Conformity Assessment of Medical Devices Under The New MDR
|
|
|
- Brandon Watts
- 6 years ago
- Views:
Transcription
1 Conformity Assessment of Medical Devices Under The New MDR (Dec. 06, 2017) Tina Lochner, Medcert Slide 1
2 Agenda Scope: MDR Article 1 and 2 MDR Conformity Assessment: MHRA* (UK Competent Authority) MDR Annex IX, X, XI Class I, IIa, IIb, III and Custom Made Devices Must Know What requirements need to be met for a conformity assessment? Summary *Medicines and Healthcare Products Agency Slide 2
3 Scope of the new MDR - Article 1egulation [1.] This Regulation lays down rules concerning the placing on the market, making available on the market or putting into service of medical devices for human use and accessories for such devices in the Union. This Regulation also applies to clinical investigations concerning such medical devices and accessories conducted in the Union. [2.] This Regulation shall also apply, as from the date of application of common specifications adopted pursuant to Article 9, to the groups of products without an intended medical purpose that are listed in Annex XVI, e.g. contact lenses; equipment for liposuction; infra-red, visible light and ultra-violet emitting equipment; equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields; substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction. [3.] Devices with both a medical and a non-medical intended purpose shall fulfil cumulatively the requirements applicable to devices with an intended medical purpose and those applicable to devices without an intended medical purpose. Slide 3
4 Scope of the new MDR - Article 2egulation Medical device means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes: diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease, diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability, investigation, replacement or modification of the anatomy or of a physiological or pathological process or state, providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations, and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means. Slide 4
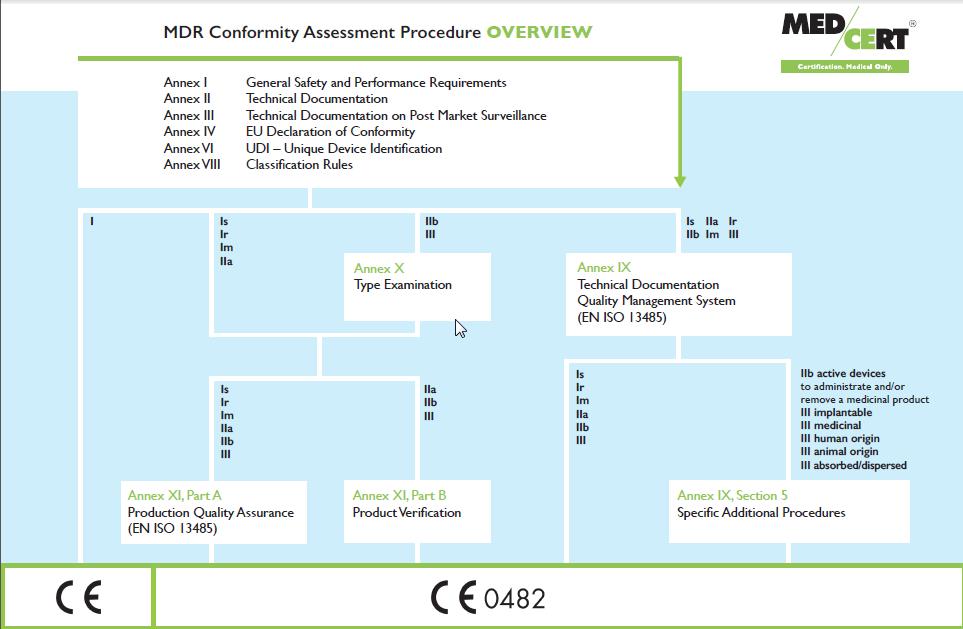
5 MDR Conformity Assessment ANNEX VIII CLASSIFICATION RULES CHAPTER I Definitions specific to classification rules CHAPTER II Implementing rules CHAPTER III Classification rules ANNEX IX CONFORMITY ASSESSMENT BASED ON A QUALITY MANAGEMENT SYSTEM AND ON ASSESSMENT OF TECHNICAL DOCUMENTATION CHAPTER I Quality Management System CHAPTER II Assessment of the technical documentation CHAPTER III Administrative provisions ANNEX X CONFORMITY ASSESSMENT BASED ON TYPE-EXAMINATION ANNEX XI CONFORMITY ASSESSMENT BASED ON PRODUCT CONFORMITY VERIFICATION PART A Production quality assurance PART B Product verification Slide 5

6 Device Classification Slide 6

7 Classification Conformity Assessment Slide 7
8 Annex IX Conformity Assessment Based On A Quality Management System And On Assessment Of Technical Documentation - Quality management system assessment: application with NB and Notified Body access to the technical documentation - Quality management system audit: including technical documentation assessment for devices selected on a representative basis per published guidance developed by the MDCG* -> Notified Body documents its rationale for the samples taken. - Assessment of the technical documentation: special assessment procedures for certain class III and class IIb devices** require that NB transmits a clinical evaluation assessment report, along with the manufacturer's clinical evaluation documentation, to the Commission for transmission to the relevant expert panel; NB to seek scientific opinion from a competent authority or EMA, change assessment, and batch verification based on official certificates issued by a Member State laboratory or a designated laboratory or description of ADME and other properties where applicable *Medical Device Coordination Group **e.g. devices incorporating a medicinal substance, devices incorporating human blood or plasma derivative or a substance that, if used separately, may be considered to be a medicinal product, devices manufactured utilizing derivatives of tissues or cells of human origin, devices manufactured utilizing animal tissue rendered non-viable or utilizing non-viable products derived from animal tissue, devices that are composed of substances or of combinations of substances that are absorbed by or locally dispersed in the human body Slide 8
9 Annex IX Conformity Assessment Based On A Quality Management System And On Assessment Of Technical Documentation - Assessment of substantial changes to the quality management system or the device-range covered - Surveillance assessment: Audits at least once every 12 months including manufacturer's suppliers and/or subcontractors; unannounced audits randomly performed at least once every five years on the site of the manufacturer and, where appropriate, of the manufacturer's suppliers and/or subcontractors combined with the periodic surveillance assessment or be performed in addition; surveillance assessment to include an assessment of the technical documentation (class IIa/IIb devices) or a test of the approved parts and/or materials that are essential for the integrity of the device, including, where appropriate, a check that the quantities of produced or purchased parts and/or materials correspond to the quantities of finished devices (class III). Slide 9
10 Annex X Assessment Based On Type-Examination EU type-examination is the procedure whereby a Notified Body ascertains and certifies that a device, including its technical documentation and relevant life cycle processes and a corresponding representative sample of the device production envisaged, fulfils the relevant provisions of this Regulation. - Application with NB - NB assessment of technical documentation - NB review the clinical evidence - NB to carry out or arrange for the appropriate assessments and the physical or laboratory tests - Change assessment Slide 10
11 Annex XI Conformity Assessment Based On Product Conformity Verification The objective of the conformity assessment based on product conformity verification is to ensure that devices conform to the type for which an EU type-examination certificate has been issued, and that they meet the provisions of this Regulation which apply to them. PART A PRODUCTION QUALITY ASSURANCE - Quality management system assessment: application with NB - Surveillance assessment as in Annex IX - Batch verification in the case of devices incorporating, as an integral part, a medicinal substance which, if used separately, would be considered to be a medicinal product derived from human blood or human plasma as in Annex IX - EU declaration of conformity by manufacturer and technical documentation assessment by NB for the devices selected on a representative basis Slide 11
