Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India
|
|
|
- Marybeth Maxwell
- 6 years ago
- Views:
Transcription
1 Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India 1
2 2
3 Content Message Foreword 1. Introduction 2. Background & Objectives 3. Applicable Regulations and Guidelines 4. Competent Authorities 5. Scope 6. Principles for Development of Similar Biologics 6.1 Selection of Reference Biologic 6.2 Manufacturing Process 6.3 Quality Based Consideration of Similar Biologics 6.4 Quality Comparability Study 7. Data Requirements for Preclinical Studies 7.1 Prerequisite before Conducting Preclinical Studies 7.2 Preclinical Studies (Pharmacodynamic and Toxicology Studies) 7.3 Immune Responses in Animals 8. Data Requirements for Clinical Trial Application 8.1 Pharmacokinetic (PK) studies 8.2 Pharmacodynamics (PD) studies 8.3 Confirmatory Safety and Efficacy Study 8.4 Safety and Immunogenicity data 8.5 Extrapolation of Efficacy and Safety Data to Other Indications 9. Data Requirements for Market Authorization Application 9.1 Approval of package insert 10. Post Market Data for Similar Biologics 10.1 Pharmacovigilance Plan 10.2 Adverse Drug Reaction (ADR) Reporting 10.3 Post Marketing Studies (Phase IV) 3
4 Annexures: 11. Application Forms 12. Archiving of Data / Retention of Samples 13. Glossary 14. References 1. Protocols on Regulatory Pathway for Recombinant Pharma Products Adopted from Mashelkar Report. 4
5 MESSAGE from DBT Secretary 5
6 Foreword from DCGI 6
7 Guidelines on Similar Biologic: Regulatory Requirements for Marketing Authorization in India 1. Introduction The Guidelines on Similar Biologic prepared by Central Drugs Standard Control Organization (CDSCO) and the Department of Biotechnology (DBT) lay down the regulatory pathway for a Similar Biologic claiming to be Similar to an already authorized Reference Biologic. A Similar Biologic product is that which is similar in terms of quality, safety and efficacy to an approved Reference Biological product based on comparability. These guidelines address the regulatory pathway regarding manufacturing process and safety, efficacy and quality aspects for Similar Biologics. These guidelines also address the pre market regulatory requirements including comparability exercise for quality, preclinical and clinical studies and post market regulatory requirements for Similar Biologics. These guidelines are for the guidance of all stakeholders and are not meant to substitute or rephrase the Rules made under Drugs and Cosmetics Act, 1940 or any other relevant Acts and are subject to being in conformity with the Drugs and Cosmetics Act and Rules as may be amended from time to time. 7
8 2. Background & Objectives CDSCO is the national regulatory authority in India that evaluates safety, efficacy and quality of drugs in the country. DBT through Review Committee on Genetic Manipulation (RCGM) is responsible for overseeing the development and preclinical evaluation of recombinant DNA derived products. Presently, several organizations are actively engaged in manufacturing and marketing Similar Biologics in India. So far, these Similar Biologics were approved by RCGM and CDSCO using an abbreviated version of the pathway applicable to new drugs on a case by case basis. Since there are several such products under development in India, both regulatory agencies considered the need to publish a clear regulatory pathway outlining the requirements to ensure comparable safety, efficacy and quality of a Similar Biologic to the reference Biologic. Based on demonstration of similarity in the comparative assessment, a Similar Biologic may require reduced preclinical and clinical data package as part of submission for market authorization. The objective of this document is to provide guidelines to applicants to enable them to understand and comply with the regulatory requirements for the market authorization of Similar Biologics in India. 8
9 3. Applicable Regulations and Guidelines The Similar Biologics are regulated as per the Drugs and Cosmetics Act, 1940, the Drugs and Cosmetics Rules, 1945 (as amended from time to time) and Rules for the manufacture, use, import, export and storage of hazardous microorganisms/ genetically engineered organisms or cells, 1989 (Rules, 1989) notified under the Environment (Protection) Act, Various applicable guidelines are as follows: Recombinant DNA Safety Guidelines, 1990 Guidelines for generating preclinical and clinical data for rdna vaccines, diagnostics and other Biologicals, 1999 CDSCO guidance for industry, 2008: o o o o Submission of Clinical Trial Application for Evaluating Safety and Efficacy Requirements for permission of New Drugs Approval Post approval changes in Biological products: Quality, Safety and Efficacy Documents Preparation of the Quality Information for Drug Submission for New Drug Approval: Biotechnological/Biological Products Guidelines and Handbook for Institutional Biosafety Committees (IBSCs), 2011 Guidelines on Similar Biologics: Regulatory Requirements for Marketing authorization in India
10 4. Competent Authorities The competent authorities involved in the approval process are as follows: Institutional BioSafety Committee (IBSC) IBSC is required to be constituted by any person including research institutions handling hazardous microorganisms and/ or genetically engineered organisms. IBSC is responsible for ensuring biosafety on site, along with initial review of applications to be recommended to RCGM. IBSC is also assigned with the responsibility to review and authorize firm for exchange of aforesaid organisms for the purpose of research. Review Committee on Genetic Manipulation (RCGM) 1 RCGM is functioning from the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India. In the context of Similar Biologics, RCGM is responsible for authorizing the conduct of research and development, exchange of genetically engineered cell banks for the purpose of research and development and review of data up to preclinical evaluation. Genetic Engineering Appraisal Committee (GEAC) 1 GEAC functions under the Ministry of Environment and Forests (MoEF) as statutory body for review of applications and approval of activities where final drug product 10
11 Contains genetically modified organisms/ living modified organisms. ( 1 RCGM and GEAC are statutory committees set up as per provisions of Rules, 1989) Central Drugs Standard Control Organization (CDSCO) 2 CDSCO, headed by the Drug Controller General of India (DCGI) is the apex regulatory body under Ministry of Health & Family Welfare (MoHFW), Government of India, which is responsible for the approval of clinical trials as well as new drugs. In the context of Similar Biologics, CDSCO is responsible for clinical trial approval (also grants permission for import of drugs for clinical trial and export of clinical samples for biochemical and immunological analysis) and permission for manufacturing and marketing. Zonal CDSCO is responsible for authorizing import of drugs for examination, test and analysis for research and development. ( 2 CDSCO functions as per the provisions of the Drugs and Cosmetics Act 1940) 11
12 5. Scope These guidelines apply to Similar Biologics that contain well characterized proteins as their active substance, derived through modern biotechnological methods such as use of recombinant DNA technology. The demonstration of similarity depends upon detailed and comprehensive product characterization, preclinical and clinical studies carried out in comparison with a Reference Biologic. Similar Biologic can only be developed against the Reference Biologic that has been approved using a complete data package in India. In case the Reference Biologic is not authorized in India, it should have been approved / licensed and marketed in an ICH (The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) country. Any product can be considered as Similar Biologic, only if it is proven to be Similar using extensive quality characterization against the Reference Biologic. Further product development should only be considered once the similarity of the Similar Biologic is demonstrated in quality to a Reference Biologic. These guidelines are applicable for Similar Biologics to be developed in India or imported into the country for marketing authorization. Detailed regulatory pathways for indigenously developed and imported products 3 are given in Annexure I. ( 3 Adopted from Report of the Task Force on Recombinant Pharma, 2005, chaired by Dr R.A. Mashelkar, DG, CSIR) 12
13 6. Principles for Development of Similar Biologics Similar Biologic is developed through a sequential process to demonstrate the Similarity by extensive characterization studies revealing the molecular and quality attributes with regard to the Reference Biologic. Although the extent of preclinical and clinical evaluation of the Similar Biologic is likely to be less than that required for the Reference Biologic, it is essential that the testing of the Similar Biologic be sufficient to ensure that the product meets acceptable levels of safety, efficacy and quality to ensure public health in accordance with international guidelines. (WHO 2013). Generally, abbreviated data requirements are only possible for preclinical and /or clinical components of the development program but not for the quality components by demonstration of comparability of product ( Similarity established to the Reference Biologic) Identification of any significant differences in safety, efficacy and quality studies would mean the need for a more extensive preclinical and clinical evaluation and the product will not qualify as a Similar Biologic. In case the Reference Biologic is used for more than one indication, the Similar Biologic also qualifies for all the indications only if it is justified and if meets the 13
14 conditions set forth in the section Extrapolation of Efficacy and Safety Data to Other Indications. Justification for extrapolation of indication shall be based on comparability in quality, preclinical and clinical studies, available literature data and whether or not the same mechanism of action is involved in specific indications. 6.1 Selection of Reference Biologic Reference Biologic is an innovator s product approved after evaluation of complete dossier is critical for the development of Similar Biologic. The Reference Biologic has to be used in all the comparability exercise with respect to quality, preclinical and clinical considerations. The following factors should be considered for selection of the Reference Biologic: The Reference Biologic should be licensed / approved in India or ICH countries and should be the innovator s product. The Reference Biologic should be licensed based on a full safety, efficacy and quality data. Therefore another Similar Biologic cannot be considered as a choice for Reference Biologic. In case the Reference Biologic is not marketed in India, the Reference Biologic should have been licensed in any ICH country. The Reference Biologic product can be imported for developing the Similar Biologic for quality, pre clinical and clinical comparability. 14
15 The same Reference Biologic should be used throughout the studies supporting the safety, efficacy and quality of the product (i.e. in the development Programme for the Similar Biologic). The dosage form, strength and route of administration of the Similar Biologic should be the same as that of the Reference Biologic. The active drug substance (active ingredient) of the reference biologic and that of Similar Biologic must shown to be similar. The acceptance of an innovator product as a Reference Biologic for evaluation of Similar Biologic does not imply approval for its use in India. 6.2 Manufacturing Process The Similar Biologic manufacturer should develop the manufacturing process to yield a comparable quality product in terms of identity, purity and potency to the Reference Biologic. The manufacturing process for Similar Biologic should be validated and demonstrated to be highly consistent and robust. If the host cell line used for the production of reference Biologic is disclosed, it is desired to use the same host cell line for manufacturing Similar Biologic. Alternatively any cell line that is adequately characterized and appropriate for intended use can be used to develop a Similar Biologic, with appropriate justification in order to minimize the potential for significant changes in quality attributes (QAs) of the product and to avoid introduction of certain types of process related impurities that could impact clinical outcomes and immunogenicity. For the establishment and characterization of the cell banks, the guidelines issued by the ICH viz. Q5A 4, Q5B 5 and Q5D 6 should be referred for guidance. ( 4 ICHQ5A(R1): Viral Safety Evaluation of Biotechnology products derived from cell lines of Human or Animal Origin 15
16 5 ICHQ5B: Quality of Biotechnological Products: Analysis of the expression construct in cells used for production of R DNA derived protein products 6 ICH Q5D: Derivation and characterization of cell substrates used for production of Biotechnological/Biological products) The data requirements for review of manufacturing process at preclinical submission stage include a complete description of the manufacturing process from development and characterization of cell banks, stability of clone, cell culture/ fermentation, harvest, excipients, formulation, purification, primary packaging interactions (if different from reference Biologic), etc. and the consequences on product characteristics as indicated below: Molecular Biology Considerations The details regarding host cell cultures (including viral clearance), vectors, gene sequences, promoters etc. used in the production of Similar Biologics should be provided with appropriate drawings/figures. The detail of post translational modifications, if any (glycosylation, oxidation, deamidation, phosphorylation etc.) should be explained Upstream Process Development Upstream process should be described in detail including media components used for cell growth. At least three batches of reproducible fermentation data at pilot scale (batch size adequate to give enough purified product to generate preclinical data). Upstream process should be well controlled and monitored. 16
