Compassionate Use Navigator Information for Physicians
|
|
|
- Anabel Fletcher
- 5 years ago
- Views:
Transcription
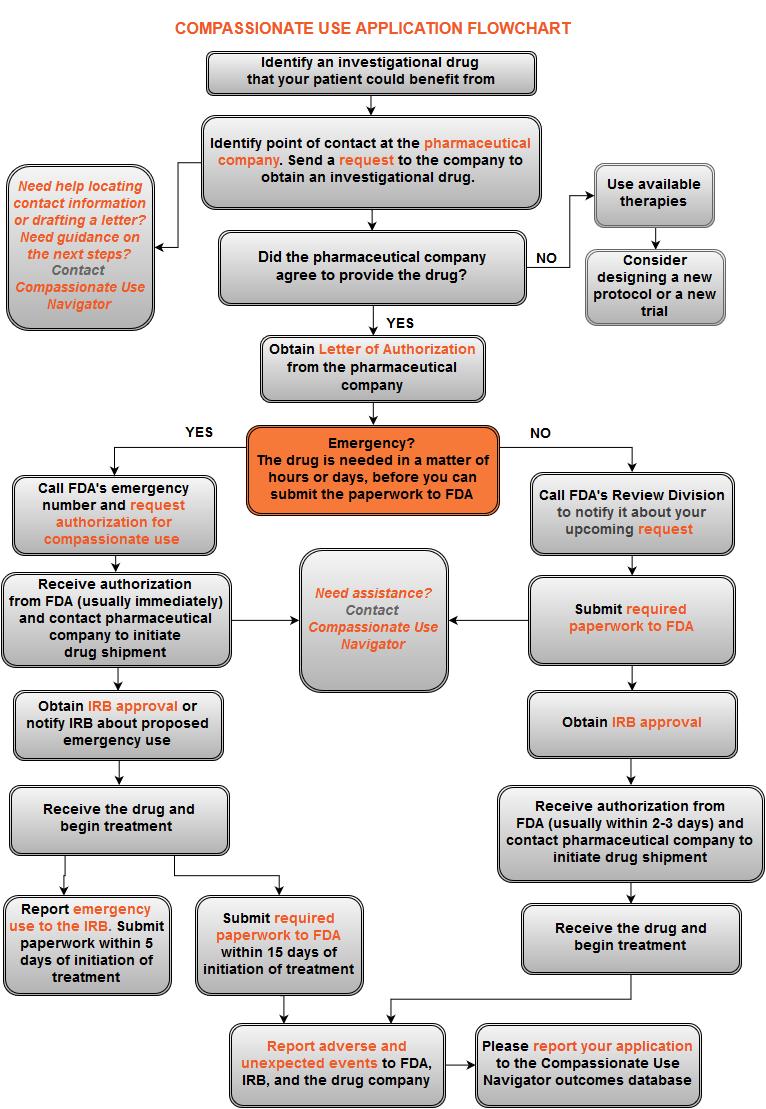
1 Compassionate Use Navigator Information for Physicians Contact: Elena Gerasimov, Program Director, As a physician, you must have wished there would be more treatment options for patients who have not responded to available therapies. Thank you for your willingness to seek access to an investigational drug for your patient through compassionate use (expanded access). Application Process Overview Identify the investigational drug and the company that makes it. Apply to drug manufacturer: Ask the company to provide the drug for your patient. Secure a Letter of Authorization from the company. Apply to the U.S. Food and Drug Administration for authorization of a compassionate use request for your patient. Apply for IRB approval. Submit required safety reports. 1
2 Apply to a Drug Manufacturer This is the first and most important step. The drug manufacturer must agree to provide the investigational drug. Without its agreement, you will not be able to proceed and your patient will not receive the drug. Who to contact? You will need to find a point of contact at the drug company to whom you will send a request to provide the drug, along with your patient s medical history. Your point of contact may be a regulatory affairs official, the chief medical officer, CEO, VP for clinical development, or it may be a general inquiries line. Some companies post a form on their website for you to complete, others provide a phone number or an address to send an inquiry. There is no standard industry application form. Most drug manufacturers will consider your request on a case-by-case basis. There is no legal limit on the time a company can take to respond to a request, and there is no requirement for companies to provide reasons for denials. If the company declines to provide the drug, FDA advises that your options are to use approved treatments, enroll the patient in a clinical trial or an ongoing expanded access program, or to consider designing a new protocol or a new trial. Obtain LOA from a drug company If the company agrees to provide the drug, ask for a letter of authorization (LOA). You will need the LOA to apply for the FDA s authorization. In most cases, manufacturers will have submitted documentation to FDA, and received permission to study the drug you are requesting. The LOA should reference this documentation, including the number of the investigational new drug (IND), or the New Drug Application (NDA) or Biologics License Application (BLA) number. These numbers are issued by FDA to the company. If a manufacturer does not have an IND, FDA requires the following information in the letter: chemistry, manufacturing, and controls information, and pharmacology and toxicology information, including a description of the manufacturing facility. The drug manufacturer may prefer that you submit to FDA a separate individual patient IND application. The company will be supplying the drug, but will not itself 2
3 be submitting the IND for your patient s access. In this case, you, not the company, will be the sponsor for that IND. If FDA authorizes the compassionate use request, it will issue another IND number to you. The IND number serves as FDA s authorization for the company to provide the drug. Upon receipt of the IND number from the FDA, you must then provide that number to the manufacturer so the company can ship the drug directly to you. 3
4 Apply for FDA Authorization After a drug manufacturer has agreed to provide an investigational drug, the next step is to obtain FDA authorization. It is a good idea to call the FDA Review Division and describe your patient s situation prior to sending a written application. An FDA official will help you determine whether your patient s condition requires you to follow a non-emergency (individual patient IND) or emergency (emergency IND) pathway, which is used when treatment of the patient must occur within a very limited number of hours or days. FDA authorizes compassionate use in 99.5% of cases. Your steps will be as follows: Call FDA s drug review division to discuss your planned submission. Send required paperwork to FDA by fax or , followed by hard copy. Obtain informed consent and IRB approval. Receive an IND number from FDA. Submit this IND number to the manufacturer. Receive the drug from the manufacturer and begin treatment. Application paperwork 1. Form 3926, Individual Expanded Access Applications. It should take about 45 minutes to complete. Although this form has not been officially approved yet, FDA will accept it. Form 3926 takes less time to complete than the currently used Form FDA 1571 and Form Letter of Authorization from the drug company. 3. A cover letter specifying that this is a request for an individual patient IND or an emergency IND and your contact information. 4. A copy of the informed consent. Information required on Form 3926 includes: a. Brief clinical history of the patient including: diagnosis, disease status, prior therapy and response to prior therapy, rationale for requesting the proposed treatment. It can include a list of available therapeutic options that would ordinarily be tried before resorting to the investigational drug, or an explanation of why the use of the investigational drug is preferable to the use of available therapeutic options. b. Proposed treatment plan including: the dose, route of administration, planned duration, monitoring procedures and modifications (e.g., dose reduction or treatment delay) for toxicity. 4
5 c. Name of the manufacturer (drug supply reference statement) and a statement that a Letter of Authorization to cross-reference an appropriate IND of the supplier or Drug Master File (DMF) of the manufacturer is included. d. Informed consent statement that states that informed consent and approval of an appropriate Institutional Review Board (IRB) will be obtained prior to initiating treatment. e. Investigator qualification statement that specifies the training, experience, and licensure of the treating physician. The first two pages of a Curriculum Vitae typically contain this information and are usually sufficient. Receive FDA approval In practice, FDA will review the paperwork for oncology patients seeking compassionate use in about 2 business days, although by law it has up to 30 days to review the application. If the request is approved, an IND number will be issued by the FDA and you will be contacted by phone or fax, and a letter will follow. It will contain the name and telephone number of the FDA official to whom questions about the application should be directed. In the unlikely situation when treatment is not allowed to proceed (i.e., a clinical hold is placed on the application), FDA will notify you by phone and in writing. Contact information for FDA The initial request may be made by fax to the appropriate drug review division with hard copies of documentation to follow. For questions about expanded access for a specific investigational drug, contact the appropriate review division, if known, or CDER s Division of Drug Information (DDI), if not known, at phone: or ; fax: ; or druginfo@fda.hhs.gov. For questions about expanded access for emergency use for investigational biologics, contact CBER s Office of Communication, Outreach, and Development at phone: or ; or industry.biologics@fda.gov. For questions about non-emergency individual expanded access for biologics, contact CBER at: or