12 Annex XI Conformity Assessment Based On Product Conformity Verification PART B PRODUCT VERIFICATION Product verification shall be understood to be the procedure whereby after examination of every manufactured device, the manufacturer, by issuing an EU declaration of conformity in accordance with Article 19 and Annex IV, shall be deemed to ensure and to declare that the devices which have been subject to the procedure set out in Sections 14 and 15 conform to the type described in the EU type-examination certificate and meet the requirements of this Regulation which apply to them. Verification by examination and testing of every product or batch verification of each batch in the case of devices incorporating, as an integral part, a medicinal substance which, if used separately, would be considered to be a medicinal product derived from human blood or human plasma. Slide 12
13 Must know The requirement for manufacturers to conduct a conformity assessment before placing their device on the market, and for higher risk devices to involve a Notified Body, is unchanged. Slide 13
14 Class I devices: Similar to the current MDD Annex VII EC declaration of conformity. - New EU Declaration of Conformity (new Article 19) prepared by manufacturer, - Fulfill general obligations of all manufacturers (new Article 10), - Involvement of a Notified Body is limited for Class I devices, and only required for sterile devices, reusable surgical instruments or devices with a measuring function. Slide 14
15 Class IIa devices: Similar to the current MDD, manufacturers of Class IIa devices have the option of following the same conformity assessment route as for Class IIb devices, the new EU MDR s Annex IX, with the Notified Body only assessing representative technical documentation. Manufacturers may choose not to follow the full quality management system approach essentially similar to the current MDD s Annex VII EC declaration of Conformity combined with either Annex IV or Annex V: - Compile the new technical documentation (new Annex II) - Manufacturer prepares the new EU declaration of conformity (new Article 19) - and then the Notified Body either: - (a) assesses the Technical Documentation of a representative sample of the devices (Annex XI, Part A, Section 10), - or (b) carries out tests to confirm the conformity of the devices (Annex XI, Part B, Section 18). Slide 15
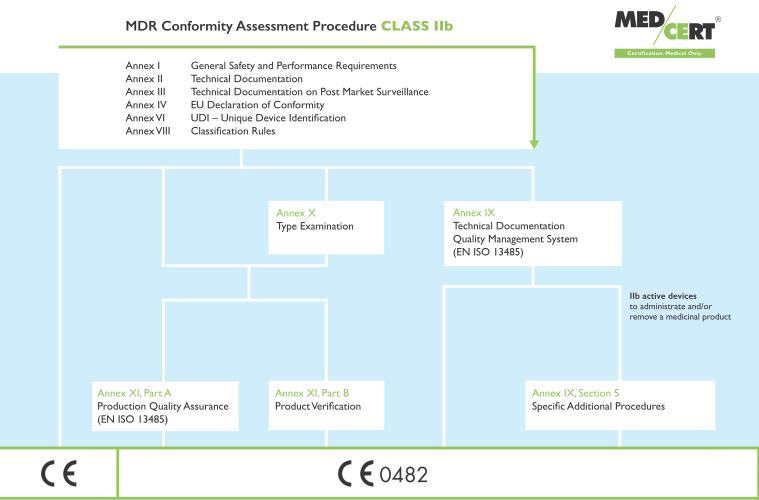
16 Class IIb devices: Similar to the current MDD, manufacturers of Class IIb devices have the option of following the same conformity assessment route as for Class III devices, the new EU MDR s Annex IX with the difference that the Notified Body is only required to assess the technical documentation of at least one representative device of each generic device group produced by the manufacturer. As with the current MDD, there are alternative routes for manufacturers of Class IIb devices who chose not to follow the full quality management system approach. These are the same as those for Class III devices with one fewer alternative available to manufacturers of Class IIb devices compared to the MDD: there is no equivalent to the MDD s Annex VI product quality assurance. Manufacturers currently following this route will have to choose an option from the new Annex XI. Either Part A, Production Quality Assurance, or Part B, Product Verification. Slide 16
17 Class III devices: The current MDD s Annex II full quality assurance route will be replaced by the new EU MDR s Annex IX conformity assessment based on quality management system assurance and assessment of the technical documentation. There are alternatives for those manufacturers who chose not to follow the full quality management system approach: the current MDD alternative for Class III devices of Annex III EC type-examination, combined with either Annex IV EC verification or Annex V production quality assurance will be replaced by the new EU MDR s Annex X conformity assessment based on type examination combined with new Annex XI conformity assessment based on product conformity verification. This is essentially identical to those of the current MDD. The new EU MDR s Annex XI conformity assessment based on product conformity verification includes both the MDD s current options; Part A being the new Production Quality Assurance route, replacing the current MDD s Annex V production quality assurance. Part B being the new Product Verification route, replacing the current MDD s Annex IV EC verification. Slide 17
18 Custom made devices: Unlike the current MDD where the requirements for custom made devices are part of the current MDD Annex VIII the new EU MDR has a dedicated Annex, Annex XIII procedure for custom made devices. However, the requirements to draw up a statement about the device and keep records etc. are fundamentally the same as in the current MDD. The exception being class III custom made devices, where a quality system assessment by a Notified Body is required; either the quality management system assessment of the new Annex IX, Chapter 1 or the Production Quality Assurance of the new Annex XI, Part A. Slide 18

19 Slide 19

20 Slide 20
21 Slide 21
22 Slide 22
23 Slide 23
24 What requirements need to be met for a conformity assessment? 1. General Safety and Performance Requirements (Annex I MDR): - Benefits must outweigh risks and achieve claimed performance supported by clinical evidence and investigation - Chemical, physical and biological properties for medical devices disclosed - [Performance characteristics for ivds disclosed] - Information supplied by manufacturer with the device; e.g. IFU and correct device labelling 2. Technical documentation (Annex II MDR) 3. Harmonized standards / common specifications (Articles 8 and 9 MDR) See Annex IX, X and XI of MDR for details Slide 24

25 Summary Slide 25
26 Slide 26






27 Visit us: ( Slide 27
Update on the IVDR. Sue Spencer
 Update on the IVDR Sue Spencer Caution The new regulations are draft the principles have now been agreed but the Annexes are subject to minor changes Further details will be added later pre and post application
Update on the IVDR Sue Spencer Caution The new regulations are draft the principles have now been agreed but the Annexes are subject to minor changes Further details will be added later pre and post application
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards. 26 th September / V. Fischer / Rev. 01
 ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
ASQ Tappan Zee Section Medical Devices, New Regulations and Standards 26 th September / V. Fischer / Rev. 01 Agenda Medical Device, General Aspects Examples Quality System(s) New Regulations & Standards
Ready or Not: The New Medical Device Regulations Are Here!
 Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Ready or Not: The New Medical Device Regulations Are Here! Felicia R Cochran, PhD, CMPP TM feliciacochran@earthlink.net FR Cochran 1 General Disclaimers This presentation represents the knowledge, professional
Regulation of software a medical device
 Regulation of software a medical device IOF-TTO Event 17.10.2017 Steve Eglem Responsible of clinical investigation @ famhp Member of the medical device European Clinical and Evaluation (CIE) working group
Regulation of software a medical device IOF-TTO Event 17.10.2017 Steve Eglem Responsible of clinical investigation @ famhp Member of the medical device European Clinical and Evaluation (CIE) working group
GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES
 MDEG - 2007-12 - II-3.3 MSOGClassIGuidance_Final GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES Foreword These guidance notes do not aim to be a definite interpretation of National Laws and/or
MDEG - 2007-12 - II-3.3 MSOGClassIGuidance_Final GUIDANCE NOTES FOR MANUFACTURERS OF CLASS I MEDICAL DEVICES Foreword These guidance notes do not aim to be a definite interpretation of National Laws and/or
European Medical Device Regulations (MDR): What To Expect
: What To Expect") European Medical Device Regulations (MDR): What To Expect MDQC March 2016 Paul Brooks Senior Vice President Healthcare Solutions Copyright 2015 BSI. All rights reserved. Sources for MDR Update Commission
European Medical Device Regulations (MDR): What To Expect MDQC March 2016 Paul Brooks Senior Vice President Healthcare Solutions Copyright 2015 BSI. All rights reserved. Sources for MDR Update Commission
The challenges of software medical device regulation.
 The challenges of software medical device regulation. david.grainger@mhra.gov.uk Introduction A brief history of software device regulation A look at the new device regulations 2 Current framework In Vitro
The challenges of software medical device regulation. david.grainger@mhra.gov.uk Introduction A brief history of software device regulation A look at the new device regulations 2 Current framework In Vitro
Medical Devices Regulation (MDR) Readiness Review
 Readiness Review") ` Medical Devices Regulation (MDR) Readiness Review Company Name Address Certification No. Date: Contact Name Job Title Telephone Email How ready are you for the Medical Devices Regulation? The MDR, which
` Medical Devices Regulation (MDR) Readiness Review Company Name Address Certification No. Date: Contact Name Job Title Telephone Email How ready are you for the Medical Devices Regulation? The MDR, which
Implications of the new MDR from a Product Testing and Certification Perspective
 Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
Implications of the new MDR from a Product Testing and Certification Perspective Helping You to Access Global Markets FAST and PREDICTABLY www.test-medical-devices.com 1 Hans Gerd Evering MDR - Implications
Due diligence in the European medical devices industry
 Due diligence in the European medical devices industry Alison Dennis, Reed Smith LLP www.practicallaw.com/0-205-5707 As the medical devices industry is highly regulated, determining a target company's
Due diligence in the European medical devices industry Alison Dennis, Reed Smith LLP www.practicallaw.com/0-205-5707 As the medical devices industry is highly regulated, determining a target company's
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS
 Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Medical Device Regulatory Framework 9 SEPTEMBER 2015 FUNDISA CONFERENCE JANE ROGERS Key Topics Definitions Essential Principles Classification Conformity Assessment Framework License to Manufacture, Import,
Medical Device Regulation Overview
 Medical Device Regulation Overview Dr Haidong Liang, PhD Clifton Medtech Consulting info@cliftonmedtech.com http://cliftonmedtech.com/ What is the issue? Existing EU Directives dating back to the 1990s
Medical Device Regulation Overview Dr Haidong Liang, PhD Clifton Medtech Consulting info@cliftonmedtech.com http://cliftonmedtech.com/ What is the issue? Existing EU Directives dating back to the 1990s
FINAL DOCUMENT. Global Harmonization Task Force (revision of GHTF/SG1/N29:2005)
") GHTF/SG1/N071:2012 FINAL DOCUMENT Global Harmonization Task Force (revision of GHTF/SG1/N29:2005) Title: Definition of the Terms Medical Device and In Vitro Diagnostic (IVD) Medical Device Authoring Group:
GHTF/SG1/N071:2012 FINAL DOCUMENT Global Harmonization Task Force (revision of GHTF/SG1/N29:2005) Title: Definition of the Terms Medical Device and In Vitro Diagnostic (IVD) Medical Device Authoring Group:
MDR ID: Definition: Applicable:
 MDR Classification: (Reference Medical Device Regulation EU 2017/745, Annex VIII) Product: Product Name 1. DURATION OF USE MDR ID: Definition: Applicable: - Invasive Device: Continue Go to 2. Invasive
MDR Classification: (Reference Medical Device Regulation EU 2017/745, Annex VIII) Product: Product Name 1. DURATION OF USE MDR ID: Definition: Applicable: - Invasive Device: Continue Go to 2. Invasive
EU MDR Timeline. Dr. Christian B. Fulda Jones Day FDLI Annual Conference Access materials at fdli.org/annual2018
 EU MDR Timeline Dr. Christian B. Fulda Jones Day EU MDR - Timeline EU has entered into force, but will only apply for products as of May 26, 2020, with transitional period through 2024/2025? Look closer!
EU MDR Timeline Dr. Christian B. Fulda Jones Day EU MDR - Timeline EU has entered into force, but will only apply for products as of May 26, 2020, with transitional period through 2024/2025? Look closer!