17 Details of upstream process kinetics data from consistency batches indicating cell growth, product formation, ph, temperature, dissolved oxygen, major nutrient consumption pattern and agitation rate. Concentration to be defined in terms of product/litre, yield and volumetric productivity. Data to verify that the specific protein yield (amount of protein per unit cell mass) remains constant for all upstream batches. Demonstrate that the overall productivity is reproducible and scalable Downstream Process Development Detail description of the methods followed for the cell harvesting and extraction of the protein. Steps involved in purification of protein. Batch size for protein purification. Description of each unit operation step during purification and recovery of protein along with quantitative recovery of product at each stage. Describe the quality of the refolded protein if the starting material is aggregated or from inclusion bodies and include details of the refolding process, specific activity at different doses, dose response curve, stability data and confirmation of solubility and absence of aggregation. Consistency of recovery in three consecutive batches of purification from three independent batches of cell culture/ fermentation. Describe post translational variation, if any. 17
18 Details of removal of impurities like product related variants & impurities, and host cell & process related impurities considered to pose a risk of Immunogenicity (EMEA 1997). Virus clearance validation studies. For clinical trial application, additional requirements are applicable as per CDSCO guidelines. A well defined manufacturing process with its associated process controls assures that an acceptable product is produced on consistent basis in accordance with Good Manufacturing Practice (GMP). Data for submission should include: Detailed description of the drug substance and drug product processes Critical and key Quality Attributes of the product Manufacturing process controls Critical process parameters Stability data Comparability of product manufactured at clinical scale against Reference Biologic Data from consistency batches and/or process validation batches as applicable. 6.3 Quality Based Considerations for Similar Biologics Analytical Methods The analytical methods should be chosen for establishing product comparability as per the critical quality attributes of the product. For certain attributes (e.g. product 18
19 aggregation) it is customary to use multiple, orthogonal methods for characterization. Extensive state of the art analytical methods should be applied to detect even slight differences in all relevant quality attributes. Indian Pharmacopoeia monograph should be followed, if available. The measurement of quality attributes in characterization should entail the use of appropriately qualified assays, which are reproducible and reliable. The methods used to measure quality attributes for batch release, stability studies and inprocess controls should be validated in accordance with ICH guidelines(ich Q2 7, Q5C 8, Q6B 9 ), as appropriate. The characterization studies should include samples of the applicant s r-dna derived product, Reference Biologic as control, known positive standard and negative control, wherever relevant. To ensure the statistical analysis, each quantitative experiment should be done at least three times and data should be represented in terms of mean and standard deviation. Appropriate statistical significance should be represented throughout the characterization data. Physicochemical and Biological characterization methods to be used for r-dna derived products are given in Annexure II. It may be noted that this Annexure II is suggestive but not limited to the specified method and the requirements may vary on case by case. ( 7 ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology 8 ICH Q5C: Stability testing of Biotechnological/Biological Products 9 ICHQ6B: Specifications: test Procedures and Acceptance criteria for Biotechnological/Biological Products) 19
20 6.3.2 Product Characterization Characterization studies for Similar Biologic include physicochemical properties, Biological activity, immunological properties, functional assays, purity (process and product related impurities etc.), contamination, strength and content. Principles outlined in the ICH Q6B guideline should be followed. Indian Pharmacopoeia Monograph should be followed, if available. i. Structural and Physicochemical Properties: The analysis of physicochemical characteristic should include determination of primary and higher order structure of the drug substance and the product along with other significant physicochemical properties. The target amino acid sequence of the Similar Biologic should be confirmed and is expected to be the same as for the Reference Biologic. Analytical methods that are used (including Biological and functional assays) should have acceptable precision and accuracy. In cases, where post translational modifications are taking place, these modifications need to be identified and quantified. In case any significant differences are found, these should be scientifically justified and critically examined in preclinical studies and clinical trials. ii. Biological Activity: Biological products may have multiple biological activities. In such cases, appropriate biological assays will be required to characterize the activity and establish the product s mechanism of action and clinical effects (in units of activity). The data from biological assays will supplement the physicochemical characterization of the product as described in the section
21 Biological assays should be validated against an international or national Reference standard, where available and appropriate. If no such standards are available, an internal Reference standard must be established as per the ICH guidelines. If the methods of bioassay(s) are documented in the specification, test(s) can be conducted accordingly. iii. Immunological Properties: The manufacturing process of Similar Biologics is known to affect the level of process related impurities and post translational modifications of the product. These characteristics may affect the immunogenicity of the product. Hence evaluation by characterization (antibody or antibody derived product); comparison to Reference Biologic with respect to specificity, affinity, binding strength and Fc function; and evaluation by animal studies should be performed. iv. Purity and Impurities: Characterization of Similar Biologic requires evaluation ofthe following via a combination of analytical procedures: Product related variants (e.g., glycoforms, isomers etc.) Product related impurities (e.g., aggregated, oxidized or deamidated product) Host cell related impurities (e.g., host cell protein, host cell DNA etc.) Process related impurities (residual media components, resin leachates etc.) 21
22 Differences observed in the purity and impurity profiles of the Similar Biologic relative to the Reference Biologic should be evaluated to assess their potential impact on safety and efficacy. Where the Similar Biologic exhibits different impurities, those impurities should be identified and characterized when possible. Depending on type and amount of the impurity, conduct of preclinical and clinical studies will help to confirm that there is no adverse impact on safety and efficacy of the Similar Biologic Specifications Specifications of Similar Biologic (for drug substance and drug product) are established around quality attributes (QAs) with the intent of ensuring consistency in product quality and comparability to Reference Biologic according to relevant guideline (ICH Q6B). Methods used for setting specifications may or may not be the same as the analytical methods used for product characterization and for establishing product comparability. Acceptance limits should be set based on Reference Biologic data and data from sufficient number of batches from preclinical or clinical batches, which must be in line with international norms Stability The shelf life and storage condition of drug substance and drug product should be assigned based on real time stability studies. Stability studies on drug substance and drug product should be carried out using containers and conditions that are 22
23 representative of the actual storage containers and conditions, according to relevant guidelines (e.g. ICH Q1 A(R2), ICH Q5C, WHO TRS 822). Side by side accelerated and stress stability studies comparing the Similar Biologic to the Reference Biologic will be of value in determining the Similarity of the products by showing comparable degradation profiles. 6.4 Quality Comparability Study The quality comparison between Similar Biologic and Reference Biologic is essential. The applicant should submit a full quality dossier as per CDSCO guidance for industry, 2008 including the results of comparability exercise for the Similar Biologic with the Reference Biologic before the applicant proposes to take the Similar Biologic to clinical development. First three consecutive standardized batches which have been used to demonstrate consistency of the manufacturing process should be used. Head to head characterization studies are required to compare the Similar Biologic and the Reference Biologic at active drug product level. It is required to assure that the molecular structure of active drug substance present in the Similar Biologic is comparable to active drug substance present in Reference Biologic. However, in cases where the required analyses of quality attributes of the active substance of the Reference Biologic can be made at the finished product stage, testing of the isolated active ingredient may not be needed. Differences between the Similar Biologic and the Reference Biologic should be evaluated for their potential impact 23
24 on safety and efficacy of the Similar Biologic and additional characterization studies may be necessary. Minor differences between Similar Biologic and reference Biologic in each quality component may be there. However, appropriate data should be submitted to verify that these differences do not impact on the safety and efficacy. The quality comparison between the Similar Biologic and the reference Biologic should be governed by Quality Attributes (QAs), which employ state of the art high resolution analytical techniques and methods that are sensitive enough to detect the possibilities of changes to the product. From the perspective of establishing Similarity, Quality Attributes of a Similar Biologic may be considered in two categories; Critical Quality Attributes (CQA) and Key Quality Attributes (KQA) : 1) Critical Quality Attributes (CQA) are those Quality Attrib utes which have direct impact on the clinical safety or efficacy. All attributes that directly impact the known mechanism(s) of action of the molecule fall in this category. CQAs must be controlled within limits that need to be established based on the Reference Biologic. 2) Key Quality Attributes (KQA) are those Quality Attributes which are not known to impact clinical safety and efficacy but are considered relevant from a product and process consistency perspective. Attributes that do not impact the known mechanism(s) of action of the molecule fall in this category. KQAs must necessarily 24
25 be controlled within acceptable limits; however it may acceptable to have slight differences in comparison to the Reference Biologic. The list of routine analytical tests to be included for a comprehensive quality comparability exercise of Critical and Key Quality Attributes is given in Annexure II. This is intended as a guidance, and proposes a framework to establish analytical similarity that incorporates molecular structure, function and heterogeneity. It may be noted that is only indicative and a specific determination will need to be made for each biologic molecule. 7. Data Requirements for Preclinical Studies 7.1 Prerequisite before Conducting Preclinical Studies The applicant has to comply with the RCGM requirements like demonstration of consistency of the process and product, product characterization and product specifications. The applicant should submit the data generated along with the following basic clinical information and preclinical study protocols to RCGM for obtaining permission. The toxicology studies should be initiated after the approval of RCGM. The basic information about the Reference Biologic and Similar Biologic may include the following: Basic information about the Reference Biologic Information about the drug, route of administration, absorption and elimination rate, therapeutic index, dose, vehicle, mode of administration, dose response etc. 25