6 Mailing addresses (courier service can be used) For a drug An original and two photocopies: Food and Drug Administration Center for Drug Evaluation and Research Central Document Room 5901-B Ammendale Road Beltsville, MD For a biologic product An original application and a CD with a copy: Food and Drug Administration Center for Biologics Evaluation and Research Document Control Center New Hampshire Avenue WO71, G112 Silver Spring, MD Emergency requests Your application process will be quicker if the patient needs the drug on an emergency basis because you may proceed before submitting paperwork to FDA or an IRB. FDA defines an emergency as the case in which: the physician considers the product is immediately needed for the patient s serious or life-threatening condition; no satisfactory alternative standard therapy is available; the patient cannot receive the product through any existing clinical trials or expanded access protocols; access to the drug is needed before the written application can be submitted (approximately within the next hours). Your steps will be as follows: Call FDA at the emergency numbers below. Receive an IND number from FDA. Submit this IND number to the manufacturer. Obtain informed consent. Notify an IRB of planned emergency use if there is no time to obtain IRB approval. Receive the drug from the manufacturer and begin treatment. Apply for IRB approval within 5 days of the beginning of treatment. Submit required paperwork to FDA within 15 days of the beginning of treatment. About 30-50% of compassionate use requests FDA receives are considered emergency requests. 6
7 Emergency contact information at FDA Emergency requests may be submitted over the phone, by or by fax. You will need to explain your patient s situation and agree to submit the required paperwork within 15 working days. An FDA official may provide verbal authorization for compassionate use over the phone. During normal business hours contact Center for Drug Evaluation and Research, Division of Drug Information (DDI) (8am-4:30pm EST weekdays): Phone: or Fax: druginfo@fda.hhs.gov. After hours or if you can t reach anyone at DDI contact the Emergency Call Center (after 4:30pm EST weekdays and all day on weekends) at: or Submit IND number to drug manufacturer and await shipment of the drug Upon receipt of the IND number from the FDA, you must then submit the IND number to the manufacturer, and the company will ship the drug directly to you. The IND is considered active as soon as the number was issued. 7
8 Apply to IRB You must apply for an Institutional Review Board s (IRB) approval for compassionate use prior to initiating treatment. You should specify in the application that the primary intent is treatment, not clinical research. If your institution does not have an IRB you can: Use a for-profit IRB. This is a private IRB that reviews research proposals at a cost. Partner with an IRB from your local hospital/institution. Universities that are affiliated with hospitals will most likely have an IRB. Start an IRB at your institution. Some drug companies may require an IRB approval letter before the drug will be shipped. If treatment cannot be delayed, some IRBs have sent manufacturers a written statement indicating that the IRB is aware of the proposed use and considers it to meet the requirements for emergency compassionate use. Although this is not a formal IRB approval, the acknowledgment letter has been acceptable to drug companies. In an emergency life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval, by law you may begin treatment and report the emergency use of the investigational drug to an IRB within 5 working days (21 CFR ). Some IRBs have specific procedures for approving emergency requests. However, you should notify the IRB right away and follow your institution s procedures for such cases. Requirements of IRBs differ, but as a general rule you should the following to the IRB: description of case and treatment plan, including justification for compassionate use; a proposed consent form; any available documentation from the FDA regarding the emergency IND. Obtain informed consent In general, the consent should satisfy federal requirements under 21 CFR 50. In a rare situation when it may not be possible to obtain consent from the child s legal guardian, federal regulations (21 CFR (a)) may allow for a consent waiver. 8
9 Safety Reports Requirements FDA requires all sponsors of IND applications, including physicians who administer an investigational drug to a patient, to report adverse events. What to report? As a treating physician you must report any suspected adverse reaction to treatment that is both serious and unexpected in an IND Safety Report. Before submitting an IND safety report, you need to ensure the event meets all three of the definitions: Suspected adverse reaction; Serious; Unexpected. If the adverse event does not meet all three of the definitions, it should not be submitted as an IND safety report. Adverse reaction is an adverse event where there is a reason to conclude that the drug caused the event. A suspected adverse reaction is an adverse event for which there is a reasonable possibility that it is caused by the drug. For the purposes of safety reporting, reasonable possibility means there is evidence to suggest a causal relationship between the drug and the adverse event. Serious adverse event refers to an event that results in the following outcomes: death; a life-threatening adverse event (places the patient at immediate risk of death); in-patient hospitalization or prolongation of an existing hospitalization; a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions. Unexpected adverse event refers to an event that is not consistent with previously described risk information, or occurs at a specificity or severity that has not been previously observed. This definition is also used in the absence of an investigator s brochure in which observed adverse events are described. Deciding whether an adverse event meets the definition of a suspected adverse reaction can be difficult, but this decision is critical to avoid the submission of uninformative IND safety reports. You should evaluate the available information and decide whether there is a reasonable possibility that the drug caused the adverse event. Any relevant additional information which becomes available and is necessary to evaluate the suspected adverse reaction that pertains to a 9
