Obtaining Pre-Study Approvals for Clinical Trials K-30 Module 5 Navigating the Pre-Award Process
|
|
|
- Morgan Ball
- 6 years ago
- Views:
Transcription
1 Obtaining Pre-Study Approvals for Clinical Trials K-30 Module 5 Navigating the Pre-Award Process Helene Orescan, J.D. Bishoy Anastasi, MBA, CCRP David Geffen School of Medicine at UCLA Industry Sponsored Clinical Trials February 7,
2 K30 Program The Clinical Research Curriculum Award (CRCA) is designed to attract talented individuals to the challenges of clinical research and to provide them with the critical skills that are needed to translate basic discoveries into clinical treatments. Challenges, i.e. Regulatory-Approvals 2
3 Clinical Research and Clinical Trials Human Research Clinical Research Clinical Trials Investigator Initiated Protocol Sponsor Initiated Protocol 3
4 What is a Clinical Trial? September 2006 At UCLA a Clinical Trial is defined as: The controlled, clinical testing in human subjects of investigational new drugs, devices, treatments, or diagnostics, or comparisons of approved drugs, devices, treatments, or diagnostics, to assess their safety, efficacy, benefits, costs, adverse reactions, and/or outcomes. Such studies may be conducted under an industry-authored protocol or an investigator-authored protocol. Financial support for a clinical trial must be provided by a for-profit entity. 4
5 What is Not Considered a Clinical Trial per UCLA Policy? Retrospective chart review Laboratory Research (i.e. bench and basic science studies) Federally Funded Clinical Research 5
6 Indirect Costs Facilities & Administrative costs (F&A) also known as overhead or Indirect Costs Industry-sponsored Clinical Trials (both, sponsor-initiated & investigator-initiated) all costs subject to 26% Rate (with the exception of mandatory IRB fees) Other Clinical Research (not a Clinical Trial ) conducted on-campus select costs subject to 54% Rate 6
7 Sponsor-Initiated Clinical Trials Industry Sponsor authors the protocol Industry Sponsor holds the IND/IDE Industry Sponsor funds the study Industry Sponsor assumes most liability 7
8 Investigator-Initiated Clinical Trials Investigator authors the protocol Investigator usually holds the IND/IDE Industry sponsor funds the study University generally assumes some liability 8
9 Approvals Required to Conduct Clinical Trials Institutional Review Board (IRB) Contract/Grant Sponsor Regulatory Budget and Coverage Analysis Conflict of Interest Review Committee (CIRC)* Medical Radiation Safety* Internal Scientific Peer Review Committee* Reimbursement Review* Value Analysis Committee (VAC)* *applicable to certain clinical trials check with your contracts officer. TIP: Avoid delays, submit applications/doc s in parallel (IRB/contract/budget/etc.) 9
10 IRB/Human Subjects Protection Approvals UCLA Institutional Review Board (IRB) Functions under : Federal Guidelines (CFR) Federal-Wide Assurance (FWA) UCLA policy and guidelines UC policy and guidelines Committees own decisions 10


11 IRB and Related Approvals Web-IRB Tutorial: Quick Reference Guides 11
12 How you can help expedite the pre-award process Simultaneous IRB & Contract submissions Know your contacts At the IRB At the Sponsor At Industry Clinical Trials Proactive completion of paperwork Initial vs. Final 12
13 Contracts & Grants Industry-Sponsored Clinical Trials Clinical Trial agreements (IIRP and sponsor-initiated) funded by industry (for-profit) sponsors Office of Contract & Grant Administration (OCGA)-Agreements related to Research (but not Clinical Trials), agreements with Gov t or non-profit organizations e.g. NIH, foundations, etc. Office of Intellectual Property & Industry Sponsored Research (OIP-ISR) License agreements for University held patents, copyrights, etc., & research agreements funded by industry sponsors. 13
14 Clinical Trials Contracting Clinical trial agreements/contracts contain many important legal terms & conditions Authority to sign a contract is restricted to certain University officials Principal Investigators are not authorized to sign contracts that bind the University Signing of a contract is contingent upon final IRB approval 14
15 Industry Clinical Trials Our Role in Protecting You & the Institution Contract negotiation and execution (mandatory for all clinical trials before study initiation/enrollment) Budget Coverage Analysis review and approval required prior to contract execution Assurance that financial terms in the Informed Consent Form, Budget, and Contract are consistent. 15
16 Available Resources and Services Industry Clinical Trials Reviews, Negotiates and Executes Clinical Trial Agreements with industry (for-profit) sponsors. Reviews Conflict of Interest Forms submit to CIRC if required Provides Clinical Research Administrators (CRAs) for Support with Budget development and Coverage Analysis approval. 16
17 Industry Clinical Trials Current Assignment Directory OCT_Contracting_Directory.pdf 17
18 Minimum Documents REQUIRED to Initiate Contract Review Current Protocol Draft Clinical Trial Agreement Draft Goldenrod (only PI signature is required) Financial Disclosures 18
19 Financial Disclosure Process & CIRC Form State or Federal Requirement 700U State of California Form 700U Addendum Institutional Form Who Discloses Principal Investigator PI & Key Personnel What to Disclose $500+ $10,000+ Positive Disclosure Supplement CIRC Review CIRC must review and approve disclosures prior to contract sign-off. 19

20 700-U 700-U Addendum 20

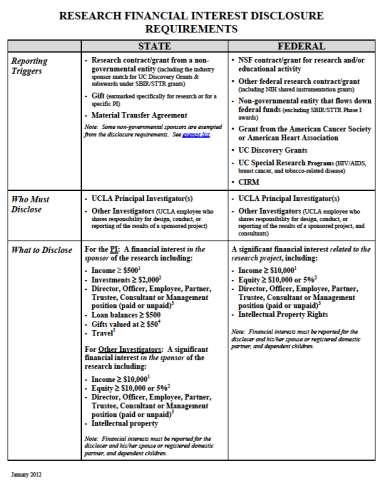
21 Financial Disclosure Instructions 21
22 Why do we need financial disclosure forms? It s the law! Institution must manage financial interests. Positive disclosure(s) may affect Informed Consent Form language. 22
23 Financial Disclosure Process & CIRC Important Facts: Disclosures are effective for a 12-month period (700U = report 12 months prior, 700U Addendum = report 12 months prior & future) CIRC Meeting & Submission Deadlines Industry Contracts office must receive disclosure documents at least three (3) business days before CIRC case submission deadline Missed deadline can cause a significant delay (30 days minimum) 23

24 2012 CIRC Meeting Deadlines 24
25 Financial Disclosure Process & CIRC Important Facts to Note: If a Clinical Research Organization (CRO) is executing the contract, disclosure forms need to be completed for both the study Sponsor and CRO. Contract Amendments that add funding or change the Principal Investigator, also require completion of new 700U/700U-Addendum forms. 25