The EU s In Vitro Diagnostics (IVD) Regulation:
 Regulation:") The EU s In Vitro Diagnostics (IVD) Regulation: a summary of the regulatory requirements for software and genetic tests The European Union (EU) s In Vitro Diagnostics (IVD) Regulation (of 5 April 2017)
The EU s In Vitro Diagnostics (IVD) Regulation: a summary of the regulatory requirements for software and genetic tests The European Union (EU) s In Vitro Diagnostics (IVD) Regulation (of 5 April 2017)
MDR. Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation
 MDR Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation Suzanne Halliday, D.Phil. Jaishankar Kutty, Ph.D. Ronald Rakos, Ph.D BSI Roadshow, October 2017
MDR Device Classification Conformity Assessment Safety & Performance Requirements Technical Documentation Suzanne Halliday, D.Phil. Jaishankar Kutty, Ph.D. Ronald Rakos, Ph.D BSI Roadshow, October 2017
GENERAL AND ORGANISATIONAL REQUIREMENTS
 NBOG working document applicable for MDR and IVDR WD 2017-1 Draft list of documents to be submitted in the application for designation as a notified body under Regulation (EU) 2017/745 and Regulation (EU)
NBOG working document applicable for MDR and IVDR WD 2017-1 Draft list of documents to be submitted in the application for designation as a notified body under Regulation (EU) 2017/745 and Regulation (EU)
TERMS AND DEFINITIONS
 TERMS AND DEFINITIONS Advisory notice A notice issued by the organization, subsequent to delivery of the medical device, to provide supplementary information and/or to advise what action should be taken
TERMS AND DEFINITIONS Advisory notice A notice issued by the organization, subsequent to delivery of the medical device, to provide supplementary information and/or to advise what action should be taken
Medical Devices. LATVIA LAWIN Klavins & Slaidins
 Medical Devices LATVIA LAWIN Klavins & Slaidins CONTACT INFORMATION Sarmis Spilbergs LAWIN Klavins & Slaidins Elizabetes 15, Riga, LV 1010, Latvia +371 67814848 sarmis.spilbergs@lawin.lv www.lawin.com
Medical Devices LATVIA LAWIN Klavins & Slaidins CONTACT INFORMATION Sarmis Spilbergs LAWIN Klavins & Slaidins Elizabetes 15, Riga, LV 1010, Latvia +371 67814848 sarmis.spilbergs@lawin.lv www.lawin.com
GS1 Ireland Healthcare User Group (HUG) Information Day
 Information Day") GS1 Ireland Healthcare User Group (HUG) Information Day Regulatory update Medical Devices and the impact for Irish Healthcare Sinead Duggan, HPRA 28 th March 2017 29/03/2017 2 Medical Devices Regulation
GS1 Ireland Healthcare User Group (HUG) Information Day Regulatory update Medical Devices and the impact for Irish Healthcare Sinead Duggan, HPRA 28 th March 2017 29/03/2017 2 Medical Devices Regulation
EMA perspective on increasing focus on review of device components of medicinal products
 EMA perspective on increasing focus on review of device components of medicinal products TOPRA Annual Human Medicines Symposium 2 4 October 2017 - Victoria Park Plaza Hotel, London Presented by Armin Ritzhaupt,
EMA perspective on increasing focus on review of device components of medicinal products TOPRA Annual Human Medicines Symposium 2 4 October 2017 - Victoria Park Plaza Hotel, London Presented by Armin Ritzhaupt,
IVD Regulation 2017/746
 IVD Regulation 2017/746 Dr. Anne Van Nerom Famhp 2017-06-13 Recast-symposium Auditorium Storck (Eurostation II) Rue Juliette Wytsmanstraat 14 1050 Brussels Belgium T +32 2 642 51 11 F +32 2 642 50 01 email:
IVD Regulation 2017/746 Dr. Anne Van Nerom Famhp 2017-06-13 Recast-symposium Auditorium Storck (Eurostation II) Rue Juliette Wytsmanstraat 14 1050 Brussels Belgium T +32 2 642 51 11 F +32 2 642 50 01 email:
MEDICAL DEVICE. Technical file.
 MEDICAL DEVICE Technical file www.icaro-research.eu ICARO MDTF v1.0 1 Mar 2016 1. Do you plan to launch your medical device in Europe? If you re reading this, chances are good that you re considering introducing
MEDICAL DEVICE Technical file www.icaro-research.eu ICARO MDTF v1.0 1 Mar 2016 1. Do you plan to launch your medical device in Europe? If you re reading this, chances are good that you re considering introducing
Classification under the IVD Regulation
 Classification under the IVD Regulation Nick Baker Head of IVD Notified Body LRQA Ltd Improving performance, reducing risk IVD regulation shift in regulatory scrutiny Under the current IVD directive 10-15%
Classification under the IVD Regulation Nick Baker Head of IVD Notified Body LRQA Ltd Improving performance, reducing risk IVD regulation shift in regulatory scrutiny Under the current IVD directive 10-15%
Changes to the Medical Devices Directive and affect on Manufacturers
 TÜV Product Service Ltd Webinar 18 th November 2009 Changes to the Medical Devices Directive and affect on Manufacturers Henry Sibun Manager, Medical & Health Services UK CONTENTS / 1. Introduction 1.
TÜV Product Service Ltd Webinar 18 th November 2009 Changes to the Medical Devices Directive and affect on Manufacturers Henry Sibun Manager, Medical & Health Services UK CONTENTS / 1. Introduction 1.
HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737)
") MDA/GL No 1: July 2013 Guidelines for implementation of medical device regulatory system HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737) [Appendix 4 Schedule 3 Medical
MDA/GL No 1: July 2013 Guidelines for implementation of medical device regulatory system HOW TO APPLY FOR MEDICAL DEVICE REGISTRATION UNDER MEDICAL DEVICE ACT 2012 (ACT 737) [Appendix 4 Schedule 3 Medical
GETTING READY FOR THE EUROPEAN UNION (EU) MEDICAL DEVICE REGULATION (MDR)
 MEDICAL DEVICE REGULATION (MDR)") GETTING READY FOR THE EUROPEAN UNION (EU) MEDICAL DEVICE REGULATION (MDR) Caroline Leab Abbott Director of Regulatory Affairs David Wolf PTC Program Director of Medical Device Strategy liveworx.com NEW
GETTING READY FOR THE EUROPEAN UNION (EU) MEDICAL DEVICE REGULATION (MDR) Caroline Leab Abbott Director of Regulatory Affairs David Wolf PTC Program Director of Medical Device Strategy liveworx.com NEW
Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations
 Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations Paul Brooks Vice President Healthcare, Americas Presentation to:
Update on Regulatory Environment- Europe Experience with 2007/47/EC M5 & Discussions on Possible Recast of EU Medical Device Regulations Paul Brooks Vice President Healthcare, Americas Presentation to:
Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994
 Regulations, 1994") Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994 SUR-G0006-2 27 AUGUST 2010 This guide does not purport to be an interpretation of law and/or
Guide for Class I Manufacturers on Compliance with European Communities (Medical Devices) Regulations, 1994 SUR-G0006-2 27 AUGUST 2010 This guide does not purport to be an interpretation of law and/or
The New EU IVDR. Overview of the Main Changes & Clinical Data Requirements. Advance Regulatory Consulting Ltd.
 Overview of the Main Changes & Clinical Data Requirements Advance Regulatory Consulting Ltd. : Timeline: Entry in to force Q2 (Apr) 2017 Adoption: +6m NB s apply for designation IVDs classified as Class
Overview of the Main Changes & Clinical Data Requirements Advance Regulatory Consulting Ltd. : Timeline: Entry in to force Q2 (Apr) 2017 Adoption: +6m NB s apply for designation IVDs classified as Class
DRAFT MEDICAL DEVICE GUIDANCE DOCUMENT
 November 2015 DRAFT DRAFT MEDICAL DEVICE GUIDANCE DOCUMENT REQUIREMENTS FOR LABELLING OF MEDICAL DEVICES Medical Device Authority MINISTRY OF HEALTH MALAYSIA Contents Page Preface... iii 1 Introduction.
November 2015 DRAFT DRAFT MEDICAL DEVICE GUIDANCE DOCUMENT REQUIREMENTS FOR LABELLING OF MEDICAL DEVICES Medical Device Authority MINISTRY OF HEALTH MALAYSIA Contents Page Preface... iii 1 Introduction.