10 previously submitted IND safety report must be submitted as a Followup IND Safety Report. When to report? Serious and unexpected adverse events that are fatal or lifethreatening must be reported to FDA as soon as possible, and no later than 7 days after they occur. Suspected adverse reactions or other safety information that are not serious or life-threatening must be reported no later than 15 calendar days after the event. A Follow-Up IND Safety Report should be submitted as soon as new information is available, and no later than 15 calendar days after you receive the information. If the safety report submitted within 7 calendar days is complete, an additional submission within 15 days from day zero is not required. At the conclusion of treatment, you must provide FDA with a written summary of the results of the expanded access use, including adverse effects, the patient s response, drug disposition, and other relevant information. When submitting this summary report, you may request FDA to close the IND application. If the investigational treatment continues for a year or longer, there is a requirement to make an annual report to FDA for any year during which your patient has received the investigational drug. You will need to submit a brief report of the progress within 60 days of the anniversary from the date when the original IND application went into effect. Who to report to? In addition to reporting to the FDA, you need to report to the drug company and your IRB. The FDA believes that the pharmaceutical company is better positioned than the individual investigator to assess the overall safety of the investigational drug because it has access to serious adverse event reports from multiple study sites and multiple studies and is able to aggregate and analyze these reports. The company is also more familiar with the drug s mechanism of action, class effects, and other information. For these reasons, investigators must immediately report any serious adverse event to the company, whether or not the investigator considers the event to be drug related. Drug companies can impose additional reporting requirements when they provide the drug. You might also have an obligation to report adverse events to your IRB. In general, when an adverse event meets the criteria for reporting it to FDA, it should also be considered an unanticipated problem and reported to the IRB. 10
11 How to report? Safety reports to FDA must be submitted by , fax or telephone to the Review Division that authorized the expanded access request. Use FDA Form 3500A together with Form Protocol amendments After an IND application has gone into effect, you may amend the application as needed. You are expected to submit protocol amendments for new protocols or changes to existing protocols before implementation of any changes. Amendments could include any increase in drug dosage or duration of therapy beyond that described in the current protocol, change in the design of a protocol, addition or elimination of a test or procedure, or addition of a new investigator. If the amendment concerns change in application sponsor or addition of a new investigator, Form 1572 should be included in addition to Form More information Please use the Reporting Requirements Chart for links to reporting forms and deadlines. For detailed explanation of the definitions, requirements, and procedures related to IND application safety reports and the responsibilities of IND applications sponsors with regard to such reporting, see Guidance for Industry and Investigators: Safety Reporting Requirements for INDs and BA/BE Studies. 11
12 Help us build a registry of requests share your case To develop effective compassionate use policies, it is necessary to understand the unmet need. Currently, only the number of requests that were granted by both the companies and FDA is known. There is no data on the numbers of applications submitted for pediatric patients, or applications that were denied. Let us know to which companies you have made compassionate use requests. Let us know the point of contact in the company that you used, whether the company responded and in how much time, and whether your request was ultimately granted. And, please let us know how your patient did was there any possible benefit from the drug? Were there unexpected toxicities? Please help us build a registry of compassionate use application outcomes by answering a few questions for our database, or Elena Gerasimov, Program Director, at Elena@kidsvcancer.org. Thank you! 12
13 13
FDA approval of emergency expanded access use may be requested by telephone, facsimile, or other means of electronic communications.
 Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of an Individual Patient with an Investigational Drug or a REMS-restricted, Approved Drug Guidelines:
Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of an Individual Patient with an Investigational Drug or a REMS-restricted, Approved Drug Guidelines:
Guidelines: c. Providing the investigational drug for the requested use will not interfere with the initiation, conduct, or completion of clinical
 Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of Intermediate-size Patient Populations with an Investigational Drug or a REMS-restricted, Approved
Guidelines for the Submission of an Expanded Access IND to Permit Diagnosis, Monitoring or Treatment of Intermediate-size Patient Populations with an Investigational Drug or a REMS-restricted, Approved
IND Development Process Published on ResearchGo UCLA (
 IND Development Process IND Development Overview The Pre-IND Process The IND Study Protocol Preparing the Initial IND Submission Filing the IND Maintaining the IND IND Templates, Education and Useful Links
IND Development Process IND Development Overview The Pre-IND Process The IND Study Protocol Preparing the Initial IND Submission Filing the IND Maintaining the IND IND Templates, Education and Useful Links
How to put together an IND application
 How to put together an IND application Judit Milstein, Chief, Project Management Staff Judit.milstein@fda.hhs.gov Eithu Lwin, Regulatory Health Project Manager Eithu.Lwin@fda.hhs.gov Division of Transplant
How to put together an IND application Judit Milstein, Chief, Project Management Staff Judit.milstein@fda.hhs.gov Eithu Lwin, Regulatory Health Project Manager Eithu.Lwin@fda.hhs.gov Division of Transplant
Expanded Access and the Individual Patient IND
 Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
MANUAL: Administrative Policy & Procedure Manual POLICY:
 SJMHS Locations: St. Joseph Mercy Ann Arbor, St. Joseph Mercy Chelsea, St. Joseph Mercy Livingston, St. Mary Mercy Livonia MANUAL: Administrative Policy & Procedure Manual POLICY: Expanded access is the
SJMHS Locations: St. Joseph Mercy Ann Arbor, St. Joseph Mercy Chelsea, St. Joseph Mercy Livingston, St. Mary Mercy Livonia MANUAL: Administrative Policy & Procedure Manual POLICY: Expanded access is the
Expanded Access. to Investigational Drugs & Biologics. for Treatment Use
 SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
SJMHS Research Compliance Office Guidance Document Expanded Access to Investigational Drugs & Biologics for Treatment Use May 2015 1 Expanded Access to Investigational Drugs & Biologics for Treatment Use