26 Here are some tips to help you navigate the pre-award clinical trial process. 26
27 Simultaneous Submission Process Clinical Trial Sponsor PI (Principal Investigator) Contracts Submission -Min. Documents, incl. Financial Disclosure Forms to CO Budget Development Industry Contracts CRA Review IRB Submission CIRC Review & Approval CO Negotiates CTA Negotiate Budget w/ Sponsor Budget Finalized Scientific Review ISPRC, MSRC, CTRC Final Docs To CO CTA Fully Executed IRB & Other Approvals To ORDM/EFM for Fund Number 27
 Negotiated (Industry Contracts Bill")

28 What s Involved? Confidentiality Disclosure Agreement (CDA) Negotiated (Industry Contracts Bill Wu) Feasibility Questionnaire & thorough Protocol Review Initial Go / No-Go Decision Budget Development and Negotiation Regulatory Submission and Approvals Clinical Trial Agreement (CTA) Negotiation Coverage Analysis and Approval 28
29 Phase 1 - Go / No-Go Adequate Subject Population to conduct Study? Adequate Resources to conduct Study? Personnel(PI and staff time) Physical Space Other ancillary support Pharmacy Radiology CTRC 29
30 Phase 2 Regulatory Approvals at UCLA IRB Submission & Approval Financial Disclosures Contract Review & Negotiation Budget Negotiation & Coverage Analysis Other Institutional Approvals (if applicable) RAD safety, VAC, Reimbursement Review 30

31 Clinical Trial Budgets 31
32 To Develop A Budget That Covers All Costs: Understand the Protocol Differentiate between routine care and research procedures & services Analyze Costs Personnel Hidden Costs Administrative Patient care Indirect Costs Develop appropriate payment criteria 32

33 Can the Budget pull the weight? 33
34 Budgeting: Key Budget Elements Cost Per Patient (CPP) Budget All protocol required procedures and services identified (budget should mirror protocol visit schedule). Procedural costs (reduced research rates available online for outpt services) PI Effort compensation for time/effort managing study. Research Staff Effort study coord, nurse, and/or data manager. Overhead/Indirect Costs Screening and randomization costs. Invoiceable Items Non-refundable Start-Up Costs IRB Prep and IRB Review Fees Setup Fees for Rx, RAD, CTRC, etc.) Ongoing drug dispensing costs Additional Procedural Costs Unscheduled visit, additional labs/tests, blood/tissue samples, biopsy, etc. Study Admin & Maintenance fees SAEs Annual Renewal Fees (IRB, Rx, etc.) Protocol Amendment fees Record Retention Additional Supplies Shipping Fees Misc. Screen Failures, Re-consent, etc. 34

35 Cost Per Patient Budget 35
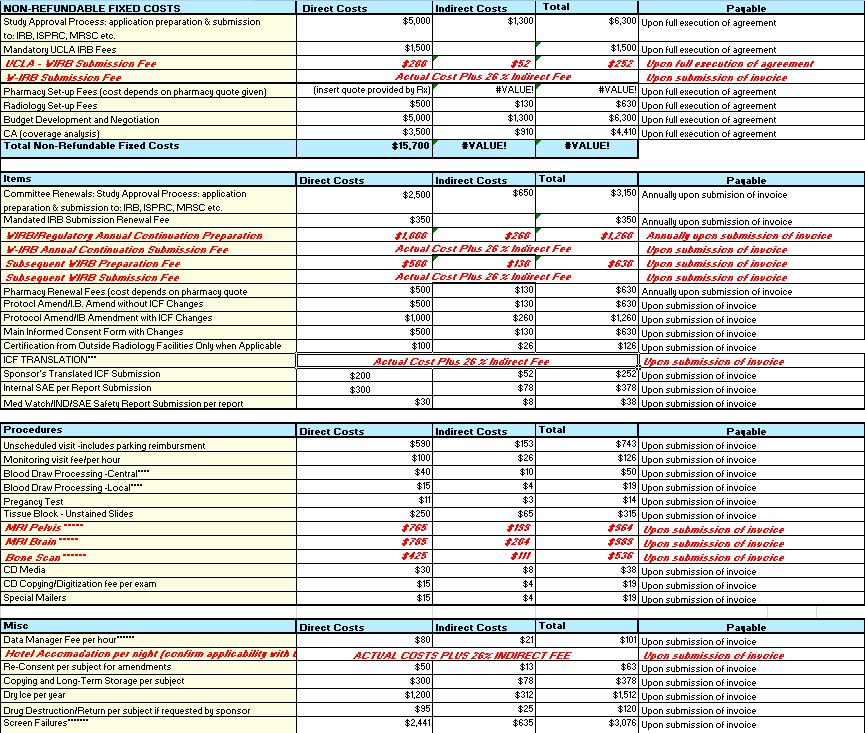
36 Invoiceable Items 36
37 Budgeting: A Process Determination of the true cost to perform clinical trial at UCLA. Careful consideration of routine care versus research-induced costs. Necessary pre-approval/pre-authorizations for devices/certain standard of care charges. Budget approval by someone with fiduciary responsibility or as required by department. 37
38 Coverage Analysis (CA) Budget Coverage Analysis is conducted on all qualifying trials. CA documents the process of identifying the procedural costs which may be billed to insurance as routine care, vs. research costs, which will be paid for by the sponsor, for procedures/services performed as a result of a subject s participation in a clinical trial. Routine Care = Billable to Insurance Research Procedure/Service = Must be provided for by Sponsor. 38
39 Coverage Analysis What is a Qualifying Trial? In order to be covered, the service must be part of a trial that meets all of the following criteria to be considered a qualifying trial : Evaluates a Medicare benefit an item or service that falls within a Medicare Benefit category and is not statutorily excluded from coverage (e.g. cosmetic surgery, hearing aids); and Has therapeutic intent conversely not designed exclusively to test toxicity or disease pathology; and Enrolls diagnosed beneficiaries trials must enroll patients with diagnosed disease rather than healthy volunteers (but may also enroll a healthy control group); and Has desirable characteristics: Trials funded by NIH, CDC, AHRQ, CMS, DOD and VA; or Trials supported by centers or cooperative groups funded by NIH, CDC, AHRQ, CMS, DOD and VA; or Trials conducted under an IND reviewed by the FDA; or IND exempt under 21 CFR 312.2(b)(1) 39
40 Clinical Research Administrator s (CRAs) Role Provide support to investigators during the preaward process and beyond: Budget Review Coverage Analysis and Approval Billing compliance guidance Resource for Pre-Study requirements Current CRAs: Bishoy Anastasi, MPH, MBA, CCRP Silvia Estrada, MPA, CCRC 40
41 Other Approvals Radiology Reduced Research Rates Use Contact Us link to register for a username/password Outpatient/Inpatient Laboratories Contact Nerissa Juse, Njuse@mednet.ucla.edu Investigational Pharmacy Contact Bill Hirokawa or Christina Shin, x UCLA CTRC (formerly GCRC) 41

42 CTSI CTRC website 42
43 Other Institutional Approvals Some examples: Conflict of Interest Review Committee (CIRC) Reviews positive disclosures of financial interest (and makes recommendations regarding award acceptance Environment, Health & Safety (EH&S) Medical Radiation Safety Committee Research use of isotopes or radiation machines in humans Institutional Biosafety Committee Research use of infectious substances Research use of recombinant DNA Internal Scientific Peer Review Committee (ISPRC) Cancer and cancer prevention studies Billing/Reimbursement Review 43
44 Sponsor Regulatory Documents CV(s) Current Medical License(s) FDA Form 1572 Lab Normals and CLIA certificates Delegation of Authority Logs IRB/ICF Approvals Financial Disclosure Forms (FDFs) for Sponsor (independent of Institutional FDFs) 44
45 Final Documents REQUIRED for Contract Execution Final Protocol Final Budget Coverage Analysis Final Goldenrod Financial Disclosure Forms (and online supplement, if applicable) CIRC Approval (if applicable) IRB Approval, including approved ICF PI Exception Letter (if applicable) Other documents specific to your department DOM: Investigator Responsibilities, Other Support Summary 45
46 Top Reasons for Delays in Contract Execution: Incomplete and/or Missing Forms Financial Disclosure Forms (700U/700U Addendum) Online Supplement (for positive disclosure) Goldenrod (missing signatures or e-sign (wet-ink signatures required)) P.I. Exception Letter (must be signed by department chair and Dr. Rome (Associate Dean of Research)) Budget Draft vs. Final Budget Investigator Approval Coverage Analysis Approval 46