The Medical Device Coordination Group: a new Authority Under EU Device Regulations
 The Medical Device Coordination Group: a new Authority Under EU Device Regulations By Robert D. Cumming and Nancy W. Mathewson, Esq. This article discusses the organization and functions of the Medical
The Medical Device Coordination Group: a new Authority Under EU Device Regulations By Robert D. Cumming and Nancy W. Mathewson, Esq. This article discusses the organization and functions of the Medical
Guide for Ethics Committees on Clinical Investigation of Medical Devices
 Guide for Ethics Committees on Clinical Investigation of Medical Devices AUT-G0044-2 04 JUNE 2010 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes
Guide for Ethics Committees on Clinical Investigation of Medical Devices AUT-G0044-2 04 JUNE 2010 This guide does not purport to be an interpretation of law and/or regulations and is for guidance purposes
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155
 MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
MEDICAL DEVICE CLINICAL INVESTIGATIONS AND ISO 14155 EXECUTIVE SUMMARY Medical device regulations around the world generally require manufacturers of most types of medical devices to supply data as part
Recommendations on Medical Device and IVD Field Safety Corrective Actions and Recalls using Unique Device Identifiers & GS1 Standards
 Recommendations on Medical Device and IVD Field Safety Corrective Actions and Recalls using Unique Device Identifiers & GS1 Standards January 2017 Contributors Name Andy Crosbie (Sub group chair) David
Recommendations on Medical Device and IVD Field Safety Corrective Actions and Recalls using Unique Device Identifiers & GS1 Standards January 2017 Contributors Name Andy Crosbie (Sub group chair) David
Medical Device Epidemiology Introduction
 Medical Device Epidemiology Introduction October 24, 2014 Mary E Ritchey, PhD Pre-Conference Educational Session International Conference on Pharmacoepidemiology Course Overview Topic Time Welcome and
Medical Device Epidemiology Introduction October 24, 2014 Mary E Ritchey, PhD Pre-Conference Educational Session International Conference on Pharmacoepidemiology Course Overview Topic Time Welcome and
UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS
 UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS Executive Summary European Medicines Agency (EMA) regulations for licensing of medical devices include the use of authorized
UNANNOUNCED EU MEDICAL DEVICE AUDITS BY NOTIFIED BODIES: IMPACT ON SUPPLIERS Executive Summary European Medicines Agency (EMA) regulations for licensing of medical devices include the use of authorized
Recast Medical Device directives Impacts on materiovigilance
 Recast Medical Device directives Impacts on materiovigilance Journée Vigilance 23.03.2017 Valérie Nys Revision of the EU Medical Devices Legislation Directive 90/385/EEC on active implantable medical devices
Recast Medical Device directives Impacts on materiovigilance Journée Vigilance 23.03.2017 Valérie Nys Revision of the EU Medical Devices Legislation Directive 90/385/EEC on active implantable medical devices
VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE
 VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE Post-Market Surveillance dal punto di vista dell O.N in vista dei nuovi Regolamenti (MDR/IVDR) Francesco
VIGILANZA E SORVEGLIANZA POST- COMMERCIALIZZAZIONE VIGILANZA E SORVEGLIANZA POST-COMMERCIALIZZAZIONE Post-Market Surveillance dal punto di vista dell O.N in vista dei nuovi Regolamenti (MDR/IVDR) Francesco
Technical Documentation
 Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
Technical Documentation Helga Seiler M.Sc. Vision Science and Business (Optometry) Manager RA Disclaimer 2 The following list of information is not exhaustive The information and views given in the following
MedDev Rev 4 Medical Devices Regulation. Clinical Evidence Requirements Key Changes and Clarifications. Alan Eller 21 March 2017
 MedDev 2.7.1 Rev 4 Medical Devices Regulation Clinical Evidence Requirements Key Changes and Clarifications Alan Eller 21 March 2017 Copyright 2016 BSI. All rights reserved. 1 Clinical Evidence Requirements
MedDev 2.7.1 Rev 4 Medical Devices Regulation Clinical Evidence Requirements Key Changes and Clarifications Alan Eller 21 March 2017 Copyright 2016 BSI. All rights reserved. 1 Clinical Evidence Requirements
Regulating Cell Therapy: An Ex-Regulators Perspective
 Regulating Cell Therapy: An Ex-Regulators Perspective Christopher A Bravery 1 Introduction Regulation of Healthcare products in the EU What is cell therapy? How does it fit into existing framework? Is
Regulating Cell Therapy: An Ex-Regulators Perspective Christopher A Bravery 1 Introduction Regulation of Healthcare products in the EU What is cell therapy? How does it fit into existing framework? Is
ISO INTERNATIONAL STANDARD
 INTERNATIONAL STANDARD ISO 11607-1 First edition 2006-04-15 Packaging for terminally sterilized medical devices Part 1: Requirements for materials, sterile barrier systems and packaging systems Emballages
INTERNATIONAL STANDARD ISO 11607-1 First edition 2006-04-15 Packaging for terminally sterilized medical devices Part 1: Requirements for materials, sterile barrier systems and packaging systems Emballages
ISO INTERNATIONAL STANDARD. Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process
 INTERNATIONAL STANDARD ISO 10993-1 Fourth edition 2009-10-15 Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process Évaluation biologique des dispositifs
INTERNATIONAL STANDARD ISO 10993-1 Fourth edition 2009-10-15 Biological evaluation of medical devices Part 1: Evaluation and testing within a risk management process Évaluation biologique des dispositifs
Regulations and Directives Past, Present, Future
 Regulations and Directives Past, Present, Future Haris Memić, Almir Badnjević and Zijad Džemić Abstract Regulations are binding acts which are obligatory in the European Union. All members of the European
Regulations and Directives Past, Present, Future Haris Memić, Almir Badnjević and Zijad Džemić Abstract Regulations are binding acts which are obligatory in the European Union. All members of the European
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL GUIDELINES ON MEDICAL DEVICES
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Competitiveness in pharmaceuticals, medical devices, cosmetics MEDDEV. 2.11/1 rev.1 April 2005 GUIDELINES ON MEDICAL DEVICES
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Competitiveness in pharmaceuticals, medical devices, cosmetics MEDDEV. 2.11/1 rev.1 April 2005 GUIDELINES ON MEDICAL DEVICES
CE Marking for Medical devices
 CE Marking of Medical Devices Bart Mersseman Head of Notified Body SGS Belgium Thursday Octobere 12 th 2017 Vlaamse dag van de CE-markering CE Marking for Medical devices Prologue: Legislation in Europe
CE Marking of Medical Devices Bart Mersseman Head of Notified Body SGS Belgium Thursday Octobere 12 th 2017 Vlaamse dag van de CE-markering CE Marking for Medical devices Prologue: Legislation in Europe