Investigational New Drug Development Steps for CRCs
 Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
POLICY #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional
3.1. Overall Principal Investigator (PI), who holds the IND and is the Sponsor.
, who holds the IND and is the Sponsor.") SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
SOP #: RCO-100 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational New Drug Application (IND) and who is the Sponsor of the research has additional responsibilities
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES
 IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
IDENTIFYING & MANAGING GCP COMPLIANCE RISKS FOR THE PHARMACEUTICAL, BIOTECH & DEVICE INDUSTRIES Karen Weaver Epstein, Becker & Green Health Care & Life Sciences Practice APPLICABLE REGULATIONS 21 CFR 312
Subpart H [Reserved] Subpart I Expanded Access to Investigational Drugs for Treatment. 21 CFR Ch. I ( Edition)
![Subpart H [Reserved] Subpart I Expanded Access to Investigational Drugs for Treatment. 21 CFR Ch. I ( Edition)](/thumbs/89/100764462.jpg "Subpart H [Reserved] Subpart I Expanded Access to Investigational Drugs for Treatment. 21 CFR Ch. I ( Edition)") 312.300 (c) Disposition of unused drug. The person who ships the drug under paragraph (a) of this section shall assure the return of all unused supplies of the drug from individual investigators whenever
312.300 (c) Disposition of unused drug. The person who ships the drug under paragraph (a) of this section shall assure the return of all unused supplies of the drug from individual investigators whenever
Unpacking Expanded Access to Investigational Drugs and Biologics
 Unpacking Expanded Access to Investigational Drugs and Biologics When All Else Fails Richard Klein Office of Health and Constituent Affairs Food and Drug Administration September 13, 2016 FDA Drug Approval
Unpacking Expanded Access to Investigational Drugs and Biologics When All Else Fails Richard Klein Office of Health and Constituent Affairs Food and Drug Administration September 13, 2016 FDA Drug Approval
Investigator-Initiated INDs
 Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Guidance for IRBs, Clinical Investigators and Sponsors
 Guidance for IRBs, Clinical Investigators and Sponsors IRB Responsibilities for Reviewing the Qualifications of Investigators, Adequacy of Research Sites, and the Determination of Whether an IND/IDE is
Guidance for IRBs, Clinical Investigators and Sponsors IRB Responsibilities for Reviewing the Qualifications of Investigators, Adequacy of Research Sites, and the Determination of Whether an IND/IDE is
What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance
 Vol. 9, No. 2, February 2013 Happy Trials to You What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance Reprinted from the Guide to Good Clinical Practice with permission of Thompson Publishing
Vol. 9, No. 2, February 2013 Happy Trials to You What s New in GCP? FDA Clarifies, Expands Safety Reporting Guidance Reprinted from the Guide to Good Clinical Practice with permission of Thompson Publishing
USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH
 Policy P.11 Written By: B. Laurel Elder Created: June 3, 2013 Edited Replaces 10-9-2009 Version P.11.4 USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH A. Procedure Contents this procedure
Policy P.11 Written By: B. Laurel Elder Created: June 3, 2013 Edited Replaces 10-9-2009 Version P.11.4 USE OF INVESTIGATIONAL DRUGS OR BIOLOGICS IN HUMAN SUBJECTS RESEARCH A. Procedure Contents this procedure
Agency Information Collection Activities; Submission for Office of Management and Budget
 This document is scheduled to be published in the Federal Register on 02/12/2019 and available online at https://federalregister.gov/d/2019-01962, and on govinfo.gov 4164-01-P DEPARTMENT OF HEALTH AND
This document is scheduled to be published in the Federal Register on 02/12/2019 and available online at https://federalregister.gov/d/2019-01962, and on govinfo.gov 4164-01-P DEPARTMENT OF HEALTH AND
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)
 SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)") POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
Clinical Trials and the Code of Federal Regulations. Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014
 Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
To document the review procedures for a submission regarding compassionate/treatment use of investigational drugs, biologics and devices.
 UNIVERSITY OF TENNESSEE GRADUATE SCHOOL OF MEDICINE INSTITUTIONAL REVIEW BOARD COMPASSIONATE/TREATMENT USE OF MEDICAL DRUGS, BIOLOGICS AND DEVICES I. PURPOSE To document the review procedures for a submission
UNIVERSITY OF TENNESSEE GRADUATE SCHOOL OF MEDICINE INSTITUTIONAL REVIEW BOARD COMPASSIONATE/TREATMENT USE OF MEDICAL DRUGS, BIOLOGICS AND DEVICES I. PURPOSE To document the review procedures for a submission
US FDA Expedited Programs and Expanded Access
 US FDA Expedited Programs and Expanded Access Ke Liu, MD, PhD Chief, Oncology Branch Division of Clinical Evaluation, Pharmacology and Toxicology Office of Tissues and Advanced Therapies Center for Biologics
US FDA Expedited Programs and Expanded Access Ke Liu, MD, PhD Chief, Oncology Branch Division of Clinical Evaluation, Pharmacology and Toxicology Office of Tissues and Advanced Therapies Center for Biologics
IND IND ACKNOWLEDGEMENT
 DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration Silver Spring MD 20993 IND 110513 IND ACKNOWLEDGEMENT Multidisciplinary Association for Psychedelic Studies Attention: Rick Doblin,
DEPARTMENT OF HEALTH AND HUMAN SERVICES Food and Drug Administration Silver Spring MD 20993 IND 110513 IND ACKNOWLEDGEMENT Multidisciplinary Association for Psychedelic Studies Attention: Rick Doblin,
University of California, Irvine Human Research Protections Standard Operating Policies and Procedures
 University of California, Irvine Human Research Protections Standard Operating Policies and Procedures Policy Number: 41 Title: Investigational Drugs, Agents, and Biologics Date of Last of Revision: 07/28/2006;
University of California, Irvine Human Research Protections Standard Operating Policies and Procedures Policy Number: 41 Title: Investigational Drugs, Agents, and Biologics Date of Last of Revision: 07/28/2006;
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
SWOG
 SWOG http://swog.org Page 1 of 5 pages Original Release Date: July 1985 Departments Affected: All Revision Date: April 2018 Introduction SERIOUS ADVERSE EVENTS The timely reporting of serious adverse events
SWOG http://swog.org Page 1 of 5 pages Original Release Date: July 1985 Departments Affected: All Revision Date: April 2018 Introduction SERIOUS ADVERSE EVENTS The timely reporting of serious adverse events
MCW Office of Research Standard Operating Procedure
 MCW Office of Research Standard Operating Procedure USE AND STORAGE OF INVESTIGATIONAL DRUGS AND BIOLOGICS Unit: Applies to: Human Research Protections Program (HRPP), Office of Research MCW/FH Faculty