47 After IRB approval and contract execution Running the study Subject recruitment and screening Study visits Data management Regulatory compliance Adverse event reporting Maintaining IRB approval Protocol Amendments Financial management Account set-up and receipt of initial funds Expenditure transactions Sponsor invoicing and accounts receivables management 47
48 Additional Resources Industry Contracts Website: For Patient/Community For Faculty/Staff Contact us 48
49 ClinicalTrials.ucla.edu 49
50 Clinical Trials Website Log-In: User name: Password: Please note: Site access is limited to on-campus IT connections. 50
51 Industry Clinical Trials Training Opportunities Current Workshops offered through Campus Human Resources (lms.ucla.edu) Clinical Trial Budgets 101 Intro to development & negotiation. Thursday, February 9 th, :30am Upcoming Workshops: Clinical Trial Budgets 201 Performing Coverage Analysis Clinical Trial Budgets 301 Advanced development & negotiation. Focus: Investigator-Initiated budgets. Focused training can be developed upon group/department request. 51
52 Industry Clinical Trial Services Available Upon Request Budget Development & Negotiation* Pre-Study Support Advice & Consultation (including protocol feasibility assessment no charge) (*Fee-based services that can be written into budgets and passed through to the sponsor.) 52
53 Industry Clinical Trials Contact Info Helene Orescan, J.D. Director DGSOM Dean s Office, Industry Clinical Trials Horescan@mednet.ucla.edu Bishoy Anastasi, MPH, MBA, CCRP CRA Supervisor DGSOM Dean s Office, Industry Clinical Trials Banastasi@mednet.ucla.edu
ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016
 UCLA CTSI WELCOME TO ONLINE TRAINING FOR CLINICAL RESEARCH COORDINATORS ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016 Objectives Define the sponsor of a clinical trial and learn
UCLA CTSI WELCOME TO ONLINE TRAINING FOR CLINICAL RESEARCH COORDINATORS ROLE OF THE RESEARCH COORDINATOR Study Startup Best Practices May 2016 Objectives Define the sponsor of a clinical trial and learn
Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals
 ORIENTATION FOR NEW CLINICAL RESEARCH COORDINATORS Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals Overall Agenda for Orientation Module 1: Introduction
ORIENTATION FOR NEW CLINICAL RESEARCH COORDINATORS Presented by NC TraCS Institute UNC Office of Clinical Trials UNC Network for Research Professionals Overall Agenda for Orientation Module 1: Introduction
Clinical Trial Basics:
 Clinical Trial Basics: Components and Responsibilities Pre-Award Aida Nana Ama Manu, Project Coordinator Four Main Components Non-Disclosure Agreements Institutional Review Board Application Clinical Trial
Clinical Trial Basics: Components and Responsibilities Pre-Award Aida Nana Ama Manu, Project Coordinator Four Main Components Non-Disclosure Agreements Institutional Review Board Application Clinical Trial
Clinical & Translational Research, What is it and Why Should I Care?
 Clinical & Translational Research, What is it and Why Should I Care? Ann Smith, Operations Administrative Manager, Clinical & Translational Sciences Institute What is Clinical Research? Research Human
Clinical & Translational Research, What is it and Why Should I Care? Ann Smith, Operations Administrative Manager, Clinical & Translational Sciences Institute What is Clinical Research? Research Human
Guidelines for Setting Up a Regulatory Binder
 Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Guidelines for Setting Up a Regulatory Binder Prepare for an audit Georgamaly Estronza, MS UPR - Medical Sciences Campus IRB Administrator - Compliance Officer Email: georgamaly.estronza@upr.edu Introduction
Clinical Research at MSU
 Financial Administration Development Program: Clinical Research at MSU Kristen Burt, Office of Regulatory Affairs (ORA) Sharon Schooley, Clinical & Translational Sciences Institute (CTSI) February 24,
Financial Administration Development Program: Clinical Research at MSU Kristen Burt, Office of Regulatory Affairs (ORA) Sharon Schooley, Clinical & Translational Sciences Institute (CTSI) February 24,
Clinical Trials Series Part II
 Clinical Trials Series Part II Agenda Recap December Presentation J. Schmelz Example: New CT from HSC Investigator Non Cancer Clinical Trial J. Bates, P. Miranda Example: New CT from External Entity Non
Clinical Trials Series Part II Agenda Recap December Presentation J. Schmelz Example: New CT from HSC Investigator Non Cancer Clinical Trial J. Bates, P. Miranda Example: New CT from External Entity Non
ROLE OF THE RESEARCH COORDINATOR Investigational New Drug Application-Sponsor Responsibilities 21CFR Part , subpart D
 Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Investigational
Regulatory Binder: Set-up and Maintenance
 Regulatory Binder: Set-up and Maintenance Introduction Federal and state regulations, institutional policy, and good clinical and research practices require investigators to maintain documents related
Regulatory Binder: Set-up and Maintenance Introduction Federal and state regulations, institutional policy, and good clinical and research practices require investigators to maintain documents related
Clinical Trials Management for Molecular Diagnostics. April 2016
 Clinical Trials Management for Molecular Diagnostics April 2016 Clinical Operations Responsibilities Accrue samples that have proper informed consent for use Retrospective cohorts Remnant samples Prospective
Clinical Trials Management for Molecular Diagnostics April 2016 Clinical Operations Responsibilities Accrue samples that have proper informed consent for use Retrospective cohorts Remnant samples Prospective
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1
 Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
Regulatory Documentation and Submissions for C2012 Clinical Trials DCP SOP #1 Phone: 650.691.4400 Fax: 650.691.4410 Email: regulatory.ccsainc.com COMPLIANCE & STANDARDIZATION Rationale for Revision of
HCCA 13 TH ANNUAL COMPLIANCE INSTITUTE LAS VEGAS, NV APRIL 29, 2009
 HCCA 13 TH ANNUAL COMPLIANCE INSTITUTE LAS VEGAS, NV APRIL 29, 2009 MEDICARE COVERAGE ANALYSIS WORKSHOP: THE HOW TO OF MEDICARE COVERAGE IN RESEARCH Suzanne LivPage, J.D. Director, Clinical Research Initiation