New EU Regulation on medical devices
 New EU Regulation on medical devices Summary of key changes and challenges for implementation Adrian Bartlett Medical Devices, EU Policy Manager Nickie Colson Policy Advisor, EU & International Team Content
New EU Regulation on medical devices Summary of key changes and challenges for implementation Adrian Bartlett Medical Devices, EU Policy Manager Nickie Colson Policy Advisor, EU & International Team Content
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior
 Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Panel Discussion: European Medical Device Regulations Preparing for the Storm Moderator: Lenita Y. Sims Spears, Senior Quality Consultant/Senior Regulatory and Compliance Counsel, BioTeknica Panelists:
Conformity Assessment of Own Brand Labelling
 NBRG/306/06 Co-ordination of Title: Chapter: Conformity Assessment of Own Brand Labelling 2.5.5 Conformity assessment for particular product groups Text: Key words: manufacturer means the natural or legal
NBRG/306/06 Co-ordination of Title: Chapter: Conformity Assessment of Own Brand Labelling 2.5.5 Conformity assessment for particular product groups Text: Key words: manufacturer means the natural or legal
Biological evaluation of medical devices --
 Translated English of Chinese Standard: GB/T16886.1-2011 Translated by: www.chinesestandard.net Wayne Zheng et al. Email: Sales@ChineseStandard.net ICS 11.040.01 C 30 NATIONAL STANDARD OF THE PEOPLE S
Translated English of Chinese Standard: GB/T16886.1-2011 Translated by: www.chinesestandard.net Wayne Zheng et al. Email: Sales@ChineseStandard.net ICS 11.040.01 C 30 NATIONAL STANDARD OF THE PEOPLE S
IVDR Workshop Diagnostik Akademie, Sven Hoffmann Global Head of Technical Competence Center IVD TÜV Rheinland LGA Products GmbH
 IVDR Workshop Diagnostik Akademie, 19.10.2017 Sven Hoffmann Global Head of Technical Competence Center IVD TÜV Rheinland LGA Products GmbH Agenda Kapitel Thema Dauer Referent 1 Clinical Evidence 2 Performance
IVDR Workshop Diagnostik Akademie, 19.10.2017 Sven Hoffmann Global Head of Technical Competence Center IVD TÜV Rheinland LGA Products GmbH Agenda Kapitel Thema Dauer Referent 1 Clinical Evidence 2 Performance
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS
 FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
FREQUENTLY ASKED QUESTIONS DURING ELECTRO-MEDICAL DEVICE MARKET ACCESS Helping You to Access Global Markets FAST and PREDICTABLY Vishal Thakker, MEng(Hons), AMIMechE Scheme Manager/Product Specialist BSI
AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5. The Declaration of Conformity
 AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5 The Declaration of Conformity 30 October 2003 DISCLAIMER This document is provided for guidance only. It should not be relied upon to address every
AUSTRALIAN MEDICAL DEVICES GUIDANCE DOCUMENT NUMBER 5 The Declaration of Conformity 30 October 2003 DISCLAIMER This document is provided for guidance only. It should not be relied upon to address every
Flexible, robust solutions from BSI. An In Vitro Diagnostic Notified Body. Expertise and experience. IVD regulatory solutions
 Flexible, robust solutions from BSI An In Vitro Diagnostic Notified Body Expertise and experience IVD regulatory solutions Updated May 2017 Your guide to the In Vitro Diagnostic Directive The purpose of
Flexible, robust solutions from BSI An In Vitro Diagnostic Notified Body Expertise and experience IVD regulatory solutions Updated May 2017 Your guide to the In Vitro Diagnostic Directive The purpose of
PART 1 DEVICE MASTER FILE
 Form-No.: 01 03 00 19e Proposal for a General Product Documentation 0.1 Cover page (company, title, No., release, signature etc.) 0.2 Content (e. g. as document master file) 0.3 General information about
Form-No.: 01 03 00 19e Proposal for a General Product Documentation 0.1 Cover page (company, title, No., release, signature etc.) 0.2 Content (e. g. as document master file) 0.3 General information about
The new EU Regulations on medical devices and first steps of their implementation
 The new EU Regulations on medical devices and first steps of their implementation TOPRA Annual Medical Devices Symposium 2017 London, 3 October 2017 Erik HANSSON Deputy Head of Unit Health Technology and
The new EU Regulations on medical devices and first steps of their implementation TOPRA Annual Medical Devices Symposium 2017 London, 3 October 2017 Erik HANSSON Deputy Head of Unit Health Technology and
POSITION PAPER Moving from the MDD to the MDR
 A summary of Key Changes regarding Sterile Packaging and considerations on recommended changes to standards Introduction EN ISO 11607 specifies the requirements and test methods for materials, preformed
A summary of Key Changes regarding Sterile Packaging and considerations on recommended changes to standards Introduction EN ISO 11607 specifies the requirements and test methods for materials, preformed
FINAL DOCUMENT. International Medical Device Regulators Forum. Medical Device Regulatory Audit Reports
 FINAL DOCUMENT International Medical Device Regulators Forum Title: Authoring Group: Medical Device Regulatory Audit Reports IMDRF MDSAP Working Group Date: 2 October 2015 Toshiyoshi Tominaga, IMDRF Chair
FINAL DOCUMENT International Medical Device Regulators Forum Title: Authoring Group: Medical Device Regulatory Audit Reports IMDRF MDSAP Working Group Date: 2 October 2015 Toshiyoshi Tominaga, IMDRF Chair
Clinical Evaluation and Clinical Investigation update MDR
 Clinical Evaluation and Clinical Investigation update MDR December 5 th, 2017 Carine Cochereau, Cardinal Health Slide 1 Directives: obligation for Member State (MS) to implement provisions into their national
Clinical Evaluation and Clinical Investigation update MDR December 5 th, 2017 Carine Cochereau, Cardinal Health Slide 1 Directives: obligation for Member State (MS) to implement provisions into their national
Medical Device Directive
 Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
Medical Device Directive WG9 - IEC/SC 62A ISO/TC 184/SC 2 Joint Working Group 9 Saeed Zahedi 4 th of July 2012 Blatchford Copyright 2012 Commercial in confidence Definition and Requirements MDD is law,
ASEAN AGREEMENT ON MEDICAL DEVICE DIRECTIVE
 ASEAN AGREEMENT ON MEDICAL DEVICE DIRECTIVE The Governments of Brunei Darussalam, the Kingdom of Cambodia, the Republic of Indonesia, the Lao People's Democratic Republic, Malaysia, the Republic of the
ASEAN AGREEMENT ON MEDICAL DEVICE DIRECTIVE The Governments of Brunei Darussalam, the Kingdom of Cambodia, the Republic of Indonesia, the Lao People's Democratic Republic, Malaysia, the Republic of the
PROPOSED REVISED DOCUMENT
 SG1(PD)/N011R20 PROPOSED REVISED DOCUMENT Global Harmonization Task Force Title: Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical
SG1(PD)/N011R20 PROPOSED REVISED DOCUMENT Global Harmonization Task Force Title: Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical
WELMEC European cooperation in legal metrology
 WELMEC 8.6 Issue 1 WELMEC European cooperation in legal metrology Measuring Instruments Directive 2004/22/EC Presumption of Conformity of the Quality System of Manufacturers with Module D or H 1 when EN
WELMEC 8.6 Issue 1 WELMEC European cooperation in legal metrology Measuring Instruments Directive 2004/22/EC Presumption of Conformity of the Quality System of Manufacturers with Module D or H 1 when EN
ISO INTERNATIONAL STANDARD
 INTERNATIONAL STANDARD ISO 11737-2 Second edition 2009-11-15 Sterilization of medical devices Microbiological methods Part 2: Tests of sterility performed in the definition, validation and maintenance
INTERNATIONAL STANDARD ISO 11737-2 Second edition 2009-11-15 Sterilization of medical devices Microbiological methods Part 2: Tests of sterility performed in the definition, validation and maintenance
The importance of implementing a continuous cycle of improvement for medical devices. Roles for various stakeholders
 The importance of implementing a continuous cycle of improvement for medical devices. Roles for various stakeholders Robert Geertsma, Arjan van Drongelen Robert.Geertsma@RIVM.nl RIVM National Institute
The importance of implementing a continuous cycle of improvement for medical devices. Roles for various stakeholders Robert Geertsma, Arjan van Drongelen Robert.Geertsma@RIVM.nl RIVM National Institute
Medical devices utilizing animal tissues and their derivatives. Part 1: Application of risk management
 Provläsningsexemplar / Preview INTERNATIONAL STANDARD ISO 22442-1 Second edition 2015-11-01 Medical devices utilizing animal tissues and their derivatives Part 1: Application of risk management Dispositifs
Provläsningsexemplar / Preview INTERNATIONAL STANDARD ISO 22442-1 Second edition 2015-11-01 Medical devices utilizing animal tissues and their derivatives Part 1: Application of risk management Dispositifs
MedDev and NB-MED Recommendations
 Titel of 2.1 Scope, field of application, explanation of terms Guidelines relating to the application of AIMD and MDD: Definition of medical devices, accessory and manufacturer Guidelines relating to the
Titel of 2.1 Scope, field of application, explanation of terms Guidelines relating to the application of AIMD and MDD: Definition of medical devices, accessory and manufacturer Guidelines relating to the
Brussels, C(2017) 8179 final. Guidelines
 8179 final. Guidelines") EUROPEAN COMMISSION Brussels, 8.12.2017 C(2017) 8179 final Guidelines Detailed Commission guidelines on good manufacturing practice for investigational medicinal products for human use, pursuant to the
EUROPEAN COMMISSION Brussels, 8.12.2017 C(2017) 8179 final Guidelines Detailed Commission guidelines on good manufacturing practice for investigational medicinal products for human use, pursuant to the
MEDICAL DEVICE GUIDANCE
 Effective from 1 January 2013 MEDICAL DEVICE GUIDANCE GN-15: Guidance on Medical Device Product Registration Revision 5 CONTENTS 1. INTRODUCTION... 4 1.1. Scope... 5 1.2. Definitions... 5 2. RISK CLASSIFICATION
Effective from 1 January 2013 MEDICAL DEVICE GUIDANCE GN-15: Guidance on Medical Device Product Registration Revision 5 CONTENTS 1. INTRODUCTION... 4 1.1. Scope... 5 1.2. Definitions... 5 2. RISK CLASSIFICATION
Surveillance and Medical Devices
 Surveillance and Medical Devices Nicole Pratt Senior Research Fellow Medicine and Device Centre for Research Excellence Contact: Prof Libby Roughead UniSA, GPO Box 2471, Adelaide SA 5001 Libby.roughead@unisa.edu.au
Surveillance and Medical Devices Nicole Pratt Senior Research Fellow Medicine and Device Centre for Research Excellence Contact: Prof Libby Roughead UniSA, GPO Box 2471, Adelaide SA 5001 Libby.roughead@unisa.edu.au
Pharmabiotics: a Regulatory Hurdle in Europe
 Pharmabiotics: a Regulatory Hurdle in Europe Dr. Magali Cordaillat-Simmons PRI Executive Scientist Raleigh, NC, USA September 8th, 2014 PHARMABIOTICS: A REGULATORY HURDLE IN EUROPE I. Introduction to Pharmabiotics
Pharmabiotics: a Regulatory Hurdle in Europe Dr. Magali Cordaillat-Simmons PRI Executive Scientist Raleigh, NC, USA September 8th, 2014 PHARMABIOTICS: A REGULATORY HURDLE IN EUROPE I. Introduction to Pharmabiotics
The rules governing medicinal products in the European Union. Presentation and content of the dossier Edition
 The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
The rules governing medicinal products in the European Union Volume 2B Notice to Applicants Medicinal products for human use Presentation and content of the dossier 1998 Edition EUROPEAN COMMISSION Directorate
GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN MAKING AN ASSESSMENT OF CLINICAL INVESTIGATION NOTIFICATION
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Cosmetics and Medical Devices MEDDEV 2.7.2 December 2008 GUIDELINES ON MEDICAL DEVICES GUIDE FOR COMPETENT AUTHORITIES IN
Post market Surveillance ISO EU Medical Device Regulation
 Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
Post market Surveillance ISO13485 2016 EU Medical Device Regulation Patrick Caines, Ph.D. Baxter Healthcare 15 June 2017 Agenda Post market Regulatory Requirements ISO 13485 2016 Summary of key changes
GUIDELINES ON MEDICAL DEVICES
 EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single Market : regulatory environment, standardisation and New Approach Pressure equipment, medical devices, metrology MEDDEV. 2.11/1 rev.1 February
EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single Market : regulatory environment, standardisation and New Approach Pressure equipment, medical devices, metrology MEDDEV. 2.11/1 rev.1 February
Update on Recast of IVD Directive
 Update on Recast of IVD Directive www.pei.de IVD Classification In-house Assays EU Reference Laboratories C M Nübling, PEI Concerns identified by the European Commission in the Medical Devices Roadmap
Update on Recast of IVD Directive www.pei.de IVD Classification In-house Assays EU Reference Laboratories C M Nübling, PEI Concerns identified by the European Commission in the Medical Devices Roadmap
INSTRUCTIONS FOR CERTIFICATION OF FACTORY PRODUCTION CONTROL
 . August 2017 were approved by Manager of Product Certification Bureau of the Polish Register of Shipping on 25 August 2017 Copyright by, 2017. GDAŃSK, AUGUST 2017 1/12 CONTENTS 1. Factory Production Control
. August 2017 were approved by Manager of Product Certification Bureau of the Polish Register of Shipping on 25 August 2017 Copyright by, 2017. GDAŃSK, AUGUST 2017 1/12 CONTENTS 1. Factory Production Control
Post Market Surveillance (including PMCF): common non compliances
: common non compliances") Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
Post Market Surveillance (including PMCF): common non compliances Jayanth Katta Ph.D Product Specialist & Certification Manager, General Devices Team, Healthcare 1 Overview EU PMS Requirements for Medical
Understanding Clinical Equivalence