26 Bioequivalence range, if available. Tissue specific localization, if available. Available toxicity data on Reference Biologic. Mode of action. Basic information about the Similar Biologic Known / proposed clinical use Target population (Age, sex, pregnancy, lactating, children etc.) Dosage (frequency and intervals) units Route / alternate routes of administration Final formulation + adjuvants, additives etc. Toxicology data of adjuvants Diluents Presentation e.g. pre filled syringe, cartridge, vial The application to RCGM should be accompanied by approval of Institutional Biosafety Committee (IBSC) of the developer (copy of the minutes should be submitted), and approval of Institutional Animal Ethics Committee (IAEC), if available. The applicant should also provide details of the proposed site for conduct of toxicity testing and personnel to be involved e.g. study director, principal investigator, pathologist, other Investigators and quality assurance officer at the site. Status of GLP certification of proposed facility should also be provided. 7.2 Preclinical Studies (Pharmacodynamic and Toxicology Studies) The preclinical studies should be conducted prior to the initiation of any clinical studies. These preclinical studies should be comparative in nature and designed to 26
27 detect differences if any, between the Similar Biologic and Reference Biologic. The preclinical study design may vary depending upon the clinical parameters such as therapeutic index, the type and number of indications applied. The approach adopted should be fully justified in the preclinical overview. Preclinical studies should be conducted with the final formulation of the Similar Biologic intended for clinical use and for the Reference Biologic unless otherwise justified. The dosage form, dose, strength and route of administration of the Similar Biologic should be the same as that of the Reference Biologic and in case of any differences in these parameters, it should be justified. The following studies are required for preclinical evaluation: Pharmacodynamic Studies i. In vitro studies: Comparability of test and Reference Biologic should beestablished by in vitro cell based bioassay (e.g. cell proliferation assays /cytotoxicity / neutralizing / receptor binding assays). ii. In vivo studies: In vivo evaluation of Biological/ pharmacodynamic activity may be dispensable if in vitro assays are available, which are known to reliably reflect the clinically relevant pharmacodynamic activity of the Reference Biologic. In cases where the in vitro assays do not reflect the pharmacodynamics, In vivo studies should be performed as applicable Toxicological Studies 27
28 In case of in vivo toxicity studies, at least one repeat dose toxicity study in pharmacologically relevant animal model should be conducted to ensure and confirm the potential toxicity (including toxicokinetics) of the molecule. The duration of the study would be generally not less than 28 days with 14 days recovery period. However the duration may vary depending on the dosage and other parameters on case by case basis. Regarding the animal models to be used, the applicant should provide the scientific justification for the choice of animal model(s) based on the data available in scientific literature. However if the pharmacologically relevant animal species is not available and has been appropriately justified, the toxicity studies need to be undertaken in two species i.e. one rodent and other non rodent species, as per the requirements of Schedule Y with due permission from the RCGM. Regarding the route of administration, in cases when the pharmacologically relevant animal model is used, the route of administration would include only the intended route, whereas in all other cases, Schedule Y should be followed. The dose should be calculated based on the therapeutic dose of the Reference Biologic. If required a pilot dose response study should be conducted prior to initiating the toxicity studies. Generally there would be three levels of doses (viz. low, medium and high) used in the animal toxicology studies corresponding to 1X, 2X and 5X of human equivalent dose or higher test dose for repeat dose toxicity studies. In the toxicity study the Similar Biologic should be compared with 28
29 Reference Biologic at least at 1X of human equivalent dose (HED). Any difference in the levels of doses should be justified and approved prior to the studies. Regarding the schedule of administration, the therapeutic schedules may be used as the basis. Depending on the route of administration, local tolerance should be evaluated. If feasible, this evaluation may be performed as a part of above mentioned repeat dose toxicity study. Accordingly the study groups of animals in repeat dose toxicity testing will consist of: i. Historical Control (Optional) ii. Vehicle Control iii. Vehicle Control for recovery group iv. Formulation without protein (for vaccines) if multiple adjuvants each to be checked independently v. 1X Similar Biologic for study duration (lowest dose) vi. 1X Reference Biologic for study duration vii. 2X Medium dose Similar Biologic viii. 5X High dose Similar Biologic ix. Similar Biologic with a recovery group going beyond the end of study period for 7 to 14 days The protocols and the study reports should provide complete details of various steps in the toxicity testing as indicated below: 29
30 Procedures prior to euthanasia e.g. blood drawing, body weight, etc. Events immediately after euthanasia, necropsy, gross description, organ weights and organs sampled for histopathology. Biochemical parameters Equipment and methods used units of measurement and expression. Haematology procedures and parameters method to be used (automated or manual). Statistical methods used. Bone marrow either examined as an aspirate /smear or on histopathology section. In case of histopathological observations, the applicants should consider the following points: Every observation considered as deviation from described normal histology needs to be documented and the incidence of each of these in the different groups should be denoted Whether such a feature is significant or not can be decided on review of statistical significance or dose response or if it is within or outside the normal range of values in case of biochemical and haematological observations. If all organs from all animals were not examined e.g. in 5 animals only 4 livers were examined, the reason for the 1 liver not being examined should be documented. In case of premature death or morbidity the proposed course of action is to be included in the protocol. 30
31 Other toxicity studies, including safety pharmacology, reproductive toxicity, mutagenicity and carcinogenicity studies are not generally required for evaluation of a Similar Biologic unless warranted by the results from the repeat dose toxicological studies. The final report of the study should reflect all the aspects approved in the protocol and the following additional sections/documents: RCGM approval of protocol and test centre IBSC approval of report IAEC approval for animal use and for the procedures QA statement Signatures of study director and all investigators who were involved in the study All quality analytical reports on the test material and vehicle Animal feed and animal health certifications Protocol deviations if any Discussion on the results Individual animal data, summary data and any other data like computer analysis outputs etc. Conclusion 7.3 Immune Responses in Animals 31
32 Antibody response to the Similar Biologic should be compared to that generated by the reference Biologic in suitable animal model. The test serum samples should be tested for reaction to host cell proteins. For evaluating immune toxicity of the Similar Biologic under study, the results of local tolerance (part of repeat do se or standalone test) should be analyzed with the observations regarding immunogenicity in sub chronic study. Therefore, the immunogenicity testing should be included as part of the sub chronic repeat dose study while developing the protocols. The other parameters for evaluating immune toxicity include immune complexes in targeted tissues may be considered while evaluating histopathology observations, etc. After completion of preclinical studies the reports are submitted to RCGM for review and consideration. Other toxicity studies, including safety pharmacology, reproductive toxicity, mutagenicity and carcinogenicity studies are not generally required for evaluation of a Similar Biologic unless warranted by the results from the repeat dose toxicological studies. Based on the successful evaluation of preclinical study reports including demonstration of consistency of the process and product, product characterization, product specifications and comparison of similar biologics to reference Biologic, RCGM will recommend the DCG(I) to allow the sponsor to conduct appropriate 32
33 phase of clinical trial as per the CDSCO requirements. The applicant may submit parallel application to RCGM and office of DCG (I) seeking approval to conduct clinical trial. However, office of DCG (I) shall complete the scrutiny of application and issue permission, only after RCGM clearance was received. 8. Data Requirements for Clinical Trial Application Besides the information submitted in the preclinical application, the applicant has to submit application for conduct of clinical trial as per the CDSCO guidance for industry, The quality data submitted should indicate that there are no differences in Critical Quality Attributes (CQAs), and that all Key Quality Attributes (KQAs) are well controlled in order to allow the initiation of clinical evaluation. 8.1 Pharmacokinetic (PK) Studies This PK data should support the subsequent Phase III clinical development given that the purported Similar Biologic would be established to be similar as the Reference Biologic product. After completion of extensive characterization comparability on quality attributes, a PK study of the Similar Biologic in comparison with the Reference Biologic product may be performed in an appropriate number of: a. Normal Healthy Volunteers (NHV) and / or b. Patients The design of comparative pharmacokinetic studies should take the following factors into consideration. Half life 33
34 Linearity of PK parameters Endogenous levels and diurnal variations of Similar Biologic under study (where applicable) Conditions and diseases to be treated Route(s) of administration, and Indications Appropriate design considerations include: Single dose, comparative, PK studies Parallel arm or Cross over Multiple dose, comparative parallel arm steady state PK studies In sequential development approach, the Normal Healthy Volunteers (NHV) study is performed before the Phase III safety and efficacy study Single Dose Comparative PK Studies Dosage in the PK study should be within the therapeutic dose range of reference Biologic. Appropriate rationale for dose selection should be provided. The route of administration should be the one where the sensitivity to detect differences is the largest. Sample size should have statistical rationale (i.e. statistically justified) and comparability limits should be defined and justified prior to conducting the study. The analytical method should be validated to have satisfactory specificity, sensitivity and a range of qualification with adequate accuracy and precision. It should have capability to detect and follow the time course of the Similar Biologic 34
35 (the parent molecule and / or degradation products) in a complex Biological matrix that contains many other proteins. Differences in elimination kinetics between Similar Biologic and reference Biologic e.g. clearance and elimination half life should be explored. Similarity in terms of absorption / bioavailability should not be the only parameters of interest. A parallel arm design study is more appropriate for Similar Biologics with a long half life or for proteins for which formation of antibodies is likely or if study is being done in patients. In case of short half life, cross over design may be considered with a scientific justification Multiple Dose Comparative PK Studies Multiple dose, comparative, parallel arm steady state PK studies are required for a Similar Biologic that is used in a multiple dose regimen, where markedly higher or lower concentrations are expected at steady state than that expected from single dose data PK measurements, and where time dependence and dose dependence of PK parameters cannot be ruled out. In case multi dose comparative PK studies are not done adequate justification should be provided. 8.2 Pharmacodynamic Studies As required for the PK studies in the Similar Biologic clinical development program, the pharmacodynamic (PD) studies should also be comparative in nature. Comparative, parallel arm or cross over, PD study in most relevant population 35
36 (patients or healthy volunteers) is required for detecting differences between Similar Biologic and Reference Biologic. If a PD marker is available in healthy volunteers, PD in healthy volunteers can be done, unless considered unethical due to e.g. expected adverse events and toxicity in e.g. oncology drugs. Comparative PD studies are recommended when the PD properties of the Reference Biologic are well characterized with at least one PD marker validated for a clinical outcome of the molecule. The relationship between dose / exposure, the relevant PD marker(s) and response / efficacy of the Reference Biologic should be well established and used to justify the design. The acceptance ranges for the demonstration of Similarity in PD parameters should be predefined and appropriately justified. The parameters investigated in PD studies should be clinically relevant and surrogate markers should be clinically validated. PD studies may be combined with PK studies, in which case the PK/PD relationship should be characterized. If PD marker is not available and the PK can be done in patients then the PK study can be combined with phase III clinical study. The PD study can also be a part of Phase III clinical trials wherever applicable. 8.3 Confirmatory Safety and Efficacy Study The establishment of in vitro, pre clinical and PK/PD Similarity as described in earlier section is important as robust, high quality processes, a comprehensive quality comparison and comparative preclinical and PK/PD studies help in demonstrating the Similarity of the Similar Biologic in these settings. 36