HCCA 13 TH ANNUAL COMPLIANCE INSTITUTE LAS VEGAS, NV APRIL 29, 2009 MEDICARE COVERAGE ANALYSIS WORKSHOP: THE HOW TO OF MEDICARE COVERAGE IN RESEARCH Suzanne LivPage, J.D. Director, Clinical Research Initiation
Office for Human Subject Protection. University of Rochester
 POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
POLICY 1. Purpose Outline the responsibilities and regulatory requirements when conducting human subject research that involves the use of drugs, agents, biological products, or nutritional products (e.g.,
Sponsoring an IND? Ignorance is Not Always Blissful
 Translational Science 2013 Navigating The FDA Regulatory Landscape April 19, 2013 Washington D.C. Sponsoring an IND? Ignorance is Not Always Blissful Lisa A. Speicher, Ph.D Director, Clinical Trials Office
Translational Science 2013 Navigating The FDA Regulatory Landscape April 19, 2013 Washington D.C. Sponsoring an IND? Ignorance is Not Always Blissful Lisa A. Speicher, Ph.D Director, Clinical Trials Office
Compliance and Quality Monitoring: What, Why, When, and How
 Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
Compliance and Quality Monitoring: What, Why, When, and How Jeanna Julo, BA, BA, CCRP Assistant Director, Clinical Data Management & Quality Controls, Auditing & Training Research Institute, University
ClinicalTrials.gov REGISTRATION REQUIREMENTS
 ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
ClinicalTrials.gov REGISTRATION REQUIREMENTS Background: What is it? ClinicalTrials.gov is a public registry that provides easy access to information on clinical studies, both clinical trials and observational
1. POLICY STATEMENT: 2. BACKGROUND:
 POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
POLICY #: RCO-201 Page: 1 of 5 1. POLICY STATEMENT: All Food and Drug Administration (FDA) regulated research conducted under an Investigational New Drug Application (IND) and managed within DF/HCC requires
Objectives Discuss the importance of proper data collection. Identify the types of data collected for clinical trials. List potential source documents
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research January 24, 2011 Diane St. Germain, RN, MS, CRNP Nurse Consultant Division of Cancer Prevention National
Introduction to Clinical Research
 Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
Introduction to Clinical Research What is Clinical Research? Clinical research is medical research that involves people like you. People volunteer to participate in carefully conducted investigations that
FDA Sponsor and Investigator Responsibility Checklist
 FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
FDA Sponsor and Investigator Responsibility Checklist Principal Investigator: Study Name: CPHS #: IND/IDE #: Name of IND/IDE holder: The following checklist is created based on the Sponsor and Investigator
Standard Operating Procedures Guidelines for Good Clinical Practice
 SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
SOP # CRSC-105 Effective Date 10-22-2013 Version # 1 Version Date 7-30-2013 Standard Operating Procedures Guidelines for Good Clinical Practice Purpose: This SOP outlines the steps required to follow FDA
FDA Audit Preparation
 Duke University Office of Audit, Risk and Compliance (OARC) FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Associate Compliance Officer, Research Compliance Assurance (RCA) External audits
Duke University Office of Audit, Risk and Compliance (OARC) FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Associate Compliance Officer, Research Compliance Assurance (RCA) External audits
3.0 HSC Relation to Other KUMC Committees
 3.0 HSC Relation to Other KUMC Committees It is the policy of the HSC to work in conjunction with other KUMC Committees to provide protections to research subjects. The HSC coordinates reviews with other
3.0 HSC Relation to Other KUMC Committees It is the policy of the HSC to work in conjunction with other KUMC Committees to provide protections to research subjects. The HSC coordinates reviews with other
Objectives. The Regulatory Binder = Investigator Site File= Trial Center File 8/16/2010. Essential Documents: Maintaining the Site's Regulatory Binder
 Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
Essential Documents: Maintaining the Site's Regulatory Binder How to Manage the Essential Documents for the Investigator and Study Site Objectives Describe the importance of maintaining the Regulatory
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)
 SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312)") POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
POST-IRB APPROVAL FDA DRUG (IND) SPONSOR AND INVESTIGATOR RESPONSIBILITY (21 CFR312) Purpose: Investigators who initiate and submit an IND application to the FDA assume the responsibilities of both the
Clinical trials: Prerequisites
 Clinical trials: Prerequisites Albiruni R Abdul Razak Staff Medical Oncologist, Princess Margaret Cancer Centre/ Mount Sinai Hospital Assistant Professor, University of Toronto Disclosures Research Funding
Clinical trials: Prerequisites Albiruni R Abdul Razak Staff Medical Oncologist, Princess Margaret Cancer Centre/ Mount Sinai Hospital Assistant Professor, University of Toronto Disclosures Research Funding
Multi-Site Coordination Process. Drafted by: Ester Dimayuga Page 1 of 18
 Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Multi-Site Coordination Process Drafted by: Ester Dimayuga Page 1 of 18 MULTI-SITE COORDIATIO (MSC) REGULATOR PROCESS CTO-Regulatory notifies MSC of trial with UMCC as coordinating center MSC prepares
Human Research Protection Program Good Clinical Practice Guidance for Investigators Regulatory File Essential Documents
 Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Guidance for s Regulatory File Essential s All principal investigators must maintain a regulatory binder or file system, which contains all study documentation. These records may be reviewed at the time
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices
 Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
Clinical Research with Drugs/Biologics and Devices & Good Clinical Practices Jason Jobson, BLS, CCRP Research Compliance Officer Oklahoma City VA Medical Center October 2017 Goals Investigational New Drug
Regulatory Document Guidelines for DMID Clinical Studies. Version Oct-2005
 Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
Regulatory Document Guidelines for DMID Clinical Studies Version 3.0-5-Oct-2005 1 Regulatory File Document Guidelines Purpose: To aid DMID supported Investigators in establishing a file of essential documents