MCW Office of Research Standard Operating Procedure USE AND STORAGE OF INVESTIGATIONAL DRUGS AND BIOLOGICS Unit: Applies to: Human Research Protections Program (HRPP), Office of Research MCW/FH Faculty
Formal Meetings Between the FDA and Biosimilar Biological Product Sponsors or Applicants Guidance for Industry
 Formal Meetings Between the FDA and Biosimilar Biological Product Sponsors or Applicants Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration Center for Drug
Formal Meetings Between the FDA and Biosimilar Biological Product Sponsors or Applicants Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration Center for Drug
I. Purpose. II. Definitions. Last Approval Date
 Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
Expanded Access to Investigational Imaging Drugs
 Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
Expanded Access to Investigational Imaging Drugs Phillip B. Davis, MD FDA/CDER/ODEIV/DMIP 6/09/2016 1 Overview Expanded Access (EA) Defined Requirements for all EA authorizations Types of EA Individual
Structure and Mandate of FDA
 Structure and Mandate of FDA Leonard Sacks, M.D. Office of Medical Policy Center for Drug Evaluation and Research FDA FDA Clinical Investigator Training Course November 13, 2018 Mission of regulatory agencies
Structure and Mandate of FDA Leonard Sacks, M.D. Office of Medical Policy Center for Drug Evaluation and Research FDA FDA Clinical Investigator Training Course November 13, 2018 Mission of regulatory agencies
Expanded Access and the Individual Patient IND
 Office of Regulatory Affairs and Quality Expanded Access and the Individual Patient IND Research Wednesdays July 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office
Office of Regulatory Affairs and Quality Expanded Access and the Individual Patient IND Research Wednesdays July 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office
Investigational New Drug Applications: two cases
 Investigational New Drug Applications: two cases April 15, 2009 Carine M. Lenders, M.D., M.S. Medical Director, NFL program Director, Pediatric Nutrition Support Services Research Staff, General Pediatrics
Investigational New Drug Applications: two cases April 15, 2009 Carine M. Lenders, M.D., M.S. Medical Director, NFL program Director, Pediatric Nutrition Support Services Research Staff, General Pediatrics
11.0 FDA-Regulated Research
 11.0 FDA-Regulated Research The HSC evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
11.0 FDA-Regulated Research The HSC evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
FDA 101 Session: Care and Feeding of an Investigational New Drug (IND)
") FDA 101 Session: Care and Feeding of an Investigational New Drug (IND) CAPT Kofi Ansah, PharmD, MS, MBA, RAC CAPT William Bender, RPh, MSHCA, RAC CDR Keith Kiedrow, PharmD, MS, RAC CDR Hiren Patel, PharmD,
FDA 101 Session: Care and Feeding of an Investigational New Drug (IND) CAPT Kofi Ansah, PharmD, MS, MBA, RAC CAPT William Bender, RPh, MSHCA, RAC CDR Keith Kiedrow, PharmD, MS, RAC CDR Hiren Patel, PharmD,
DRAFT GUIDANCE. This guidance document is being distributed for comment purposes only. Document issued on: July 14, 2011
 Draft Guidance for Industry and Food and Drug Administration Staff In Vitro Companion Diagnostic Devices DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Document issued
Draft Guidance for Industry and Food and Drug Administration Staff In Vitro Companion Diagnostic Devices DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Document issued
GUIDELINE REGARDING COLLECTION, VERIFICATION, AND SUBMISSION OF THE REPORTS OF ADVERSE EVENTS / REACTIONS OCCURRING IN CLINICAL DRUG TRIALS
 1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
1. PURPOSE This guideline is about the collection, verification, and submission of the reports of adverse events / reactions occurring in clinical drug trials, and code breaking methods. 2. DEFINITIONS
RESEARCH ETHICS BOARD GUIDELINES SUBMISSION OF RESEARCH PROJECTS
 1. Role of the Research Ethics Board The mandate of the Research Ethics Board is to safeguard the rights, safety and well being of all research participants. The REB reviews and approves research projects
1. Role of the Research Ethics Board The mandate of the Research Ethics Board is to safeguard the rights, safety and well being of all research participants. The REB reviews and approves research projects
Human Research Protection Program Policy
 Page 1 of 11 EMERGENCY USE OF AN INVESTIGATIONAL DRUG, BIOLOGICAL PRODUCT OR DEVICE IN HUMAN SUBJECTS RESEARCH POLICY This policy does not limit the authority of a physician to provide emergency medical
Page 1 of 11 EMERGENCY USE OF AN INVESTIGATIONAL DRUG, BIOLOGICAL PRODUCT OR DEVICE IN HUMAN SUBJECTS RESEARCH POLICY This policy does not limit the authority of a physician to provide emergency medical
Guidance for Clinical Investigators, Sponsors, and IRBs
 Guidance for Clinical Investigators, Sponsors, and IRBs Adverse Event Reporting to IRBs Improving Human Subject Protection u.s. Department of Health and Human Services Office of the Commissioner (OC) Center
Guidance for Clinical Investigators, Sponsors, and IRBs Adverse Event Reporting to IRBs Improving Human Subject Protection u.s. Department of Health and Human Services Office of the Commissioner (OC) Center
REGULATORY ISSUES: HOW TO APPLY FOR AN IND. Penny Jester and Maaike Everts
 REGULATORY ISSUES: HOW TO APPLY FOR AN IND Penny Jester and Maaike Everts Outline 2 What is the purpose of an IND? What types of INDs are there? When do you need one? How do you apply for one? How do you
REGULATORY ISSUES: HOW TO APPLY FOR AN IND Penny Jester and Maaike Everts Outline 2 What is the purpose of an IND? What types of INDs are there? When do you need one? How do you apply for one? How do you
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
11.0 FDA-Regulated Research Research Involving Investigational Drugs and Biologics
 11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
Making Sense of Reporting Obligations to the IRB. fully accredited since 2006
 Making Sense of Reporting Obligations to the IRB fully accredited since 2006 Webinar Housekeeping Questions & Answers Feel free to submit questions at any point during the webinar using the chat box
Making Sense of Reporting Obligations to the IRB fully accredited since 2006 Webinar Housekeeping Questions & Answers Feel free to submit questions at any point during the webinar using the chat box
11.0 FDA-Regulated Research Research Involving Investigational Drugs and Biologics
 11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
DRUG ORDERING AND MAINTENANCE
 AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter mainly apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter mainly apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
Clinical trial applications in the EU and US
 Clinical trial applications in the EU and US Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Symposium Strasbourg 17-18 March 2005 Overview of the presentation