37 In order to eliminate any residual risk, a comparative phase III clinical trial may also be required in order to establish the comparability with respect to clinical safety and efficacy. Only in exceptional cases i.e. if there are no residual uncertainties left after comparing Similar Biologic and Reference Biologic at the analytical, nonclinical and PK/PD level, an additional comparative safety and efficacy trial is not needed. Information to establish comparative safety and efficacy in relevant patient population is mandatory for all Similar Biologics. Comparative clinical trials are critical to demonstrate the similarity in safety and efficacy profiles between the Similar Biologic and Reference Biologic with few exceptions (e.g. recombinant human soluble insulin products for which only comparative clinical safety study is required). The study should be conducted in a sensitive and homogenous patient population with appropriate sensitive primary end points as per requirement of a Phase III clinical trial. The design of the studies and the clinical comparability of the primary efficacy endpoints are important and should be given careful consideration and should be scientifically justified on clinical grounds. Equivalence, non inferiority or comparability Phase III clinical trials may be conducted based on comparability established during physicochemical characterization, preclinical and PK/PD studies, after approval of design and protocol by CDSCO. However, the comparability Phase III clinical trials intended for seeking marketing approval of similar biologics falling under the category of new drugs as per Drugs and Cosmetics rules, 1945 shall be conducted in accordance with the Indian Good Clinical Practice (GCP) guidelines, generally, in not less than hundred evaluable patients in test arm to evaluate the safety, efficacy and comparability. Based on the results of such Clinical trials, the marketing approval may be considered if safety, efficacy and comparability is established. Further, Phase IV clinical trials may be required to be conducted, generally, in more than two hundred patients in continuation of comparability clinical trials. 37
38 The nature, severity and frequency of adverse events should be compared between the similar biologic and reference biologic and should be based on safety data from a sufficient number of patients treated for an acceptable period of time and efforts should be made to ensure that comparative clinical studies have a sufficient number of patients treated for acceptable period of time in order to allow detection of significant differences in safety between similar biologic and reference biologic as per the protocol. One or more adequately powered, randomized, parallel group, blinded confirmatory clinical safety and efficacy trials are desirable based on the comparability established during preclinical and PK / PD studies. More than one safety and efficacy study may be required and the similar biologic will be treated as a stand alone product if the similar biologic is not comparable to reference biologic in preclinical evaluations conducted and /or the PK/PD studies have not demonstrated comparability Waiver of safety and efficacy study The confirmatory clinical safety and efficacy study can be waived if all the below mentioned conditions are met: i. Structural and functional comparability of Similar Biologic and Reference Biologic can be characterized to a high degree of confidence by physicochemical and in vitro techniques ii. The Similar Biologic is comparable to Reference Biologic in all preclinical evaluations conducted 38
39 iii. PK / PD study has demonstrated comparability of PD markers validated for clinical outcomeand has preferentially been done in an in patient setting with safety measurement (including meaningful immunogenicity assessment) for adequate period justified by the applicant and efficacy/pd measurements iv. A comprehensive post marketing risk management plan has been presented that will gather additional safety data with a specific emphasis on gathering immunogenicity data. The confirmatory clinical safety and efficacy study cannot be waived especially for large molecular weight biologics like Monoclonal antibodies. In case the safety and efficacy study is waived all the indications approved for reference product may be granted based on comparable quality, non clinical as well as convincing PK/PD data. Wherever the phase III trial is waived, the immunogenicity should have been gathered in the PK/PD study and will also need to be generated during postapproval Phase IV study. The confirmatory clinical safety and efficacy study cannot be waived if there is no reliable PD marker validated for clinical outcome Non comparative safety and efficacy study For a product which is found Similar in pre clinical, in vitro characterization having established PK methods and a PD marker that is surrogate of efficacy, the residual risk is significantly reduced in the Phase I study if equivalence is demonstrated for 39
40 both PK and PD. Phase III clinical trials of such a Similar Biologics product may be waived as noted above or, where considered necessary, an appropriate single arm study in at least 100 evaluable subjects may be carried out in the most sensitive indication to address any residual uncertainty. 8.4 Safety and Immunogenicity Data Both pre approval and post approval assessment of safety is desired to be conducted for a Similar Biologic. Regarding pre approval safety assessment, comparative pre approval safety data including the immunogenicity data is required for all Similar Biologics including those for which confirmatory clinical trials have been waived. This pre approval safety data is primarily intended to provide assurance of the absence of any unexpected safety concerns. Comparative safety data based on adequate patient exposure (both numbers and time) must, in conjunction with the published data on the Reference Biologic provide assurance of absence of any unexpected safety concerns and in conjunction with the proposed non comparative post marketing study provide a comprehensive approach to the evaluation of safety of the Similar Biologic. Post approval safety data requirements are elaborated in section 10.3 From a safety and Immunogenicity perspective, if the firm conducts pre approval studies that included more than 100 patients on the proposed Similar Biologic drug, the number of patients in phase IV study can be modified accordingly so that the 40
41 safety data (from both Phase III and IV) is derived from a minimum of 300 patients treated with the Similar Biologics. 8.5 Extrapolation of Efficacy and Safety Data to Other Indications Extrapolation of the safety and efficacy data of a particular clinical indication (for which clinical studies has been done) of a Similar Biologic to other clinical indications may be possible if following conditions are met: Similarity with respect to quality has been proven to Reference Biologic Similarity with respect to preclinical assessment has been proven to Reference Biologic Clinical safety and efficacy is proven in one indication Mechanism of action is same for other clinical indications Involved receptor(s) are same for other clinical indications However, new indications not mentioned by innovator will needs to be covered by a separate application. 9. Data Requirements for Market Authorization Application The applicant should submit application for market authorization as per CDSCO guidance document for industry, For cases where commercial manufacturing is performed either at a different scale and/or with a different process as compared to that used for manufacturing phase III clinical trial batches, then information on 41
42 comparability of quality needs to be additionally submitted with appropriate justification and will be dealt with on a case to case basis. The package insert of the Similar Biologic will be a combination of data and information from both clinical studies and post marketing experience with the reference product and the Biosimilar product studies. 10. Post Market Data for Similar Biologic Although Similar Biologics are not new drug products and their risk is expected to be Similar to the Reference Biologic, since a Similar Biologic is authorized based on a reduced preclinical and clinical data package, it is important to establish a formal Risk Management Plan to monitor and detect both known inherent safety concerns and potential unknown safety signals that may arise from the Similar Biologic. The risk management plan should consist of the following: 10.1 Pharmacovigilance Plan The clinical studies done on Similar Biologic prior to market authorization are limited in nature so the rare adverse events are unlikely to be encountered. Hence comprehensive pharmacovigilance plan should be prepared by manufacturer to further evaluate the clinical safety in all the approved indications in the post marketing phase. The pharmacovigilance plan should include the submission of periodic safety update reports (PSURs). The PSURs shall be submitted every six months for the first two years after approval of the Similar Biologic is granted to 42
43 the applicant. For subsequent two years the PSURs need to be submitted annually to DCGI office as per the Schedule Y Adverse Drug Reaction (ADR) Reporting All cases involving serious unexpected adverse reactions must be reported to the licensing authority within 15 days of initial receipt of the information by the applicant as per Schedule Y Post Marketing studies (Phase IV Study) Finally, in order to further reduce the residual risk of the Similar Biologic, additional safety data may need to be collected after market approval through a pre defined single arm study of generally, more than 200 evaluable patients and compared to historical data of the Reference product. The study should be completed preferably within 2 years of the marketing permission / manufacturing license unless otherwise justified. The primary aim of the post marketing phase IV study is safety and hence following parameters should be considered for the post marketing phase IV study protocol Primary endpoint: Safety Secondary endpoint: Efficacy and Immunogenicity 43
44 The phase IV protocol should be submitted along with marketing authorization application for approval. The clinical studies done on Similar Biologic prior to market authorization are limited in nature so post marketing studies should be conducted and the reports be submitted to DCGI. The plan of post market studies should be captured in Pharmacovigilance plan and update on the studies should be submitted to the CDSCO. Regarding post marketing safety and immunogenicity study at least one noncomparative post marketing clinical study with focus on safety and immunogenicity (on case by case basis) should be performed. This study must be designed to confirm that the Similar Biologic does not have any concerns with regards to the therapeutic consequences of unwanted immunogenicity. If immunogenicity is evaluated in clinical studies, it is not mandatory to carry out additional non comparative immunogenicity studies in post marketing studies. The immunogenicity of the Similar Biologic should be evaluated using appropriately designed studies with state of the art methods, taking into consideration the potential impact on both safety and efficacy. Rationale on the strategy for testing immunogenicity should be provided. 44
45 Assay methods should be validated and should be able to characterize antibody content (concentration or titer) as well as the type of antibodies formed. Of most concern are those antibodies that have potentially serious impact on safety and efficacy, such as neutralizing antibodies and antibodies with cross reactivity. When neutralizing antibodies are detected in patients in clinical studies (either pre approval clinical studies or post approval clinical studies), the impact of the antibodies on the PK/PD parameters of the Similar Biologic should be analyzed, where the data is available. Furthermore an assessment of the impact of the neutralizing antibodies and cross reacting antibodies (if applicable) on the overall safety and efficacy of the Similar Biologic should be conducted. Exceptions: In the case of Similar Biologic that can be evaluated for rare diseases, the clinical trial population size can be reduced as per the rarity and severity of the disease as well as the limitation of access to therapeutic options. 45
46 11. Application Forms Various application forms for submitting request to regulatory agencies are as under: Stage Agency Application Approval involved Manufacturing License for State FDA Form 30 Form 29 test, analysis and examination (After CDSCO NOC) Import license for test, CDSCO zonal Form 12 Form 11 analysis and examination Cell bank import / export / RCGM Form B1/B3/ transfer / received B5/B7 Carrying out Research and RCGM Form C1 Development Preclinical studies RCGM Form C3a permission Submission of Preclinical RCGM Form C5a study report Clinical Trial CDSCO Form 44 CT Permission letter Import /Manufacturing and CDSCO Form 44 Form 45A/ marketing permission (separate for 46A (bulk DS and DP) product and 46
47 Form 45/46 (Finished product) Manufacturing License State FDA/ Form 27 D Form 28 D CDSCO (counter signature) Registration certificate for CDSCO Form 40 Form 41 / import (with Form 45 schedule DI and DII) / Form 44 Marketing permission / CDSCO Form 8 & 9 Form 10 License for imported product The applicant should comply with the established pharmacopoeia requirements while testing the excipients and as well as Biological product for which monograph is available in Indian Pharmacopoeia. Refer D&C act for the application format. 47