VCU Faculty Held IND and IDE Procedure Handbook
 VCU Faculty Held IND and IDE Procedure Handbook Contents A. Introduction... 3 B. Purpose of Institutional Oversight... 3 C. Applicability... 4 D. University Oversight of Clinical Investigations Being Conducted
VCU Faculty Held IND and IDE Procedure Handbook Contents A. Introduction... 3 B. Purpose of Institutional Oversight... 3 C. Applicability... 4 D. University Oversight of Clinical Investigations Being Conducted
FDA Audit Preparation
 Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
Duke University Ethics and Compliance Office FDA Audit Preparation Margaret M. Groves, JD, CRA, CCRP, CHRC Director, CTQA Agenda External audits Best practices to get ready for audits 2 External Audits
US Special Operations Command Human Research Protection Office
 S- US Special Operations Command Human Research Protection Office Human Research Protocol Submission Form for Headquarters Level Administrative Review of Extramural* Research PURPOSE: All United States
S- US Special Operations Command Human Research Protection Office Human Research Protocol Submission Form for Headquarters Level Administrative Review of Extramural* Research PURPOSE: All United States
Source And Regulatory Documentation for DMID Clinical Studies
 Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
Source And Regulatory Documentation for DMID Clinical Studies Walt Jones RN, MPH Nurse Consultant Clinical Monitoring Coordinator OCRA, DMID, NIAID November, 2007 Source Data Defined All information in
Regulatory Binder Guidance
 Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
Regulatory Binder Guidance What is the purpose of a regulatory binder? Achieve and maintain regulatory compliance Ensure protection of human subjects and high standards of research Guidance for organization
The Research Services Organization (RSO) of the Academic Health Center
 of the Academic Health Center") Annual Report 2005 The Research Services Organization (RSO) of the Academic Health Center A Summary of the Eighth Year of Operations Submitted by: Mark S. Paller, M.D., M.S. Director, Research Services
Annual Report 2005 The Research Services Organization (RSO) of the Academic Health Center A Summary of the Eighth Year of Operations Submitted by: Mark S. Paller, M.D., M.S. Director, Research Services
Research 101. Health Care Compliance Association Regional Conference Columbus, Ohio May 8, 2015
 Research 101 Health Care Compliance Association Regional Conference Columbus, Ohio May 8, 2015 Jennifer Lanter, MSPH, BSN, RN, CCRC Director, Research & Special Billing Office Agenda Research Compliance
Research 101 Health Care Compliance Association Regional Conference Columbus, Ohio May 8, 2015 Jennifer Lanter, MSPH, BSN, RN, CCRC Director, Research & Special Billing Office Agenda Research Compliance
Work Instruction - Research Billing Risk
 THE UNIVERSITY OF TEXAS HEALTH SCIENCE CENTER AT SAN ANTONIO Work Instruction - Research Billing Risk Velos eresearch Version 9.2 Version: 2.0, 04/03/2015 Revision History Version/Amendment #: Version
THE UNIVERSITY OF TEXAS HEALTH SCIENCE CENTER AT SAN ANTONIO Work Instruction - Research Billing Risk Velos eresearch Version 9.2 Version: 2.0, 04/03/2015 Revision History Version/Amendment #: Version
Clinical trials: Prerequisites
 Clinical trials: Prerequisites Albiruni R Abdul Razak Staff Medical Oncologist, Princess Margaret Cancer Centre/ Mount Sinai Hospital Assistant Professor, University of Toronto Disclosures Research Funding
Clinical trials: Prerequisites Albiruni R Abdul Razak Staff Medical Oncologist, Princess Margaret Cancer Centre/ Mount Sinai Hospital Assistant Professor, University of Toronto Disclosures Research Funding
Recruitment Innovation Initiative
 Recruitment Innovation Initiative Financial Considerations 1 Develop a Financial Plan Project first year revenue Types of Studies Payment Industry research usually funded quarterly In contract, negotiate
Recruitment Innovation Initiative Financial Considerations 1 Develop a Financial Plan Project first year revenue Types of Studies Payment Industry research usually funded quarterly In contract, negotiate
Investigational New Drug Development Steps for CRCs
 Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
Investigational New Drug Development Steps for CRCs Natalie Nardone, PhD Program Manager, Department of Medicine CRC Instructor, Office of Clinical Research Contact: natalie.nardone@ucsf.edu Learning Objectives
Investigator Manual Revised August 19, 2013
 Investigator Manual Revised August 19, 2013 Investigator Manual Page 2 of 31 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection
Investigator Manual Revised August 19, 2013 Investigator Manual Page 2 of 31 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection
Defining research compliance Protocol review committees Conflicts of interest Export Controls
 Defining research compliance Protocol review committees Conflicts of interest Export Controls What is Research Compliance? Conformance with Federal, State, and Local Regulations and University Policies
Defining research compliance Protocol review committees Conflicts of interest Export Controls What is Research Compliance? Conformance with Federal, State, and Local Regulations and University Policies
Research and the EHR: Process Improvement Through Integration
 Research and the EHR: Process Improvement Through Integration Session 160, March 8, 2018 Arash Naeim, MD PhD, Chief Medical Research Officer, UCLA Health System Marti Arvin, VP of Audit Strategy, CynergisTek
Research and the EHR: Process Improvement Through Integration Session 160, March 8, 2018 Arash Naeim, MD PhD, Chief Medical Research Officer, UCLA Health System Marti Arvin, VP of Audit Strategy, CynergisTek
ClinicalTrials.gov Registration Guide
 ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
UC DAVIS OFFICE OF RESEARCH AAHRPP Preparation UC Davis Human Research Part III Investigator Manual. Cindy Gates IRB Administration
 UC DAVIS OFFICE OF RESEARCH AAHRPP Preparation UC Davis Human Research Part III Investigator Manual Cindy Gates IRB Administration What is the purpose of the INVESTIGATOR MANUAL? The manual is designed
UC DAVIS OFFICE OF RESEARCH AAHRPP Preparation UC Davis Human Research Part III Investigator Manual Cindy Gates IRB Administration What is the purpose of the INVESTIGATOR MANUAL? The manual is designed
