational analysis and prediction of protein- RNA interactions Michael Terribilini Iowa State University Follow this and additional works at:
Graduate Theses and Dissertations Graduate College 2008 Computational analysis and prediction of protein- RNA interactions Michael Terribilini Iowa State University Follow this and additional works at:
Exploring Suboptimal Sequence Alignments and Scoring Functions in Comparative Protein Structural Modeling
 Exploring Suboptimal Sequence Alignments and Scoring Functions in Comparative Protein Structural Modeling Presented by Kate Stafford 1,2 Research Mentor: Troy Wymore 3 1 Bioengineering and Bioinformatics
Exploring Suboptimal Sequence Alignments and Scoring Functions in Comparative Protein Structural Modeling Presented by Kate Stafford 1,2 Research Mentor: Troy Wymore 3 1 Bioengineering and Bioinformatics
Hierarchy of protein loop-lock structures: a new server for the decomposition of a protein structure into a set of closed loops
 Hierarchy of protein loop-lock structures: a new server for the decomposition of a protein structure into a set of closed loops Simon Kogan 1*, Zakharia Frenkel 1, 2, **, Oleg Kupervasser 1, 3*** and Zeev
Hierarchy of protein loop-lock structures: a new server for the decomposition of a protein structure into a set of closed loops Simon Kogan 1*, Zakharia Frenkel 1, 2, **, Oleg Kupervasser 1, 3*** and Zeev
Recursive protein modeling: a divide and conquer strategy for protein structure prediction and its case study in CASP9
 Recursive protein modeling: a divide and conquer strategy for protein structure prediction and its case study in CASP9 Jianlin Cheng Department of Computer Science, Informatics Institute, C.Bond Life Science
Recursive protein modeling: a divide and conquer strategy for protein structure prediction and its case study in CASP9 Jianlin Cheng Department of Computer Science, Informatics Institute, C.Bond Life Science
Genetic Algorithm for Predicting Protein Folding in the 2D HP Model
 Genetic Algorithm for Predicting Protein Folding in the 2D HP Model A Parameter Tuning Case Study Eyal Halm Leiden Institute of Advanced Computer Science, University of Leiden Niels Bohrweg 1 2333 CA Leiden,
Genetic Algorithm for Predicting Protein Folding in the 2D HP Model A Parameter Tuning Case Study Eyal Halm Leiden Institute of Advanced Computer Science, University of Leiden Niels Bohrweg 1 2333 CA Leiden,
Distributions of Beta Sheets in Proteins With Application to Structure Prediction
 PROTEINS: Structure, Function, and Genetics 48:85 97 (2002) Distributions of Beta Sheets in Proteins With Application to Structure Prediction Ingo Ruczinski, 1,2 * Charles Kooperberg, 2 Richard Bonneau,
PROTEINS: Structure, Function, and Genetics 48:85 97 (2002) Distributions of Beta Sheets in Proteins With Application to Structure Prediction Ingo Ruczinski, 1,2 * Charles Kooperberg, 2 Richard Bonneau,
Improving Remote Homology Detection using a Sequence Property Approach
 Wright State University CORE Scholar Browse all Theses and Dissertations Theses and Dissertations 2009 Improving Remote Homology Detection using a Sequence Property Approach Gina Marie Cooper Wright State
Wright State University CORE Scholar Browse all Theses and Dissertations Theses and Dissertations 2009 Improving Remote Homology Detection using a Sequence Property Approach Gina Marie Cooper Wright State
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur. Lecture - 5 Protein Structure - III
 Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 5 Protein Structure - III This is lecture number three on protein structure. (Refer Slide Time:
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 5 Protein Structure - III This is lecture number three on protein structure. (Refer Slide Time:
A NOVEL FRAMEWORK FOR PROTEIN STRUCTURE PREDICTION. A Dissertation presented to the Faculty of the Graduate School University of Missouri-Columbia
 A NOVEL FRAMEWORK FOR PROTEIN STRUCTURE PREDICTION A Dissertation presented to the Faculty of the Graduate School University of Missouri-Columbia In partial fulfillment of the requirements for the Degree
A NOVEL FRAMEWORK FOR PROTEIN STRUCTURE PREDICTION A Dissertation presented to the Faculty of the Graduate School University of Missouri-Columbia In partial fulfillment of the requirements for the Degree
Comparative Modeling Part 1. Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center
 Comparative Modeling Part 1 Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center Function is the most important feature of a protein Function is related to structure Structure is
Comparative Modeling Part 1 Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center Function is the most important feature of a protein Function is related to structure Structure is