Clinical trial applications in the EU and US Alain Patat, M.D. Translational Development Wyeth Research Paris, France AGAH-Club Phase 1 1 st Symposium Strasbourg 17-18 March 2005 Overview of the presentation
Understanding Reporting Obligations to the IRB
 Understanding Reporting Obligations to the IRB fully accredited since 2006 May 13 & 15, 2014 Mitchell E. Parrish, JD, RAC, CIP Regulatory Attorney 2 3 WEBINAR HOUSEKEEPING Questions & Answers Feel free
Understanding Reporting Obligations to the IRB fully accredited since 2006 May 13 & 15, 2014 Mitchell E. Parrish, JD, RAC, CIP Regulatory Attorney 2 3 WEBINAR HOUSEKEEPING Questions & Answers Feel free
INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND)
") INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND) (The field numbers below correspond to the numbered boxes on the Form FDA 1571) Field 1: NAME OF SPONSOR The sponsor
INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND) (The field numbers below correspond to the numbered boxes on the Form FDA 1571) Field 1: NAME OF SPONSOR The sponsor
INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND)
") INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND) (The field numbers below correspond to the numbered boxes on the Form FDA 1571) Field 1: NAME OF SPONSOR The sponsor
INSTRUCTIONS FOR FILLING OUT FORM FDA 1571 INVESTIGATIONAL NEW DRUG APPLICATION (IND) (The field numbers below correspond to the numbered boxes on the Form FDA 1571) Field 1: NAME OF SPONSOR The sponsor
IND Exemption, Preparation and Maintenance. Bruce K. Burnett, PhD, RAC (US, EU) Director, Regulatory Affairs Duke University
 Director, Regulatory Affairs Duke University") IND Exemption, Preparation and Maintenance Bruce K. Burnett, PhD, RAC (US, EU) Director, Regulatory Affairs Duke University 1 Templates and Guidance Best Practices Templates/Instructions http://tiny.cc/d7bpt
IND Exemption, Preparation and Maintenance Bruce K. Burnett, PhD, RAC (US, EU) Director, Regulatory Affairs Duke University 1 Templates and Guidance Best Practices Templates/Instructions http://tiny.cc/d7bpt
Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry
 Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions
Formal Meetings Between the FDA and Sponsors or Applicants of BsUFA Products Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions
Streamlining IRB Procedures for Expanded Access
 Streamlining IRB Procedures for Expanded Access Marjorie A. Speers, Ph.D. Executive Director, WCG Foundation Richard Klein Director, FDA Patient Liaison Program Office of Health and Constituent Affairs
Streamlining IRB Procedures for Expanded Access Marjorie A. Speers, Ph.D. Executive Director, WCG Foundation Richard Klein Director, FDA Patient Liaison Program Office of Health and Constituent Affairs
FDA Decisions for Investigational Device Exemption (IDE) Clinical Investigations
 Clinical Investigations") Draft Guidance for Industry, Clinical Investigators, Institutional Review Boards, and Food and Drug Administration Staff FDA Decisions for Investigational Device Exemption (IDE) Clinical Investigations
Draft Guidance for Industry, Clinical Investigators, Institutional Review Boards, and Food and Drug Administration Staff FDA Decisions for Investigational Device Exemption (IDE) Clinical Investigations
Objectives Discuss the importance of proper data collection. Identify the types of data collected for clinical trials. List potential source documents
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Role of Academic Investigators in Drug Development
 DTRCS Regulatory Education Seminar, June 12, 2007 Role of Academic Investigators in Drug Development Howard Lee, MD, PhD Associate Adjunct Professor Director, Center for Drug Terminology Sponsor Investigator
DTRCS Regulatory Education Seminar, June 12, 2007 Role of Academic Investigators in Drug Development Howard Lee, MD, PhD Associate Adjunct Professor Director, Center for Drug Terminology Sponsor Investigator
The Right Prescription for Working with Investigational Drug Service at BMC
 The Right Prescription for Working with Investigational Drug Service at BMC Andrew Schoch Pharmacy Intern Northeastern University Class of 2010 Hyeseon Hong, Pharm.D. IDS Pharmacy Manager What is Investigational
The Right Prescription for Working with Investigational Drug Service at BMC Andrew Schoch Pharmacy Intern Northeastern University Class of 2010 Hyeseon Hong, Pharm.D. IDS Pharmacy Manager What is Investigational
Chin Koerner Executive Director US Regulatory and Development Policy
 Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
Chin Koerner Executive Director US Regulatory and Development Policy Novartis Pharmaceuticals Corporation 1700 Rockville Pike Suite 510 Rockville, MD 20852 Tel 301.468.5607 Fax 301.468.5614 Email: Chin.Koerner@novartis.com
1201 Maryland Avenue SW, Suite 900, Washington, DC ,
 1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org December 28, 2010 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1201 Maryland Avenue SW, Suite 900, Washington, DC 20024 202-962-9200, www.bio.org December 28, 2010 Dockets Management Branch (HFA-305) Food and Drug Administration 5600 Fishers Lane, Rm. 1061 Rockville,
1 Purpose. 2 Procedure. Title: FDA-Regulated Research. SOP Number: 1301 Effective Date: June 2, Previous Version Dates:
 Previous Version Dates: Title: FDA-Regulated Research SOP Number: 1301 Effective Date: June 2, 2017 1 Purpose FDA regulations apply to research that involves a FDA-regulated test article in a clinical
Previous Version Dates: Title: FDA-Regulated Research SOP Number: 1301 Effective Date: June 2, 2017 1 Purpose FDA regulations apply to research that involves a FDA-regulated test article in a clinical
Guidance for Industry Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans
 Guidance for Industry Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans DRAFT GUIDANCE This guidance document is being distributed
Guidance for Industry Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans DRAFT GUIDANCE This guidance document is being distributed
Environmental Assessment: Questions and Answers Regarding Drugs With Estrogenic, Androgenic, or Thyroid Activity Guidance for Industry
 Environmental Assessment: Questions and Answers Regarding Drugs With Estrogenic, Androgenic, or Thyroid Activity Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment
Environmental Assessment: Questions and Answers Regarding Drugs With Estrogenic, Androgenic, or Thyroid Activity Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment
Investigational New Drug Application
 Investigational New Drug Application Regulatory Sponsor: Funding Sponsor: Study Product: Protocol Number: Name of the Sponsor-Investigator Department Name Address Phone Number Name of Primary Funding Institution
Investigational New Drug Application Regulatory Sponsor: Funding Sponsor: Study Product: Protocol Number: Name of the Sponsor-Investigator Department Name Address Phone Number Name of Primary Funding Institution
Device research sponsors, whether companies or investigators, are held responsible for meeting the same regulations.
 POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: A DF/HCC Investigator who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities that
POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: A DF/HCC Investigator who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities that
3.1. Overall Principal Investigator (PI), who holds the IDE and/or is the Sponsor
, who holds the IDE and/or is the Sponsor") POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities
POLICY #: RCO-101 Page: 1 of 11 1. POLICY STATEMENT: An Overall Principal Investigator (PI) who holds an Investigational Device Exemption (IDE) or who is the Sponsor of the research has additional responsibilities
ROLE OF THE RESEARCH COORDINATOR Investigational New Drug Application-Sponsor Responsibilities 21CFR Part , subpart D
 Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Guidance for Industry
 Guidance for Industry Cooperative Manufacturing Arrangements for Licensed Biologics Additional copies of this guidance are available from the Office of Communication, Training and Manufacturers Assistance
Guidance for Industry Cooperative Manufacturing Arrangements for Licensed Biologics Additional copies of this guidance are available from the Office of Communication, Training and Manufacturers Assistance
MRCTCenter ExpandedAccess InvestigationalProducts. PracticalApproach Sponsors,Physicians, InstitutionalReview Boards. Boards. Framework Framework
 MRCT MRCTCenter Center Expanded ExpandedAccess Accesstoto Investigational InvestigationalProducts Products A APractical PracticalApproach Approachfor for Sponsors, Sponsors,Physicians, Physicians, and
MRCT MRCTCenter Center Expanded ExpandedAccess Accesstoto Investigational InvestigationalProducts Products A APractical PracticalApproach Approachfor for Sponsors, Sponsors,Physicians, Physicians, and
SERIOUS ADVERSE EVENTS
 EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
EVENTS Introduction Timely reporting of Serious Adverse Events (SAEs) is required by regulations of the Food and Drug Administration (FDA) and the National Cancer Institute (NCI). Such reporting is not
Report on the Performance of Drug and Biologics Firms in Conducting Postmarketing
 This document is scheduled to be published in the Federal Register on 11/28/2016 and available online at https://federalregister.gov/d/2016-28442, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
This document is scheduled to be published in the Federal Register on 11/28/2016 and available online at https://federalregister.gov/d/2016-28442, and on FDsys.gov 4164-01-P DEPARTMENT OF HEALTH AND HUMAN
Joint Research Office of Doncaster & Bassetlaw
 Page 1 of 11 Hospitals NHS, Rotherham Doncaster & South Humber NHS and NHS Signatures: Role Name Function Date (DD-MM-YYYY) Signature Author Amy Beckitt Clinical Research Development Manager Reviewer Dr
Page 1 of 11 Hospitals NHS, Rotherham Doncaster & South Humber NHS and NHS Signatures: Role Name Function Date (DD-MM-YYYY) Signature Author Amy Beckitt Clinical Research Development Manager Reviewer Dr
Maximize the Collection of Real- World Data in Expanded Access Programs
 Maximize the Collection of Real- World Data in Expanded Access Programs Jose Ricardo Perez, MD. Executive Medical Director Novartis Oncology East Hanover, NJ Disclaimer: The opinions expressed in this
Maximize the Collection of Real- World Data in Expanded Access Programs Jose Ricardo Perez, MD. Executive Medical Director Novartis Oncology East Hanover, NJ Disclaimer: The opinions expressed in this
UT SOUTHWESTERN MEDICAL CENTER AT DALLAS INSTITUTIONAL REVIEW BOARD. Emergency Use of an Investigational Drug, Biologic or Device
 Emergency Use of an Investigational Drug, Biologic or Device Introduction The emergency use provision in the FDA regulations (21 CFR 56.102d) is an exemption from prior review and approval by the IRB for
Emergency Use of an Investigational Drug, Biologic or Device Introduction The emergency use provision in the FDA regulations (21 CFR 56.102d) is an exemption from prior review and approval by the IRB for
Florida State University Policy 7-IRB-
 Florida State University Policy 7-IRB- Title of Policy: Institutional Review Board Jurisdiction/Applicability Responsible Executive: Gary K. Ostrander Approving Official: Gary K. Ostrander Effective Date:
Florida State University Policy 7-IRB- Title of Policy: Institutional Review Board Jurisdiction/Applicability Responsible Executive: Gary K. Ostrander Approving Official: Gary K. Ostrander Effective Date:
Under this license, you are approved to manufacture aflibercept drug substance intermediate, drug substance, and formulated bulk at
 DEPARTMENT OF HEALTH AND HUMAN SERVICES Silver Spring MD 20993 Our STN: BL 125387/0 BLA APPROVAL November 18, 2011 Regeneron Pharmaceuticals, Inc. Attention: Laura Pologe, Ph.D. Associate Director, Regulatory
DEPARTMENT OF HEALTH AND HUMAN SERVICES Silver Spring MD 20993 Our STN: BL 125387/0 BLA APPROVAL November 18, 2011 Regeneron Pharmaceuticals, Inc. Attention: Laura Pologe, Ph.D. Associate Director, Regulatory
Guidance for Industry Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act
 of the PHS Act") Guidance for Industry Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act DRAFT GUIDANCE This guidance document is being distributed for comment purposes only.
Guidance for Industry Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act DRAFT GUIDANCE This guidance document is being distributed for comment purposes only.
Human Research Protection Program Policy
 Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
Adopted: 11/2005 Revised: 03/2014 Page: 1 of 6 RIGHTS AND RESPONSIBILITIES OF PRINCIPAL INVESTIGATORS IN HUMAN SUBJECTS RESEARCH POLICY Each research study will have a Principal Investigator (PI) and may
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order
 Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
Guidance for Industry and FDA Staff Procedures for Handling Post-Approval Studies Imposed by PMA Order Document issued on: [Level 2, June 15, 2009] This guidance supersedes the document issued under this
DRUG ORDERING AND MAINTENANCE
 AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter only apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
AND MAINTENANCE Disclaimer: Please note that the instructions provided in this chapter only apply to agents provided by the Pharmaceutical Management Branch (PMB), of Cancer Therapy Evaluation Program
Celldex Compassionate Use Policy
 Celldex Compassionate Use Policy Introduction Currently, none of Celldex s investigational products are available for compassionate use given their early stage of clinical development. The policy outlined
Celldex Compassionate Use Policy Introduction Currently, none of Celldex s investigational products are available for compassionate use given their early stage of clinical development. The policy outlined
FDA-Regulated Research
 Chapter 18: FDA-Regulated Research Chapter Contents 18.1 FDA-Regulated Research 18.2 Investigational Drugs 18.3 Investigational Medical Devices 18.4 Sponsor-Investigators 18.5 Humanitarian Use Devices
Chapter 18: FDA-Regulated Research Chapter Contents 18.1 FDA-Regulated Research 18.2 Investigational Drugs 18.3 Investigational Medical Devices 18.4 Sponsor-Investigators 18.5 Humanitarian Use Devices
Requirements for Humanitarian Use Device (HUD)
") Requirements for Humanitarian Use Device (HUD) 1. Definition of Humanitarian Use Device - The definition of a HUD is a device that is intended to benefit patients in the treatment and diagnosis of diseases
Requirements for Humanitarian Use Device (HUD) 1. Definition of Humanitarian Use Device - The definition of a HUD is a device that is intended to benefit patients in the treatment and diagnosis of diseases
Available online at International Journal of Current Science and Technology Vol.5, Issue, 4, pp , April, 2017
 Available online at http://www.journalijcst.com International Journal of Current Science and Technology Vol.5, Issue, 4, pp. 402-408, April, 2017 ISSN: 2320-8090 RESEARCH ARTICLE NEW APPROACH OF INVESTIGATIONAL
Available online at http://www.journalijcst.com International Journal of Current Science and Technology Vol.5, Issue, 4, pp. 402-408, April, 2017 ISSN: 2320-8090 RESEARCH ARTICLE NEW APPROACH OF INVESTIGATIONAL
Conducted Under an IND to Support a
 Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Using Foreign Clinical Trial Data not Conducted Under an IND to Support a US Application PDA Midwest Chapter Meeting March 15, 2018 2013 2017 Regulatory Compliance Associates Inc. All Rights Reserved.