4.2. Investigator Regulatory Binder: Files, usually a binder, maintained by the investigator at the investigative site.
 POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
POLICY #: RCO-203 Page: 1 of 7 1. POLICY STATEMENT: Essential regulatory documents will be on maintained for research sponsored by or conducted at Dana-Farber/Harvard Cancer Center (DF/HCC) to assure compliance
@ALSETF #EAP2015. Jess B. Rabourn CBI Expanded Access Conference July 22, 2015
 How to Win Physician Collaboration Models for Advancing Access to Lifesaving Therapies @ALSETF #EAP2015 Jess B. Rabourn CBI Expanded Access Conference July 22, 2015 How to Win Now that we want Expanded
How to Win Physician Collaboration Models for Advancing Access to Lifesaving Therapies @ALSETF #EAP2015 Jess B. Rabourn CBI Expanded Access Conference July 22, 2015 How to Win Now that we want Expanded
HUMAN RESEARCH PROTECTION PROGRAM PLAN
 HUMAN RESEARCH PROTECTION PROGRAM PLAN HRP 101 June 2017 706-542-3199 irb@uga.edu https://research.uga.edu/hso/ Table of Contents Scope... 2 Purpose... 2 Definitions... 2 Agent... 2 Clinical Trial... 2
HUMAN RESEARCH PROTECTION PROGRAM PLAN HRP 101 June 2017 706-542-3199 irb@uga.edu https://research.uga.edu/hso/ Table of Contents Scope... 2 Purpose... 2 Definitions... 2 Agent... 2 Clinical Trial... 2
Title: Department: Approved by: Director, Human Research Review and Compliance
 Title: Department: Requirements for Investigational New Drug (IND) for Human- Subjects Research Human Research Affairs Policy Type: Partners System-wide Partners System-wide Template Partners HealthCare
Title: Department: Requirements for Investigational New Drug (IND) for Human- Subjects Research Human Research Affairs Policy Type: Partners System-wide Partners System-wide Template Partners HealthCare
Work Instruction Study Registration
 THE UNIVERSITY OF TEXAS HEALTH SCIENCE CENTER AT SAN ANTONIO Work Instruction Study Registration Velos - eresearch 9.2 Version: 2.0, 4/29/2015 Revision History Version/Amendment #: Version Date: Description:
THE UNIVERSITY OF TEXAS HEALTH SCIENCE CENTER AT SAN ANTONIO Work Instruction Study Registration Velos - eresearch 9.2 Version: 2.0, 4/29/2015 Revision History Version/Amendment #: Version Date: Description:
CANCER CENTER SCIENTIFIC REVIEW COMMITTEE
 CANCER CENTER SCIENTIFIC REVIEW COMMITTEE The Clinical Scientific Review Committee (SRC) at The Medical College of Wisconsin Cancer Center plays a vital role in protocol review and monitoring to ensure
CANCER CENTER SCIENTIFIC REVIEW COMMITTEE The Clinical Scientific Review Committee (SRC) at The Medical College of Wisconsin Cancer Center plays a vital role in protocol review and monitoring to ensure
11.0 FDA-Regulated Research Research Involving Investigational Drugs and Biologics
 11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
Clinical Trials Infrastructure Workshop # 3
 Clinical Trials Infrastructure Workshop # 3 Susan Dent MD FRCPC Medical Oncologist The Ottawa Hospital Cancer Center Associate Professor of Medicine University of Ottawa Objectives To discuss the infrastructure
Clinical Trials Infrastructure Workshop # 3 Susan Dent MD FRCPC Medical Oncologist The Ottawa Hospital Cancer Center Associate Professor of Medicine University of Ottawa Objectives To discuss the infrastructure
Human Research Protection Program Policy
 Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
Page 1 of 5 REVIEW OF INVESTIGATIONAL NEW DRUG (IND)/INVESTIGATIONAL DEVICE EXEMPTION (IDE) RESEARCH IN HUMAN SUBJECTS RESEARCH POLICY It is the policy of the University of Cincinnati that studies involving
A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN
 A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN Version 1 - June 2017 A Short Guide For CT Application 1 2 A Short Guide For CT Application DEFINITIONS Clinical
A SHORT GUIDE TO THE PROCEDURE FOR A CLINICAL TRIAL APPLICATION IN THE KINGDOM OF BAHRAIN Version 1 - June 2017 A Short Guide For CT Application 1 2 A Short Guide For CT Application DEFINITIONS Clinical
CHECKLIST: Pre-review
 11-5-2014 1 of 5 The purpose of this checklist is to provide support for IRB staff conducting pre-review. This checklist is to be completed by the IRB staff, signed, dated, and retained with the IRB protocol
11-5-2014 1 of 5 The purpose of this checklist is to provide support for IRB staff conducting pre-review. This checklist is to be completed by the IRB staff, signed, dated, and retained with the IRB protocol
1.0 INTRODUCTION AND OVERVIEW
 Table of Contents 1.0 INTRODUCTION AND OVERVIEW... 3 2.0 DEFINITIONS... 4 3.0 OVERSIGHT AND ORGANIZATION... 5 3.1 CLINICAL RESEARCH OVERSIGHT COMMITTEE (CROC)... 5 3.2 CLINICAL RESEARCH SUPPORT (CRS)...
Table of Contents 1.0 INTRODUCTION AND OVERVIEW... 3 2.0 DEFINITIONS... 4 3.0 OVERSIGHT AND ORGANIZATION... 5 3.1 CLINICAL RESEARCH OVERSIGHT COMMITTEE (CROC)... 5 3.2 CLINICAL RESEARCH SUPPORT (CRS)...
Human Research Audit Program. Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance
 Human Research Audit Program Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance Monday, September 11, 2017 Objectives 1. Understand the purpose of the audit program 2. Identify
Human Research Audit Program Gabrielle Gaspard, MPH, CCRC Assistant Director, Human Research Compliance Monday, September 11, 2017 Objectives 1. Understand the purpose of the audit program 2. Identify
11.0 FDA-Regulated Research Research Involving Investigational Drugs and Biologics
 11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
11.0 FDA-Regulated Research The IRB evaluates the safety or efficacy of all drugs and devices used in research. Studies involving unapproved or investigational drugs or devices will be reviewed to ensure
Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes
 Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes Martha E. Payne, PhD, MPH Office of Research Development, Duke University School of Medicine Objectives for Today What has
Human Subjects Requirements for NIH and AHRQ Applications: Overview of Changes Martha E. Payne, PhD, MPH Office of Research Development, Duke University School of Medicine Objectives for Today What has
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd.
 Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
Audit and Regulatory Inspection Nopanan Yaibuathes Clinical Research and Compliance Manager Roche Thailand Ltd. Copyright 2009 - Pharmaceutical Research & Manufacturers Association 1 Overview Audit ICH-GCP,
and Study Initiation
 10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
and Study Initiation
 10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
10 STUDY SPECIFIC PRE-IMPLEMENTATION, SITE ACTIVATION AND STUDY INITIATION... 2 10.1 Clinical Trials Agreement... 4 10.2 Study Product Acquisition and Shipment to Sites... 5 10.3 Study-specific Preparatory,
Recommendations for Strengthening the Investigator Site Community
 Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
Recommendations for Strengthening the Investigator Site Community October 2017 CTTI MISSION: To develop and drive adoption of practices that will increase the quality and efficiency of clinical trials
Clinical Trials Office VPR CTO
 Clinical Trials Office VPR CTO Velos eresearch Mysti M. Trainer Budget Analyst - Associate Who is the VPR-CTO? Serve Under the Vice President for Research Part of the Research Administration We work closely
Clinical Trials Office VPR CTO Velos eresearch Mysti M. Trainer Budget Analyst - Associate Who is the VPR-CTO? Serve Under the Vice President for Research Part of the Research Administration We work closely
ClinicalTrials.gov Registration Guide
 ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
ClinicalTrials.gov Registration Guide The Food and Drug Administration Amendments Act (FDAAA), National Institutes of Health (NIH) and International Committee of Medical Journal Editors (ICMJE) require
Standard Operating Procedure
 Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Standard Operating Procedure Number: UM/UoM TMF/SOP08/6.0 Title: The Creation and Maintenance of Trial Master Files and Essential Documentation Version: 6.0 () Effective Date: Author: Mrs Catherine Barrow
Becoming Aware of the Requirements Surrounding Clinical Trial Disclosure (ClinicalTrials.gov)
") Becoming Aware of the Requirements Surrounding Clinical Trial Disclosure (ClinicalTrials.gov) Office of Research Compliance and Quality Assurance Yolanda P. Davis Sr. Research Compliance Officer Author:
Becoming Aware of the Requirements Surrounding Clinical Trial Disclosure (ClinicalTrials.gov) Office of Research Compliance and Quality Assurance Yolanda P. Davis Sr. Research Compliance Officer Author:
Demystifying Audits. Audits and Audit Preparation 5/23/2016. What is an Audit?
 Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
Demystifying Audits Darlene Kitterman, MBA Director, Investigator Support & Integration Services OCTRI May 26, 2016 Audits and Audit Preparation What is an Audit? A systematic and independent examination
Operationalizing Your Study At Grady
 Operationalizing Your Study At Grady June 22, 2016 Presented By: The Offices of Research & Grants Administration Learning Objectives By the end of the session, you will: Understand how to operationalize
Operationalizing Your Study At Grady June 22, 2016 Presented By: The Offices of Research & Grants Administration Learning Objectives By the end of the session, you will: Understand how to operationalize
IBM Clinical Trial Management System for Sites
 Service Description IBM Clinical Trial Management System for Sites This Service Description describes the Cloud Service IBM provides to Client. Client means the contracting party and its authorized users
Service Description IBM Clinical Trial Management System for Sites This Service Description describes the Cloud Service IBM provides to Client. Client means the contracting party and its authorized users
External IRB Review What Does it Mean for Your Institution
 External IRB Review What Does it Mean for Your Institution Wesley G Byerly, Pharm.D. Associate Vice President for Research Integrity and Regulatory Affairs University of Connecticut and UCONN Health HCCA
External IRB Review What Does it Mean for Your Institution Wesley G Byerly, Pharm.D. Associate Vice President for Research Integrity and Regulatory Affairs University of Connecticut and UCONN Health HCCA
Managing a Research Team. Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services
 Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Managing a Research Team Mandy Morneault Manager, Research Coordinator Core ITHS Clinical Services vicka@uw.edu, 206-355-5210 PI responsibilities and organizing a research team Hiring research staff Effective
Investigator Manual January 23, 2014
 Investigator Manual January 23, 2014 Investigator Manual Page 2 of 60 Table of Contents What is the purpose of this manual?... 4 What is Human Research?... 4 What is the Human Research Protection Program?...
Investigator Manual January 23, 2014 Investigator Manual Page 2 of 60 Table of Contents What is the purpose of this manual?... 4 What is Human Research?... 4 What is the Human Research Protection Program?...
Sponsor-Investigator Responsibilities In Clinical Trials
 In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
In Clinical Trials Margaret Huber, RN, BSN, CHRC Compliance Manager The lecturer has no conflicts for this presentation 9/23/2015 Objectives Define terms sponsor, investigator, and sponsor-investigator.
IRB-GCP and Timelines. Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October th, 2010
 IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
IRB-GCP and Timelines Andrew Majewski, MSc. 1 st DOLF Meeting Washington University School of Medicine St Louis, Missouri-USA October 11-14 th, 2010 1 Factors that affect Timelines Finalized Protocol Finalized
Source Documents and Regulatory Binders October 6, 2016
 Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
Source Documents and Regulatory Binders October 6, 2016 Lisa Wilson, Regulatory Lead, Clinical Trials Office and Mark Alger, CRC, Clinical Trials Office Essential Documents AKA: the stuff in the Reg Binder
PROTOCOL-SPECIFIC DOCUMENT
 PROTOCOL-SPECIFIC DOCUMENT To Collect Institutional Requirements from Relying Institutions Institutional, Local, and State Requirements Working Group of the SMART IRB Harmonization Steering Committee July
PROTOCOL-SPECIFIC DOCUMENT To Collect Institutional Requirements from Relying Institutions Institutional, Local, and State Requirements Working Group of the SMART IRB Harmonization Steering Committee July
1 Purpose. 2 Procedure. Title: FDA-Regulated Research. SOP Number: 1301 Effective Date: June 2, Previous Version Dates:
 Previous Version Dates: Title: FDA-Regulated Research SOP Number: 1301 Effective Date: June 2, 2017 1 Purpose FDA regulations apply to research that involves a FDA-regulated test article in a clinical
Previous Version Dates: Title: FDA-Regulated Research SOP Number: 1301 Effective Date: June 2, 2017 1 Purpose FDA regulations apply to research that involves a FDA-regulated test article in a clinical
Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products
 1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
1 Trial Master File / Investigator Site File Index Clinical Trials of Investigational Medicinal Products SECTION TITLE DOCUMENTS 1. Contact List Including details of relevant study site staff, responsible
IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol
 IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol Use this checklist to assemble your request for your institution s IBC to review a specific protocol. If WIRB does not already administer
IBC SERVICES SUBMISSION REQUIREMENTS Part B: Review of Protocol Use this checklist to assemble your request for your institution s IBC to review a specific protocol. If WIRB does not already administer
Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE
 Inspection: VOICE") Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute
Preparing for a United States Food and Drug Administration (FDA) Inspection: VOICE This project has been funded in whole or in part with Federal funds from the Division of AIDS (DAIDS), National Institute
The Intersection of Genomics Research and the IDE Regulation
 The Intersection of Genomics Research and the IDE Regulation Katherine Donigan, Ph.D. Personalized Medicine Staff FDA/CDRH/OIR October 19, 2017 1 In Vitro Diagnostic (IVD) Regulation Through the 1976 medical
The Intersection of Genomics Research and the IDE Regulation Katherine Donigan, Ph.D. Personalized Medicine Staff FDA/CDRH/OIR October 19, 2017 1 In Vitro Diagnostic (IVD) Regulation Through the 1976 medical
Expanded Access and the Individual Patient IND
 Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
Expanded Access and the Individual Patient IND Research Wednesdays April 26, 2017 Erika Segear Johnson, PhD, RAC Associate Director of Regulatory Affairs Office of Regulatory Affairs and Quality Office
The Johns Hopkins Institute for Clinical and Translational Research
 ICTR The Johns Hopkins Institute for Clinical and Translational Research INTRODUCTION TO CLINICALTRIALS.GOV LINDA POST, RN, BSN, CCRP RESEARCH NAVIGATOR JUNE 28, 2013 Development of this resource was supported
ICTR The Johns Hopkins Institute for Clinical and Translational Research INTRODUCTION TO CLINICALTRIALS.GOV LINDA POST, RN, BSN, CCRP RESEARCH NAVIGATOR JUNE 28, 2013 Development of this resource was supported
IRB Considerations for Investigator-Initiated Research
 November 29, 2017 IRB Considerations for Investigator-Initiated Research Robert Romanchuk, BSHS, CIP, CCRC, CCRP IRB Vice Chair, Schulman IRB Introducing advarra.com About Schulman IRB Established in 1983
November 29, 2017 IRB Considerations for Investigator-Initiated Research Robert Romanchuk, BSHS, CIP, CCRC, CCRP IRB Vice Chair, Schulman IRB Introducing advarra.com About Schulman IRB Established in 1983
2016 San Antonio Claude D. Pepper Older Americans Independence Center Pilot and Exploratory Study Core REQUEST FOR APPLICATIONS PILOT PROJECTS
 2016 San Antonio Claude D. Pepper Older Americans Independence Center Pilot and Exploratory Study Core REQUEST FOR APPLICATIONS PILOT PROJECTS Mission: The major objective of the San Antonio Claude D.