48 12. Archiving of Data / Retention of Samples: The manufacturer should establish the SOP for data archival as well as sample retention. The applicant should archive all the data (quality, preclinical and clinical documentation) for a period of at least five years after marketing approval by competent authority in India. Important samples such as test substance, vehicle, plasma / serum, tissues, paraffin blocks, microscope slides, electronic material, etc., should be retained till the period of expiry. The designated authority, which will be responsible for archiving and can be approached for inspection or retrieval if required, should be indicated in the data archival and sample retention SOP. 13. Glossary The definitions given below apply to the terms used in this guideline. They may have different meanings in other contexts. a. Comparability exercise: Comparison of a Similar Biologic with a Reference Biologic with the goal to establish Similarity in safety, efficacy and quality. b. Drug: Drug includes (as defined in Drugs and Cosmetics Act, 1940). i. all medicines for internal or external use of human beings or animals and all substances intended to be used for or in the diagnosis, treatment, mitigation or prevention of any disease or disorder in 48
49 human beings or animals, including preparations applied on human body for the purpose of repelling insects like mosquitoes; ii. such substances (other than food) intended to affect the structure or any function of human body or intended to be used for the destruction of (vermin) or insects which cause disease in human beings or animals, as may be specified from time to time by the Central Government by notification in the Official Gazette; iii. All substances intended for use as components of a drug including empty gelatine capsules; and iv. Such devices intended for internal or external use in the diagnosis, treatment, mitigation or prevention of disease or disorder in human beings or animals, as may be specified from time to time by the Central Government by notification in the Official Gazette, after consultation with the Board. c. Drug substance Any substance or mixture of substances intended to be used in the manufacture of a drug (medicinal) product and that, when used in the production of a drug, becomes an active ingredient of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, 49
50 mitigation, treatment, or prevention of disease or to affect the structure and function of the body. d. Drug product The dosage form in the final immediate packaging intended for marketing. A pharmaceutical product type that contains a drug substance, generally in association with excipients. e. Genetic engineering The technique by which heritable material, which does not usually occur or will not occur naturally in the organism or cell concerned, generated outside the organism or the cell is inserted into said cell or organism. It shall also mean the formation of new combinations of genetic material by incorporation of a cell into a host cell, where they occur naturally (self cloning) as well as modification of an organism or in a cell by deletion and removal of parts of the heritable material (Rules, 1989). f. Immunogenicity The ability of a substance to trigger an immune response or reaction (e.g., development of specific antibodies, T cell response, allergic or anaphylactic reaction). g. Impurity Any component present in the drug substance or drug product that is not the desired product, a product related substance, or excipient including buffer components. It may be either process or product related. 50
51 h. Manufacture Manufacture in relation to any drug includes any process or part of a process for producing, altering, ornamenting, finishing, packing, labelling, breaking up or otherwise treating or adopting any drug with a view to its sale or distribution but does not include the compounding or dispensing in the ordinary course of retail business; and to manufacture shall be construed accordingly. i. Reference Biologic A Reference Biologic is used as the comparator for comparability studies with the Similar Biologic in order to show Similarity in terms of safety, efficacy and quality. The Reference Biologic should be licensed / approved in India or ICH countries and should be the innovator s product. The Reference Biologic should be licensed based on a full safety, efficacy and quality data. Therefore another Similar Biologic cannot be considered as a choice for Reference Biologic. j. Pharmacovigilance The science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug related problems. k. Similar Absence of a relevant difference in the parameter of interest. l. Similar Biologic A Similar Biologic product is that which is similar in terms of quality, safety and efficacy to an approved Reference Biological product based on comparability. 51
52 14. References I. EMA guideline on Similar Biological medicinal products, London, 2014 (CHMP/437/04 Rev 1) II. III. IV. EMA Guideline on Similar Biological medicinal products containing biotechnology derived proteins as active substance: non clinical and clinical issues, London, 2014 (EMEA/CHMP/BMWP/42832/2005 Rev1) EMA guideline on Similar Biological medicinal products containing biotechnology derived proteins as active substance: non clinical and clinical issues. London, 2006 (CHMP/BMWP/42832) EMA guideline on immunogenicity assessment of biotechnology derived therapeutic proteins London, 2007 (CHMP/BMWP/14327) V. ICH guideline on preclinical safety evaluation of biotechnology derived pharmaceuticals (S6), 1997 and addendum, 2011 VI. Guideline for Safety Study of Biological Products, (KFDA, 2010) VII. World Health Organization (WHO) Guidelines on Evaluation of Similar Bio therapeutic Products (SBP), 2009 VIII. IX. World Health Organization (WHO), Guidelines on the quality, safety and efficacy of bio therapeutic protein products prepared by recombinant DNA technology, EMA DNA and Host cell protein impurities routine testing versus validation studies, 1997 X. ICH Q1 A(R2) Stability Testing of New Drug Substances and Products,

53 Annexure 1 53
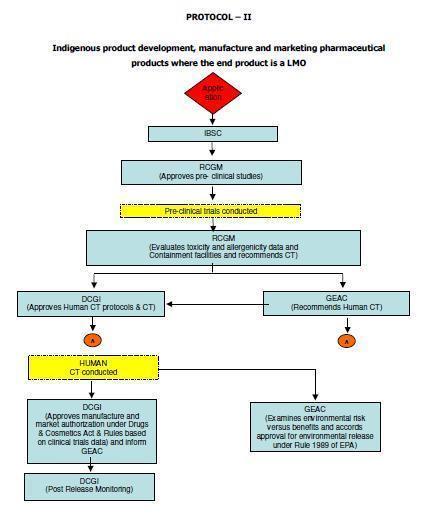
54 54
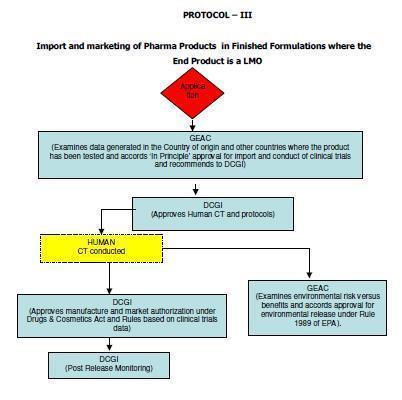
55 55
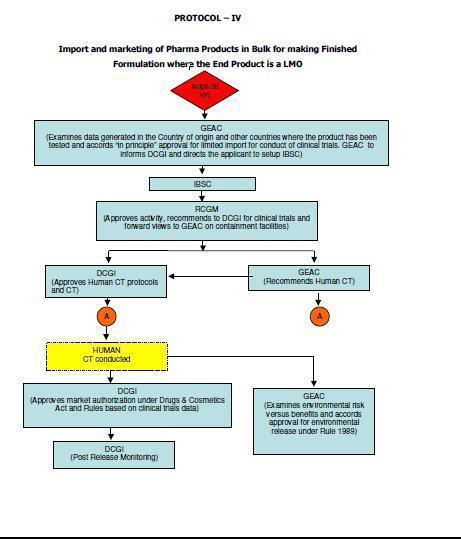
56 56
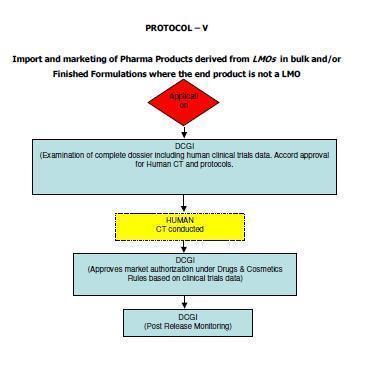
57 57
Guideline for the Quality, Safety, and Efficacy Assurance of Follow-on Biologics
 Provisional Translation (as of April 19, 2013) PFSB/ELD Notification No. 0304007 March 4, 2009 To: Prefectural Health Department (Bureau) From: Evaluation and Licensing Division, Pharmaceutical and Food
Provisional Translation (as of April 19, 2013) PFSB/ELD Notification No. 0304007 March 4, 2009 To: Prefectural Health Department (Bureau) From: Evaluation and Licensing Division, Pharmaceutical and Food
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR
European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/42832/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR
European Medicines Agency Evaluation of Medicines for Human Use
 European Medicines Agency Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BWP/49348/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL
European Medicines Agency Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BWP/49348/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/94528/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) ANNEX TO GUIDELINE
European Medicines Agency Pre-authorisation Evaluation of Medicines for Human Use London, 22 February 2006 EMEA/CHMP/BMWP/94528/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) ANNEX TO GUIDELINE
Paradigm Shift in Comparability Assessment:
 Paradigm Shift in Comparability Assessment: How Quality by Design (QbD) and Process Analytical Technology (PAT) can improve Structure-Activity Relationship (SAR) evaluation and its relevance to comparability
Paradigm Shift in Comparability Assessment: How Quality by Design (QbD) and Process Analytical Technology (PAT) can improve Structure-Activity Relationship (SAR) evaluation and its relevance to comparability
FDA Public Hearing: Approval Pathway for Biosimilar. Products. November 2-3, 2010
 FDA Public Hearing: Approval Pathway for Biosimilar and Interchangeable Biological Products November 2-3, 2010 1 The Biotechnology Industry Organization Over 1,100 members, including biotechnology companies,
FDA Public Hearing: Approval Pathway for Biosimilar and Interchangeable Biological Products November 2-3, 2010 1 The Biotechnology Industry Organization Over 1,100 members, including biotechnology companies,
Guidance for Industry
 Reprinted from FDA s website by Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product DRAFT GUIDANCE This guidance document is being distributed for comment
Reprinted from FDA s website by Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product DRAFT GUIDANCE This guidance document is being distributed for comment
GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs)
") GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs) DRAFT GUIDANCE This guidance document is for feedback purposes only Comments and suggestions regarding this draft document should be submitted within
GUIDANCE FOR INDUSTRY ON FIXED DOSE COMBINATIONS (FDCs) DRAFT GUIDANCE This guidance document is for feedback purposes only Comments and suggestions regarding this draft document should be submitted within
KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar
 KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar Kyung-Min Baek, Ph.D. Recombinant Protein Products Division Korea Food and Drug Administration(KFDA) Biopharmaceuticals A biopharmaceutical
KFDA Regulatory Framework on Biopharmaceuticals - Focus on Biosimilar Kyung-Min Baek, Ph.D. Recombinant Protein Products Division Korea Food and Drug Administration(KFDA) Biopharmaceuticals A biopharmaceutical
COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP)
") European Medicines Agency Evaluation of Medicines for Human Use London, 16 June 2005 EMEA/CHMP/94526/2005 COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP) ANNEX GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL PRODUCTS
European Medicines Agency Evaluation of Medicines for Human Use London, 16 June 2005 EMEA/CHMP/94526/2005 COMMITTEE FOR HUMAN MEDICINAL PRODUCTS (CHMP) ANNEX GUIDELINE ON SIMILAR BIOLOGICAL MEDICINAL PRODUCTS
WHO/SBP_Q&A/DRAFT/DEC 2017 ENGLISH ONLY
 0 0 0 0 WHO/SBP_Q&A/DRAFT/DEC 0 ENGLISH ONLY WHO Questions and Answers: Similar Biotherapeutic Products (Proposed document to implement the WHO guidelines on evaluation of similar biotherapeutic products,
0 0 0 0 WHO/SBP_Q&A/DRAFT/DEC 0 ENGLISH ONLY WHO Questions and Answers: Similar Biotherapeutic Products (Proposed document to implement the WHO guidelines on evaluation of similar biotherapeutic products,
Guidelines on procedures and data requirements for changes to approved biotherapeutic products
 ENGLISH ONLY EXPERT COMMITTEE ON BIOLOGICAL STANDARDIZATION Geneva, 17 to 20 October 2017 Guidelines on procedures and data requirements for changes to approved biotherapeutic products World Health Organization
ENGLISH ONLY EXPERT COMMITTEE ON BIOLOGICAL STANDARDIZATION Geneva, 17 to 20 October 2017 Guidelines on procedures and data requirements for changes to approved biotherapeutic products World Health Organization
Guidelines on procedures and data requirements for changes to approved biotherapeutic products. Proposed guidelines
 0 0 0 0 WHO/PAC for BTPs_DRAFT/ Oct 0 ENGLISH ONLY Guidelines on procedures and data requirements for changes to approved biotherapeutic products Proposed guidelines NOTE: This document has been prepared
0 0 0 0 WHO/PAC for BTPs_DRAFT/ Oct 0 ENGLISH ONLY Guidelines on procedures and data requirements for changes to approved biotherapeutic products Proposed guidelines NOTE: This document has been prepared
Stability of Biological Products
 Stability of Biological Products Dr Jurgen Lindner Principal, BioPharma Consulting & Executive Secretary, APIMAA Biological Products Functional Proteins or Polypeptides (mab s, enzymes & inhibitors, growth
Stability of Biological Products Dr Jurgen Lindner Principal, BioPharma Consulting & Executive Secretary, APIMAA Biological Products Functional Proteins or Polypeptides (mab s, enzymes & inhibitors, growth
Preparing the CMC section of IMPD for biological/biotechnology derived substances
 Preparing the CMC section of IMPD for biological/biotechnology derived substances Your Logo Dr. Una Moore Health Products Regulatory Authority, Ireland Presented by Una Moore on 16 th April 2014. Health
Preparing the CMC section of IMPD for biological/biotechnology derived substances Your Logo Dr. Una Moore Health Products Regulatory Authority, Ireland Presented by Una Moore on 16 th April 2014. Health
Comparability to establish Biosimilarity
 Comparability to establish Biosimilarity CMC Strategy Forum Europe 2014, Sorrento, Italy Jan Visser, Head Global Analytical Characterization & Bioanalytics Sandoz Biopharmaceuticals, Hexal AG, Germany
Comparability to establish Biosimilarity CMC Strategy Forum Europe 2014, Sorrento, Italy Jan Visser, Head Global Analytical Characterization & Bioanalytics Sandoz Biopharmaceuticals, Hexal AG, Germany
Baek, Kyung-min. Recombinant Protein Products Division. Ministry of Food and Drug Safety
 Baek, Kyung-min Recombinant Protein Products Division Ministry of Food and Drug Safety About Ministry of Food and Drug Safety Regulation for Biosimilar Principle of Biosimilar Approach Status of Biosimilar
Baek, Kyung-min Recombinant Protein Products Division Ministry of Food and Drug Safety About Ministry of Food and Drug Safety Regulation for Biosimilar Principle of Biosimilar Approach Status of Biosimilar
Structure and content of an IMPD. What is required for first into man trial?