Safety Reporting Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs)
") Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs) Version 1.4 Effective date: 1 December 2011 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision
Part 1: For Clinical Trials of Investigational Medicinal Products (CTIMPs) Version 1.4 Effective date: 1 December 2011 Author: Approved by: Claire Daffern, QA Manager Dr Sarah Duggan, CTU Manager Revision
Sponsor-Investigator Responsibilities In Clinical Trials
 In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
Investigator-Initiated Research
 Vol. 4, No. 6, June 2008 Can You Handle the Truth? Investigator-Initiated Research By Harvey M. Arbit Commercial vs. Non-Commercial Clinical Research Between 1986 and 2006, pharmaceutical and biotech companies
Vol. 4, No. 6, June 2008 Can You Handle the Truth? Investigator-Initiated Research By Harvey M. Arbit Commercial vs. Non-Commercial Clinical Research Between 1986 and 2006, pharmaceutical and biotech companies
ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL
 Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
Assemble Essential Documents in Trial Master File (TMF) Appendix 1 ESSENTIAL DOCUMENTS FOR THE CONDUCT OF A CLINICAL TRIAL 8.2 Before the Clinical Phase of the Trial Commences During this planning stage
Title: Review of Medical Devices Page: 1 of 5 Written by:
 THE NORTH SHORE MEDICAL CENTER Institutional Review Board (IRB) POLICIES AND PROCEDURES IRB Policy Number: 031.1 Title: Review of Medical Devices Page: 1 of 5 Written by: Approved by: Laura W. Knight,
THE NORTH SHORE MEDICAL CENTER Institutional Review Board (IRB) POLICIES AND PROCEDURES IRB Policy Number: 031.1 Title: Review of Medical Devices Page: 1 of 5 Written by: Approved by: Laura W. Knight,
THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE
 THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE Ali Mohamadi, M.D. Senior Director, Global Regulatory Patient Engagement and Policy BioMarin Pharmaceutical 30-July-2016 Goals of Today s Presentation
THE FDA, THE DRUG APPROVAL PROCESS, AND THE PATIENT VOICE Ali Mohamadi, M.D. Senior Director, Global Regulatory Patient Engagement and Policy BioMarin Pharmaceutical 30-July-2016 Goals of Today s Presentation
Expedited Reporting. Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014
 Expedited Reporting Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014 Journal of Clinical Research Best Practices Vol. 7, No. 1, January 2011 Can You
Expedited Reporting Darlene Kitterman, MBA Director, Investigator Support & Integration Services, OCTRI September 25, 2014 Journal of Clinical Research Best Practices Vol. 7, No. 1, January 2011 Can You
What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials
 Vol. 14, No. 10, October 2018 Happy Trials to You What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials While multiple, concurrently accruing patient cohorts in first-in-human (FIH)
Vol. 14, No. 10, October 2018 Happy Trials to You What s New in GCP? FDA Draft Guidance Details FIH Multiple Cohort Trials While multiple, concurrently accruing patient cohorts in first-in-human (FIH)
Human Research Protection Program Policy
 Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
13 FDA-Regulated Research
 13 FDA-Regulated Research FDA regulations apply to research that involves a FDA-regulated test article in a clinical investigation involving human subjects as defined by the FDA regulations. For FDA-regulated
13 FDA-Regulated Research FDA regulations apply to research that involves a FDA-regulated test article in a clinical investigation involving human subjects as defined by the FDA regulations. For FDA-regulated
By Certified Mail - Return Receipt Requested And By Facsimile Transmission. CBER u1u4. Warning Letter. Dear Dr. Hohmann :
 ~suv~%. x 0 ty,~q DEPARTMENT OF HEALTH &: HUMAN SERVICES Food and Drug Administration By Certified Mail - Return Receipt Requested And By Facsimile Transmission Elizabeth L. Hohmann, M.D. Massachusetts
~suv~%. x 0 ty,~q DEPARTMENT OF HEALTH &: HUMAN SERVICES Food and Drug Administration By Certified Mail - Return Receipt Requested And By Facsimile Transmission Elizabeth L. Hohmann, M.D. Massachusetts
Guidance for Industry
 Guidance for Industry Notification to FDA of Issues that May Result in a Prescription Drug or Biological Product Shortage DRAFT GUIDANCE This guidance document is being distributed for comment purposes
Guidance for Industry Notification to FDA of Issues that May Result in a Prescription Drug or Biological Product Shortage DRAFT GUIDANCE This guidance document is being distributed for comment purposes
Speed your time to market with FDA s expedited programs
 Regulatory Sciences Expediting drug approval Speed your time to market with FDA s expedited programs The faster way to marketing submission and drug approval for serious conditions and rare diseases In
Regulatory Sciences Expediting drug approval Speed your time to market with FDA s expedited programs The faster way to marketing submission and drug approval for serious conditions and rare diseases In
New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 2)
") New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 2) Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and
New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 2) Guidance for Industry DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and
IDE Submissions for Devices Used to Manufacture Cell Therapy Products
 IDE Submissions for Devices Used to Manufacture Cell Therapy Products Janice Davis-Sproul, MAS Johns Hopkins Medicine 7 th Annual Somatic Cell Therapy Symposium 1 Overview Discuss an investigatorsponsored
IDE Submissions for Devices Used to Manufacture Cell Therapy Products Janice Davis-Sproul, MAS Johns Hopkins Medicine 7 th Annual Somatic Cell Therapy Symposium 1 Overview Discuss an investigatorsponsored