2016 San Antonio Claude D. Pepper Older Americans Independence Center Pilot and Exploratory Study Core REQUEST FOR APPLICATIONS PILOT PROJECTS Mission: The major objective of the San Antonio Claude D.
Protection of Research Participants: The IRB Process and the Winds of Change
 Protection of Research Participants: The IRB Process and the Winds of Change Ethics in Patient-Oriented Research October 12, 2011 Sharon Friend Director, OHRPP Overview Charge and Function of the IRB Quick
Protection of Research Participants: The IRB Process and the Winds of Change Ethics in Patient-Oriented Research October 12, 2011 Sharon Friend Director, OHRPP Overview Charge and Function of the IRB Quick
SELF-ASSESSMENT CHECKLIST
 SELF-ASSESSMENT CHECKLIST SECTION 1: REGULATORY DOCUMENTATION Staff Documentation 1. Are all versions of the IRB approved protocol on file (including most recent)? 2. Are there CVs/biosketches of PI, Co-Is,
SELF-ASSESSMENT CHECKLIST SECTION 1: REGULATORY DOCUMENTATION Staff Documentation 1. Are all versions of the IRB approved protocol on file (including most recent)? 2. Are there CVs/biosketches of PI, Co-Is,
Administrative Policies VMC #638.1 and Procedures Manual
 Administrative Policies VMC #638.1 and Procedures Manual January 14, 2013 TO: FROM: SUBJECT: REFERENCE: VMC Employees Paul E. Lorenz Chief Executive Officer, SCVMC Research Involving Human Subjects TJC,
Administrative Policies VMC #638.1 and Procedures Manual January 14, 2013 TO: FROM: SUBJECT: REFERENCE: VMC Employees Paul E. Lorenz Chief Executive Officer, SCVMC Research Involving Human Subjects TJC,
Case Study: University of Washington s Clinical Research Billing Audit Program
 Case Study: University of Washington s Clinical Research Billing Audit Program 7 th Annual Conference for Effective Compliance Systems in Higher Education Richard A. Meeks, CCEP Agenda Background Probe
Case Study: University of Washington s Clinical Research Billing Audit Program 7 th Annual Conference for Effective Compliance Systems in Higher Education Richard A. Meeks, CCEP Agenda Background Probe
Rules of Human Experimentation
 Rules of Human Experimentation Elaine Larson CUMC IRB Chair Associate Dean for Research, School of Nursing Professor of Epidemiology, Mailman School of Public Health Oversight for Human Research Office
Rules of Human Experimentation Elaine Larson CUMC IRB Chair Associate Dean for Research, School of Nursing Professor of Epidemiology, Mailman School of Public Health Oversight for Human Research Office
I. Purpose. II. Definitions. Last Approval Date
 Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
Investigational Drugs and Biologics Page 1 of 13 I. Purpose The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics
CLINICAL AND TRANSLATIONAL RESEARCH COMMITTEE SUBMISSION APPLICATION
 CLINICAL AND TRANSLATIONAL RESEARCH COMMITTEE SUBMISSION APPLICATION FORM 1 FOR NON-INTERVENTIONAL STUDIES NOT MANAGED BY THE WINSHIP CLINICAL TRIALS OFFICE INSTRUCTIONS: This form serves as application
CLINICAL AND TRANSLATIONAL RESEARCH COMMITTEE SUBMISSION APPLICATION FORM 1 FOR NON-INTERVENTIONAL STUDIES NOT MANAGED BY THE WINSHIP CLINICAL TRIALS OFFICE INSTRUCTIONS: This form serves as application
MARKEY CANCER CENTER STANDARD OPERATING PROCEDURES. SOP No.: MCC Type: Final
 Page 1 of 11 MARKEY CANCER CENTER STANDARD OPERATING PROCEDURES SOP No.: MCC-002.03 Type: Final Title: Protocol Review and Monitoring Committee Functional Overview Approval Signature Date MCC Director
Page 1 of 11 MARKEY CANCER CENTER STANDARD OPERATING PROCEDURES SOP No.: MCC-002.03 Type: Final Title: Protocol Review and Monitoring Committee Functional Overview Approval Signature Date MCC Director
Clinical Trials and the Code of Federal Regulations. Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014
 Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
Clinical Trials and the Code of Federal Regulations Darlene Kitterman, MBA Director, Investigator Support & Integration Services September 24, 2014 The Development of Regulations 1906: Food and Drugs Act
ELEMENTS OF A DATA MONITORING PLAN
 ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
ELEMENTS OF A DATA MONITORING PLAN Definitions Data Monitoring: The regular evaluation of data and documentation collected during a study to ensure both adherence to the approved investigative plan and
Investigator-Initiated INDs
 Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Investigator-Initiated INDs Marjorie Small, RN, CCRC Office of Clinical Research 23 May 2011 PPHS/IRB Research Grand Rounds Outline of Presentation I. What is an IND? II. Code of Federal Regulations III.
Human Research Protection Program. Investigator Manual
 Human Research Protection Program Revised July 7, 2014 HRP-910 7/7/2014 2 of 10 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection
Human Research Protection Program Revised July 7, 2014 HRP-910 7/7/2014 2 of 10 Table of Contents What is the purpose of this manual?... 3 What is Human Research?... 3 What is the Human Research Protection