 What is required for first into man? The EU IMPD Thomas Sudhop, MD Scope Structure and content of an IMPD What is required for first into man trial? Only for IMPs that do not have a marketing authorisation
What is required for first into man? The EU IMPD Thomas Sudhop, MD Scope Structure and content of an IMPD What is required for first into man trial? Only for IMPs that do not have a marketing authorisation
Quality, Safety and Efficacy of Follow-on Biologics
 Quality, Safety and Efficacy of Follow-on Biologics Teruhide YAMAGUCHI Division of Biological Chemistry and Biologicals National Institute of Health Sciences 2009.9.28 London Quality, Safety and Efficacy
Quality, Safety and Efficacy of Follow-on Biologics Teruhide YAMAGUCHI Division of Biological Chemistry and Biologicals National Institute of Health Sciences 2009.9.28 London Quality, Safety and Efficacy
Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective
 Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective Patrick G. Swann, Ph.D. Deputy Director Division of Monoclonal Antibodies Office of Biotechnology Products Office of Pharmaceutical
Critical Quality Attributes for Biotechnology Products: A Regulatory Perspective Patrick G. Swann, Ph.D. Deputy Director Division of Monoclonal Antibodies Office of Biotechnology Products Office of Pharmaceutical
Development of non-substantially manipulated cell-based ATMPs 1 : flexibility introduced via the application of the risk-based approach
 3 July 2017 EMA/CAT/216556/2017 Inspections, Human Medicines, Pharmacovigilance and Committees Division Development of non-substantially manipulated cell-based ATMPs 1 : flexibility introduced via the
3 July 2017 EMA/CAT/216556/2017 Inspections, Human Medicines, Pharmacovigilance and Committees Division Development of non-substantially manipulated cell-based ATMPs 1 : flexibility introduced via the
Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials
 14 September 2017 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) Guideline on the requirements for quality documentation concerning biological investigational medicinal
14 September 2017 EMA/CHMP/BWP/534898/2008 rev. 1 Committee for Medicinal Products for Human Use (CHMP) Guideline on the requirements for quality documentation concerning biological investigational medicinal
S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers
 S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers Current version dated 8 June 2016 International Council for Harmonisation of
S9 Implementation Working Group ICH S9 Guideline: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers Current version dated 8 June 2016 International Council for Harmonisation of
Expectations for Analytical Characterisation in the Evaluation of Biosimilarity: A Regulator`s Perspective
 Expectations for Analytical Characterisation in the Evaluation of Biosimilarity: A Regulator`s Perspective Christian Mayer AGES - Austrian Agency for Health and Food Safety Analytical Technologies Europe
Expectations for Analytical Characterisation in the Evaluation of Biosimilarity: A Regulator`s Perspective Christian Mayer AGES - Austrian Agency for Health and Food Safety Analytical Technologies Europe
Copyright. Jeremiah J. Kelly (2015). All rights reserved. Further dissemination without express written consent strictly prohibited.
. All rights reserved. Further dissemination without express written consent strictly prohibited.") Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
Statutory Framework for Biologics Drugs Investigational Use Application IND Pre-Market Approval Applications 505(b)(1) NDA 505(b)(2) NDA 505(j) ANDA Over-the-Counter (OTC) Non- Rx Drugs Monograph Biologics
Documentation requirements for an initial consultation
 Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
Language : French or English Documentation requirements for an initial consultation Because of the wide range of medical devices which incorporate, as an integral part, an ancillary medicinal substance,
Guidance for Industry
 Guidance for Industry Comparability Protocols - Protein Drug Products and Biological Products - Chemistry, Manufacturing, and Controls Information DRAFT GUIDANCE This guidance document is being distributed
Guidance for Industry Comparability Protocols - Protein Drug Products and Biological Products - Chemistry, Manufacturing, and Controls Information DRAFT GUIDANCE This guidance document is being distributed
Biosimilars in the EU
 Biosimilars in the EU Information guide for healthcare professionals Prepared jointly by the European Medicines Agency and the European Commission Table of contents Foreword 2 Summary 3 Biological medicines:
Biosimilars in the EU Information guide for healthcare professionals Prepared jointly by the European Medicines Agency and the European Commission Table of contents Foreword 2 Summary 3 Biological medicines:
Injectable modified release products
 Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CPMP/EWP/280/96 Corr1) Injectable modified release products Dr Sotiris Michaleas, National Expert for the
Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CPMP/EWP/280/96 Corr1) Injectable modified release products Dr Sotiris Michaleas, National Expert for the
Guideline on requirements for the production and control of immunological veterinary medicinal products
 14 June 2012 EMA/CVMP/IWP/206555/2010 Committee for Medicinal Products for Veterinary Use (CVMP) Guideline on requirements for the production and control of immunological veterinary Draft agreed by Immunologicals
14 June 2012 EMA/CVMP/IWP/206555/2010 Committee for Medicinal Products for Veterinary Use (CVMP) Guideline on requirements for the production and control of immunological veterinary Draft agreed by Immunologicals
Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products
 Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Table of contents 1.Introduction...2 2.Scope...3 3.Discussion...4 3.1. Chemistry, manufacturing, and controls...4
Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Table of contents 1.Introduction...2 2.Scope...3 3.Discussion...4 3.1. Chemistry, manufacturing, and controls...4
MEDICINES CONTROL COUNCIL
 MEDICINES CONTROL COUNCIL FIXED DOSE COMBINATION PRODUCTS (FDC Products) FOR HIV/AIDS, TUBERCULOSIS, and MALARIA This guideline is intended to provide recommendations to applicants wishing to submit applications
MEDICINES CONTROL COUNCIL FIXED DOSE COMBINATION PRODUCTS (FDC Products) FOR HIV/AIDS, TUBERCULOSIS, and MALARIA This guideline is intended to provide recommendations to applicants wishing to submit applications
参考資料. Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products. Draft
 参考資料 1 2 3 4 5 Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Draft 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 Table of contents 1.
参考資料 1 2 3 4 5 Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products Draft 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 Table of contents 1.
ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological entities)
") May 2011 EMA/CHMP/ICH/425213/2011 ICH/ Committee for medicinal products for human use (CHMP) ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological
May 2011 EMA/CHMP/ICH/425213/2011 ICH/ Committee for medicinal products for human use (CHMP) ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/biological
Guide to the investigational medicinal product dossier
 Guide to the investigational medicinal product dossier Item type Authors Publisher Other Irish Medicines Board (IMB) Irish Medicines Board (IMB) Downloaded 1-May-2018 07:31:12 Link to item http://hdl.handle.net/10147/96980
Guide to the investigational medicinal product dossier Item type Authors Publisher Other Irish Medicines Board (IMB) Irish Medicines Board (IMB) Downloaded 1-May-2018 07:31:12 Link to item http://hdl.handle.net/10147/96980
Guidance for Industry
 Guidance for Industry Submission of Clinical Trial Application for Evaluating Safety and Efficacy Requirements for permission of New Drugs Approval Post approval changes in biological products: Quality
Guidance for Industry Submission of Clinical Trial Application for Evaluating Safety and Efficacy Requirements for permission of New Drugs Approval Post approval changes in biological products: Quality
Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd
![Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd](/thumbs/77/76275664.jpg "Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations. [mabs -monoclonal antibodies ] Peter Lloyd") Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations [mabs -monoclonal antibodies ] Peter Lloyd Head of Pharmacokinetics-Pharmacodynamics Novartis Biologics
Unique PK-PD properties of biotechnology-based therapeutics [mabs] and First In Human dose considerations [mabs -monoclonal antibodies ] Peter Lloyd Head of Pharmacokinetics-Pharmacodynamics Novartis Biologics
GUIDELINES ON SIMILAR BIOLOGICS Regulatory Requirements for Marketing Authorization in India
 Government of India Department of Biotechnology Ministry of Science & Technology Central Drugs Standard Control Organiza on Ministry of Health & Family Welfare GUIDELINES ON SIMILAR BIOLOGICS Regulatory
Government of India Department of Biotechnology Ministry of Science & Technology Central Drugs Standard Control Organiza on Ministry of Health & Family Welfare GUIDELINES ON SIMILAR BIOLOGICS Regulatory
The rules governing medicinal products in the European Union. Presentation and content of the dossier Edition
 The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
CENTRAL DRUGS STANDARD CONTROL ORGANIZATION DIRECTORATE GENERAL OF HEALTH SERVICES MINISTRY OF HEALTH AND FAMILY WELFARE GOVERMENT OF INDIA
 1 GUIDANCE DOCUMENT FOR BA/BE NOC FOR EXPORT CENTRAL DRUGS STANDARD CONTROL ORGANIZATION DIRECTORATE GENERAL OF HEALTH SERVICES MINISTRY OF HEALTH AND FAMILY WELFARE GOVERMENT OF INDIA GUIDANCE DOCUMENT
1 GUIDANCE DOCUMENT FOR BA/BE NOC FOR EXPORT CENTRAL DRUGS STANDARD CONTROL ORGANIZATION DIRECTORATE GENERAL OF HEALTH SERVICES MINISTRY OF HEALTH AND FAMILY WELFARE GOVERMENT OF INDIA GUIDANCE DOCUMENT
A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999
 EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Pharmaceuticals and cosmetics Final Revision 0 NOTICE TO APPLICANTS A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999 This guideline
EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Pharmaceuticals and cosmetics Final Revision 0 NOTICE TO APPLICANTS A GUIDELINE ON DOSSIER REQUIREMENTS FOR TYPE I VARIATIONS November 1999 This guideline
Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory Challenges
 The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
The content of this presentation reflects the opinion of the speaker and does not necessarily represent the official position of CDER Oncology Biopharmaceuticals and Preclinical Development: Evolving Regulatory
COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE 1 : DNA VACCINES NON-AMPLIFIABLE IN EUKARYOTIC CELLS FOR VETERINARY USE
 The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Veterinary Use CVMP/IWP/07/98-FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE 1 : DNA VACCINES
The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Veterinary Use CVMP/IWP/07/98-FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS NOTE FOR GUIDANCE 1 : DNA VACCINES
Annex 4. Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology
 Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology Replacement of Annex 3 of WHO Technical Report Series, No. 814 Introduction 177
Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology Replacement of Annex 3 of WHO Technical Report Series, No. 814 Introduction 177
Identification of Critical Product Quality Attributes: Impact of Product Variants on Safety and Efficacy
 Identification of Critical Product Quality Attributes: Impact of Product Variants on Safety and Efficacy Ziping Wei, Ph.D. Analytical Biochemistry MedImmune January 25, 2010 Outline Why do we need to identify
Identification of Critical Product Quality Attributes: Impact of Product Variants on Safety and Efficacy Ziping Wei, Ph.D. Analytical Biochemistry MedImmune January 25, 2010 Outline Why do we need to identify
1. Checklist for Grant of permission to manufacture/import of Bulk Drug already approved in the country
 1. Checklist for Grant of permission to manufacture/import of Bulk Drug already approved in the country S no 1. Name of Applicant with address 2. Name of Drug 3. Therapeutic Class 4. Date of Approval Documents
1. Checklist for Grant of permission to manufacture/import of Bulk Drug already approved in the country S no 1. Name of Applicant with address 2. Name of Drug 3. Therapeutic Class 4. Date of Approval Documents
Joint MHLW/EMA reflection paper on the development of block copolymer micelle medicinal products
 1 2 3 17 January 2013 EMA/CHMP/13099/2013 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Joint MHLW/EMA reflection paper on the development of block copolymer micelle Agreed by Nanomedicines
1 2 3 17 January 2013 EMA/CHMP/13099/2013 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Joint MHLW/EMA reflection paper on the development of block copolymer micelle Agreed by Nanomedicines
BIOPHARMACEUTICAL PROCESS EVALUATED FOR VIRAL CLEARANCE
 The purpose of Viral Clearance evaluation is to assess the capability of a manufacturing production process to inactivate and/or remove potential viral contaminants. Experience and knowledge in selecting
The purpose of Viral Clearance evaluation is to assess the capability of a manufacturing production process to inactivate and/or remove potential viral contaminants. Experience and knowledge in selecting
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS OF MEDICINAL PRODUCTS
 GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS OF MEDICINAL PRODUCTS") European Medicines Agency London, 24 January 2008 Doc. Ref. EMEA/CHMP/SWP/258498/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS
European Medicines Agency London, 24 January 2008 Doc. Ref. EMEA/CHMP/SWP/258498/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON THE NON-CLINICAL DEVELOPMENT OF FIXED COMBINATIONS
ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
 European Medicines Agency August 2003 CPMP/ICH/4680/02 ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
European Medicines Agency August 2003 CPMP/ICH/4680/02 ICH Topic M 4 Q Location issues for Common Technical Document for the Registration of Pharmaceuticals for Human Use Quality Questions and Answers
Vladimir Hanes, MD, USA
 GaBI Educational Workshops 20 January 2015, Sheraton Maria Isabel Hotel & Towers, Mexico City, Mexico Vladimir Hanes, MD, USA Medical Director, Oncology Global Biosimilars Development, Amgen Inc GaBI Educational
GaBI Educational Workshops 20 January 2015, Sheraton Maria Isabel Hotel & Towers, Mexico City, Mexico Vladimir Hanes, MD, USA Medical Director, Oncology Global Biosimilars Development, Amgen Inc GaBI Educational
Biosimilars Scientific Challenges and Implications
 Biosimilars Scientific Challenges and Implications Professor Paul Declerck Laboratory for Therapeutic and Diagnostic Antibodies paul.declerck@pharm.kuleuven.be Biological medicinal product A well-defined
Biosimilars Scientific Challenges and Implications Professor Paul Declerck Laboratory for Therapeutic and Diagnostic Antibodies paul.declerck@pharm.kuleuven.be Biological medicinal product A well-defined
Non-clinical Assessment Requirements
 Non-clinical Assessment Requirements Presented by: Maria Nieto-Gutierrez Safety and Efficacy of Medicines/Human Medicines Development and Evaluation An agency of the European Union Contents: Relevance
Non-clinical Assessment Requirements Presented by: Maria Nieto-Gutierrez Safety and Efficacy of Medicines/Human Medicines Development and Evaluation An agency of the European Union Contents: Relevance
The Role of Mass Spectrometry for Developing Biotherapeutics: Regulatory Perspectives
 The Role of Mass Spectrometry for Developing Biotherapeutics: Regulatory Perspectives Jun Park, Ph.D. Division of Monoclonal Antibodies Office of Biotechnology Products CDER/FDA CASSS, Applications of
The Role of Mass Spectrometry for Developing Biotherapeutics: Regulatory Perspectives Jun Park, Ph.D. Division of Monoclonal Antibodies Office of Biotechnology Products CDER/FDA CASSS, Applications of
Considerations in Setting Specifications
 EBE Concept Paper Considerations in Setting Specifications March 28, 2013 European Biopharmaceutical Enterprises (EBE), a specialised group of EFPIA Leopold Plaza Building Rue du Trône 108 BE-1050 Brussels
EBE Concept Paper Considerations in Setting Specifications March 28, 2013 European Biopharmaceutical Enterprises (EBE), a specialised group of EFPIA Leopold Plaza Building Rue du Trône 108 BE-1050 Brussels
Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for an orphan designation
 2 March 2010 EMA/COMP/15893/2009 Final Committee for Orphan Medicinal Products (COMP) Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for
2 March 2010 EMA/COMP/15893/2009 Final Committee for Orphan Medicinal Products (COMP) Recommendation on elements required to support the medical plausibility and the assumption of significant benefit for
Official Letter from the DOH
 Issued Date 2009/04/02 Issued by DOH Ref. No 0980316268 RE The DOH issued an official letter to announce the implementation of the Guideline for BA/BE Studies on April 2, 2009 (Ref. No. 0980316265). Please
Issued Date 2009/04/02 Issued by DOH Ref. No 0980316268 RE The DOH issued an official letter to announce the implementation of the Guideline for BA/BE Studies on April 2, 2009 (Ref. No. 0980316265). Please
Reflection Paper. The Role of Product-specific Monographs for Biotherapeutic Products in Pharmacopoeias
 Reflection Paper 3 October 2014 The Role of Product-specific Monographs for Biotherapeutic Products in Pharmacopoeias This reflection paper describes a scientific and regulatory rationale for a new format
Reflection Paper 3 October 2014 The Role of Product-specific Monographs for Biotherapeutic Products in Pharmacopoeias This reflection paper describes a scientific and regulatory rationale for a new format
Beth Hutchins, PhD PhRMA ICH Gene Therapy Discussion Group
 ICH Considerations on General Principles to Address the Risk of Inadvertent Germline Integration of Gene Therapy Vectors and Current Topics on Gene Therapy in USA Beth Hutchins, PhD PhRMA ICH Gene Therapy
ICH Considerations on General Principles to Address the Risk of Inadvertent Germline Integration of Gene Therapy Vectors and Current Topics on Gene Therapy in USA Beth Hutchins, PhD PhRMA ICH Gene Therapy
Preclinical Development Drugs. Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015
 Preclinical Development Drugs Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015 Product Development: Development process: File Approval Drug Discovery Preclinical Phase 1 Phase 2 Phase 3 Lifecycle
Preclinical Development Drugs Darrin Cowley PhD Executive Director Amgen BioBoot Camp 2015 Product Development: Development process: File Approval Drug Discovery Preclinical Phase 1 Phase 2 Phase 3 Lifecycle
Ph. Eur. monographs and biosimilars
 Ph. Eur. monographs and biosimilars Emmanuelle Charton, Ph. D. European Pharmacopoeia Department European Directorate for the Quality of Medicines & HealthCare 1 Place of the Ph. Eur. within the EU regulatory
Ph. Eur. monographs and biosimilars Emmanuelle Charton, Ph. D. European Pharmacopoeia Department European Directorate for the Quality of Medicines & HealthCare 1 Place of the Ph. Eur. within the EU regulatory
Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations. Audrey Chang, PhD, Senior Director Development Services
 Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations Audrey Chang, PhD, Senior Director Development Services Definition of Biologics: PHS Act, section 351 Virus, therapeutic
Overview of Biologics Testing and Evaluation: Regulatory Requirements and Expectations Audrey Chang, PhD, Senior Director Development Services Definition of Biologics: PHS Act, section 351 Virus, therapeutic
QUESTIONS AND ANSWERS TO ICH S3A: NOTE FOR GUIDANCE ON TOXICOKINETICS: THE ASSESSMENT OF SYSTEMIC EXPOSURE IN TOXICITY STUDIES FOCUS ON MICROSAMPLING
 INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE QUESTIONS AND ANSWERS TO ICH S3A: NOTE FOR GUIDANCE ON TOXICOKINETICS: THE ASSESSMENT
INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE QUESTIONS AND ANSWERS TO ICH S3A: NOTE FOR GUIDANCE ON TOXICOKINETICS: THE ASSESSMENT
Setting Specifications for Biotech Products
 Setting Specifications for Biotech Products Session 1: What to Control? Presentation by an EU Regulator Nanna Aaby Kruse, Senior Biological Assessor, member of BWP and BMWP WHAT TO CONTROL? Control of
Setting Specifications for Biotech Products Session 1: What to Control? Presentation by an EU Regulator Nanna Aaby Kruse, Senior Biological Assessor, member of BWP and BMWP WHAT TO CONTROL? Control of
SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007
 European Medicines Agency SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007 COMMENTS FROM The Biotechnology
European Medicines Agency SUBMISSION OF COMMENTS ON The Requirements for First-in-Human Clinical Trials for Potential High Risk Medicinal Products EMEA/CHMP/SWP/28367/2007 COMMENTS FROM The Biotechnology
Regulatory expectations on impurities in drug substances - Pavia, October 2, Luisa Torchio Euticals SpA
 Regulatory expectations on impurities in drug substances - Pavia, October 2, 2015 Luisa Torchio Euticals SpA An Impurity is defined as any substance or element present in a drug substance (DS) that is
Regulatory expectations on impurities in drug substances - Pavia, October 2, 2015 Luisa Torchio Euticals SpA An Impurity is defined as any substance or element present in a drug substance (DS) that is
CTA/NDA Regulatory Landscape in China. Jack Xie, PhD, DABT SOT 2016
 CTA/NDA Regulatory Landscape in China Jack Xie, PhD, DABT SOT 2016 Disclaimer The content of the following presentation represents solely author s view and may not reflect any position of Roche or China
CTA/NDA Regulatory Landscape in China Jack Xie, PhD, DABT SOT 2016 Disclaimer The content of the following presentation represents solely author s view and may not reflect any position of Roche or China
Biosimilar regulation in Republic of Korea and Asia-Pacific Economic Cooperation (APEC) developments
 developments") Biosimilar regulation in Republic of Korea and Asia-Pacific Economic Cooperation (APEC) developments Ministry of Food and Drug Safety Jeewon Joung Outline I. Korean Regulatory Framework for Biosimilar
Biosimilar regulation in Republic of Korea and Asia-Pacific Economic Cooperation (APEC) developments Ministry of Food and Drug Safety Jeewon Joung Outline I. Korean Regulatory Framework for Biosimilar
Guideline on similar biological medicinal products containing interferon beta
 1 2 3 15 December 2011 EMA/CHMP/BMWP/652000/2010 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Guideline on similar biological medicinal products containing interferon beta 7 Draft Draft
1 2 3 15 December 2011 EMA/CHMP/BMWP/652000/2010 Committee for Medicinal Products for Human Use (CHMP) 4 5 6 Guideline on similar biological medicinal products containing interferon beta 7 Draft Draft
Technical Challenges in the Development of Biosimilars. E. Morrey Atkinson, PhD Interphex May 1, 2012
 Technical Challenges in the Development of Biosimilars E. Morrey Atkinson, PhD Interphex May 1, 2012 FDA Guidance on Biosimilarity Guidance for Industry: Scientific Consideration in Demonstrating Biosimilarity
Technical Challenges in the Development of Biosimilars E. Morrey Atkinson, PhD Interphex May 1, 2012 FDA Guidance on Biosimilarity Guidance for Industry: Scientific Consideration in Demonstrating Biosimilarity
Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines
 Annex 2 Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines Introduction 61 Background 62 Scope 62 General considerations 63 Terminology 65 Part A. Manufacturing and quality
Annex 2 Guidelines on the nonclinical evaluation of vaccine adjuvants and adjuvanted vaccines Introduction 61 Background 62 Scope 62 General considerations 63 Terminology 65 Part A. Manufacturing and quality
Republic of the Philippines Department of Health OFFICE OF THE SECRETARY M a n i l a
 Republic of the Philippines Department of Health OFFICE OF THE SECRETARY M a n i l a March 15, 1989 ADMINISTRATIVE ORDER No. 67 s. 1989 SUBJECT: Revised Rules and Regulations on Registration of Pharmaceutical
Republic of the Philippines Department of Health OFFICE OF THE SECRETARY M a n i l a March 15, 1989 ADMINISTRATIVE ORDER No. 67 s. 1989 SUBJECT: Revised Rules and Regulations on Registration of Pharmaceutical
IMPURITIES IN NEW DRUG PRODUCTS
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED TRIPARTITE GUIDELINE IMPURITIES IN NEW DRUG PRODUCTS Recommended for
Regulations & Guidelines Specific to Ethics Schedule Y & CDSCO-GCP
 Regulations & Guidelines Specific to Ethics Schedule Y & CDSCO-GCP Dr. K. Bangarurajan M Pharm, PhD Deputy Drugs Controller (India) CDSCO West Zone Outline Licensing Authority India - Clinical Trial: CDSCO
Regulations & Guidelines Specific to Ethics Schedule Y & CDSCO-GCP Dr. K. Bangarurajan M Pharm, PhD Deputy Drugs Controller (India) CDSCO West Zone Outline Licensing Authority India - Clinical Trial: CDSCO
Pharmacology. Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University.
 Pharmacology Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University. 1 PHARMACODYNAMIC STUDIES A. Primary pharmacodynamics primary action in target
Pharmacology Chatchai Chinpaisal, Ph.D. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Silpakorn University. 1 PHARMACODYNAMIC STUDIES A. Primary pharmacodynamics primary action in target
Examples of regulatory expectations for analytical characterization and testing
 Examples of regulatory expectations for analytical characterization and testing AT Europe 2016, 18 March 2016 Vicki Venizelos Quality RA B.V. Leiden, the Netherlands Overview What are the regulatory expectations?
Examples of regulatory expectations for analytical characterization and testing AT Europe 2016, 18 March 2016 Vicki Venizelos Quality RA B.V. Leiden, the Netherlands Overview What are the regulatory expectations?
Guide. recombinant DNA proteins. for the elaboration of monographs on synthetic peptides and. European Pharmacopoeia
 Guide for the elaboration of monographs on synthetic peptides and recombinant DNA proteins European Pharmacopoeia European Directorate for the Quality of Medicines & HealthCare Edition Council of Europe,
Guide for the elaboration of monographs on synthetic peptides and recombinant DNA proteins European Pharmacopoeia European Directorate for the Quality of Medicines & HealthCare Edition Council of Europe,
Characterization of Biotechnology Products: A Regulatory Perspective
 Characterization of Biotechnology Products: A Regulatory Perspective Laurie Graham Acting Team Leader FDA/CDER/OPS/OBP Division of Monoclonal Antibodies WCBP 2013 1 Disclaimer The views and opinions expressed
Characterization of Biotechnology Products: A Regulatory Perspective Laurie Graham Acting Team Leader FDA/CDER/OPS/OBP Division of Monoclonal Antibodies WCBP 2013 1 Disclaimer The views and opinions expressed
AN UPDATE ON INDIAN BIOSAFETY REGULATORY SYSTEM. Dr. Vibha Ahuja Biotech Consortium India Limited
 AN UPDATE ON INDIAN BIOSAFETY REGULATORY SYSTEM Dr. Vibha Ahuja Biotech Consortium India Limited STRUCTURE OF PRESENTATION 1. Existing regulatory framework in India 2. New Acts/policies dealing with GM
AN UPDATE ON INDIAN BIOSAFETY REGULATORY SYSTEM Dr. Vibha Ahuja Biotech Consortium India Limited STRUCTURE OF PRESENTATION 1. Existing regulatory framework in India 2. New Acts/policies dealing with GM
TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE MANAGEMENT Q12
 INTERNATIONAL CONCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE
INTERNATIONAL CONCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
Revised Immunogenicity Guideline: Assays and methods- Presentation of the draft guideline and introduction of the topics for discussion
 Revised Immunogenicity Guideline: Assays and methods- Presentation of the draft guideline and introduction of the topics for discussion Robin Thorpe & Meenu Wadhwa Revised Guideline: Differences from original
Revised Immunogenicity Guideline: Assays and methods- Presentation of the draft guideline and introduction of the topics for discussion Robin Thorpe & Meenu Wadhwa Revised Guideline: Differences from original
Content Areas of the Pharmacy Curriculum Outcomes Assessment (PCOA )
") Content Areas of the Pharmacy Curriculum Outcomes Assessment (PCOA ) Area 1.0 Basic Biomedical Sciences (10%) Area 2.0 Pharmaceutical Sciences (33%) Area 3.0 Social/Behavioral/Administrative Sciences (22%)
Content Areas of the Pharmacy Curriculum Outcomes Assessment (PCOA ) Area 1.0 Basic Biomedical Sciences (10%) Area 2.0 Pharmaceutical Sciences (33%) Area 3.0 Social/Behavioral/Administrative Sciences (22%)
Current Trends and Future of Biosimilars
 Current Trends and Future of Biosimilars Jian Wang, MD, PhD Chief, Clinical Evaluation Division Biologics and Genetic Therapies Directorate Health Canada Global Bio Conference 2017 June 28-30, 2017 Seoul,
Current Trends and Future of Biosimilars Jian Wang, MD, PhD Chief, Clinical Evaluation Division Biologics and Genetic Therapies Directorate Health Canada Global Bio Conference 2017 June 28-30, 2017 Seoul,
Current Regulatory Thinking The Draft Reflection Paper On Intravenous Liposomal Product Quality Issues
 Current Regulatory Thinking The Draft Reflection Paper On Intravenous Liposomal Product Quality Issues Dr. René Thürmer BfArM - and dmedical ldevices AGAH Workshop on Liposomal Formulations Bonn / 21.
Current Regulatory Thinking The Draft Reflection Paper On Intravenous Liposomal Product Quality Issues Dr. René Thürmer BfArM - and dmedical ldevices AGAH Workshop on Liposomal Formulations Bonn / 21.
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX
 INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX BATCH Q: Quality Q1A(R2) Stability Testing of New Drug Substances
INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE GUIDELINES INDEX BATCH Q: Quality Q1A(R2) Stability Testing of New Drug Substances
National Institute of Immunology, New Delhi
 National Institute of Immunology, New Delhi QUOTATION INVITATION NOTICE For Website Ref: NII/AKS/GPL/2013 Date: 25/12/2013 Quotations are invited from technical firms specializing in either both or one
National Institute of Immunology, New Delhi QUOTATION INVITATION NOTICE For Website Ref: NII/AKS/GPL/2013 Date: 25/12/2013 Quotations are invited from technical firms specializing in either both or one
Non-clinical documentation Overview of Requirements
 3 rd EMEA-. Non-clinical Aspects Non-clinical documentation Overview of Requirements EMEA Pre-Authorisation Human Unit 3 rd EMEA-. Non-clinical Aspects Outline Overview of Legal and Regulatory requirements
3 rd EMEA-. Non-clinical Aspects Non-clinical documentation Overview of Requirements EMEA Pre-Authorisation Human Unit 3 rd EMEA-. Non-clinical Aspects Outline Overview of Legal and Regulatory requirements
PHYSICO-CHEMICAL, BIOLOGICAL OR MICROBIOLOGICAL TESTS OF MEDICINAL PRODUCTS
 RADIOPHARMACEUTICALS Guideline Title Radiopharmaceuticals Legislative basis Directives 65/65/EEC, 75/318/EEC as amended, Directive 89/343/EEC Date of first adoption December 1990 Date of entry into June
RADIOPHARMACEUTICALS Guideline Title Radiopharmaceuticals Legislative basis Directives 65/65/EEC, 75/318/EEC as amended, Directive 89/343/EEC Date of first adoption December 1990 Date of entry into June
A.1 Contents file 4 to 5 A.1 (1)
") Contents file 4 to 5 Contents file 4 to 5 A Information Contents file 4 to 5 A.2 Index file 4 to 5 A.3 List of Abbreviations A.4 Glossary A.5 Adress-Register A.6 References B Japanese Regulations B.1 MHW
Contents file 4 to 5 Contents file 4 to 5 A Information Contents file 4 to 5 A.2 Index file 4 to 5 A.3 List of Abbreviations A.4 Glossary A.5 Adress-Register A.6 References B Japanese Regulations B.1 MHW
Regulatory Perspective
 Regulatory Perspective Presented by Dr Maria Isaac MASc, MD, PhD, Pharmaceutical Medicine Physician, Psychiatrist Senior Scientific Officer An agency of the European Union Disclaimer The views expressed
Regulatory Perspective Presented by Dr Maria Isaac MASc, MD, PhD, Pharmaceutical Medicine Physician, Psychiatrist Senior Scientific Officer An agency of the European Union Disclaimer The views expressed
What are ATMP s: Why do they Require Special Scientific and Regulatory Attention? Christopher A Bravery.
 What are ATMP s: Why do they Require Special Scientific and Regulatory Attention? Christopher A Bravery cbravery@advbiols.com CONTENTS What are ATMP s Definitions What makes ATMP s different? Starting
What are ATMP s: Why do they Require Special Scientific and Regulatory Attention? Christopher A Bravery cbravery@advbiols.com CONTENTS What are ATMP s Definitions What makes ATMP s different? Starting
Guideline on good pharmacovigilance practices (GVP)
") 4 August 2016 EMA/168402/2014 Corr* Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations II: Biological medicinal products Draft finalised by the Agency in
4 August 2016 EMA/168402/2014 Corr* Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations II: Biological medicinal products Draft finalised by the Agency in
COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP)
") The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 25 July 2002 EMEA/ COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) NOTE FOR GUIDANCE ON THE
The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use London, 25 July 2002 EMEA/ COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) NOTE FOR GUIDANCE ON THE
(See rules 122A, 122B, 122D, 122DA, 122DAA and 122E)
") 503 1 [SCHEDULE Y (See rules 122A, 122B, 122D, 122DA, 122DAA and 122E) REQUIREMENTS AND GUIDELINES FOR PERMISSION TO IMPORT AND / OR MANUFACTURE OF NEW DRUGS FOR SALE OR TO UNDERTAKE CLINICAL TRIALS 1.
503 1 [SCHEDULE Y (See rules 122A, 122B, 122D, 122DA, 122DAA and 122E) REQUIREMENTS AND GUIDELINES FOR PERMISSION TO IMPORT AND / OR MANUFACTURE OF NEW DRUGS FOR SALE OR TO UNDERTAKE CLINICAL TRIALS 1.
Guideline on repeated dose toxicity
 18 March 2010 CPMP/SWP/1042/99 Rev 1 Committee for Human Medicinal Products (CHMP) Guideline on repeated dose toxicity Draft Agreed by Safety Working Party May - December 2007 Adoption by CHMP for release
18 March 2010 CPMP/SWP/1042/99 Rev 1 Committee for Human Medicinal Products (CHMP) Guideline on repeated dose toxicity Draft Agreed by Safety Working Party May - December 2007 Adoption by CHMP for release
European Medicines Agency Evaluation of Medicines for Human Use COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT
 DRAFT") European Medicines Agency Evaluation of Medicines for Human Use London, 27 July 2005 EMEA/CHMP/89249/2004 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT Guideline on the Clinical Investigation
European Medicines Agency Evaluation of Medicines for Human Use London, 27 July 2005 EMEA/CHMP/89249/2004 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT Guideline on the Clinical Investigation
Guidelines for procedures and data requirements for changes to approved vaccines
 1 1 1 1 1 1 1 1 0 1 0 1 0 1 WHO/BS.01. ENGLISH ONLY Guidelines for procedures and data requirements for changes to approved vaccines NOTE: This document has been prepared for the purpose of inviting comments
1 1 1 1 1 1 1 1 0 1 0 1 0 1 WHO/BS.01. ENGLISH ONLY Guidelines for procedures and data requirements for changes to approved vaccines NOTE: This document has been prepared for the purpose of inviting comments
VICH Topic GL2 (Validation: Methodology) GUIDELINE ON VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY
 GUIDELINE ON VALIDATION OF ANALYTICAL PROCEDURES: METHODOLOGY") The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit CVMP/VICH/591/98-FINAL London, 10 December 1998 VICH Topic GL2 (Validation: Methodology) Step 7 Consensus
The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit CVMP/VICH/591/98-FINAL London, 10 December 1998 VICH Topic GL2 (Validation: Methodology) Step 7 Consensus