 Understanding Clinical Equivalence BSI 2014 Medical Device Mini-Roadshow Ibim Tariah Ph.D Technical Director, Healthcare Solutions 1 Understanding Clinical Equivalence Review Requirements from Directives
Understanding Clinical Equivalence BSI 2014 Medical Device Mini-Roadshow Ibim Tariah Ph.D Technical Director, Healthcare Solutions 1 Understanding Clinical Equivalence Review Requirements from Directives
Guide to field safety corrective actions for medical devices and in-vitro diagnostic medical devices
 Guide to field safety corrective actions for medical devices and in-vitro diagnostic medical devices Item type Authors Publisher Report Irish Medicines Board (IMB) Irish Medicines Board (IMB) Downloaded
Guide to field safety corrective actions for medical devices and in-vitro diagnostic medical devices Item type Authors Publisher Report Irish Medicines Board (IMB) Irish Medicines Board (IMB) Downloaded
EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY. EudraLex. Volume 4
 EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY Ref. Ares(2015)4234460-12/10/2015 Medicinal products quality, safety and efficacy Brussels, 12 October 2015 EudraLex Volume 4 EU Guidelines
EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY Ref. Ares(2015)4234460-12/10/2015 Medicinal products quality, safety and efficacy Brussels, 12 October 2015 EudraLex Volume 4 EU Guidelines
The Children s Hospital of Philadelphia Committees for the Protection of Human Subjects Policies and Procedures Determination of IND/IDE Requirement
 Page: 1 of 8 I. PURPOSE II. III. IV. The purpose of this Standard Operating Procedure is to delineate when an investigator must obtain an Investigational New Drug (IND) or Investigational Device Exemption
Page: 1 of 8 I. PURPOSE II. III. IV. The purpose of this Standard Operating Procedure is to delineate when an investigator must obtain an Investigational New Drug (IND) or Investigational Device Exemption
GUIDELINES ON MEDICAL DEVICES CLINICAL EVALUATION: A GUIDE FOR MANUFACTURERS AND NOTIFIED BODIES
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE GENERAL Consumer Goods Cosmetics and Medical Devices MEDDEV. 2.7.1 Rev.3 December 2009 GUIDELINES ON MEDICAL DEVICES CLINICAL EVALUATION: A GUIDE
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE GENERAL Consumer Goods Cosmetics and Medical Devices MEDDEV. 2.7.1 Rev.3 December 2009 GUIDELINES ON MEDICAL DEVICES CLINICAL EVALUATION: A GUIDE
Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC
 Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC Institute for Testing and Certification, Inc., Czech Republic 1. Introduction
Manual for ITC's Clients 2016 Conformity assessment of in vitro diagnostic medical devices pursuant to Council Directive 98/79/EC Institute for Testing and Certification, Inc., Czech Republic 1. Introduction
Laboratorio di Tecnologie Biomediche
 Laboratorio di Tecnologie Biomediche Introduction to medical devices Carmelo De Maria carmelo.demaria@unipi.it Medical Device A Medical Device is identified by means of its INTENDED PURPOSE Intended to
Laboratorio di Tecnologie Biomediche Introduction to medical devices Carmelo De Maria carmelo.demaria@unipi.it Medical Device A Medical Device is identified by means of its INTENDED PURPOSE Intended to
MEDICAL DEVICES REGULATORY SYSTEM
 GUIDANCE DOCUMENT MEDICAL DEVICES REGULATORY SYSTEM Table of Contents PART 1: PRE-MARKET ASSESSMENT 9 SECTION 1: INTRODUCTION 9 1.1 Principles and Main Features of A Regulatory Framework of Medical Device
GUIDANCE DOCUMENT MEDICAL DEVICES REGULATORY SYSTEM Table of Contents PART 1: PRE-MARKET ASSESSMENT 9 SECTION 1: INTRODUCTION 9 1.1 Principles and Main Features of A Regulatory Framework of Medical Device
Checklist for the assessment based on the standards
 ISO & MDD & Checklist for the assessment based on the standards ISO :2016 ISO :2016 associate with EC Directive 93/42 EEC Where applicable EC Directive 93/42/EEC Annex II/V/VI Company: Audit date Auditor:
ISO & MDD & Checklist for the assessment based on the standards ISO :2016 ISO :2016 associate with EC Directive 93/42 EEC Where applicable EC Directive 93/42/EEC Annex II/V/VI Company: Audit date Auditor:
POLICY FOR THE DECONTAMINATION OF FLEXIBLE ENDOSCOPES
 POLICY FOR THE DECONTAMINATION OF FLEXIBLE ENDOSCOPES Version 2.0 August 2012 Name of Policy: Purpose of Policy: Policy for the Decontamination of Flexible Endoscopes To provide guidance on the decontamination
POLICY FOR THE DECONTAMINATION OF FLEXIBLE ENDOSCOPES Version 2.0 August 2012 Name of Policy: Purpose of Policy: Policy for the Decontamination of Flexible Endoscopes To provide guidance on the decontamination
Medical Technologies in Belgium: overview & challenges. Karel Verlinde - bemedtech
 Medical Technologies in Belgium: overview & challenges 20/02/2018 On 5 April 2017, 2 new Regulations on medical devices were adopted, and they entered into force on 25 May 2017. These replace the existing
Medical Technologies in Belgium: overview & challenges 20/02/2018 On 5 April 2017, 2 new Regulations on medical devices were adopted, and they entered into force on 25 May 2017. These replace the existing
Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers
 ZP-21 Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers This guideline supersedes guideline SÚKL PZT-16 as of November 1, 2004. The purpose of
ZP-21 Inspection of the conduct of clinical evaluations on medical devices in the premises of healthcare providers This guideline supersedes guideline SÚKL PZT-16 as of November 1, 2004. The purpose of
Information for Manufacturers on the Inspection of Manufacturing Site(s) (Assessment of the Quality Management System)
 (Assessment of the Quality Management System)") P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Inspection of Manufacturing Site(s) (Assessment of the Quality Management System) WHO Prequalification
P r e q u a l i f i c a t i o n T e a m - D i a g n o s t i c s Information for Manufacturers on the Inspection of Manufacturing Site(s) (Assessment of the Quality Management System) WHO Prequalification
CORRESPONDENTS' GUIDELINES No 3
 CORRESPONDENTS' GUIDELINES No 3 Subject: Certificate for subsequent non-interim recovery or disposal according to Article 15(e) of Regulation (EC) No 1013/2006 on shipments of waste 1. These correspondents'
CORRESPONDENTS' GUIDELINES No 3 Subject: Certificate for subsequent non-interim recovery or disposal according to Article 15(e) of Regulation (EC) No 1013/2006 on shipments of waste 1. These correspondents'
The Role of a Notified Body in Defining Risk Assessment for a Recombinant Protein Used in a Medical Device Case Study
 The Role of a Notified Body in Defining Risk Assessment for a Recombinant Protein Used in a Medical Device Case Study CMC Strategy Forum Europe 2015 Update on Medical Device/Combination Products Karin
The Role of a Notified Body in Defining Risk Assessment for a Recombinant Protein Used in a Medical Device Case Study CMC Strategy Forum Europe 2015 Update on Medical Device/Combination Products Karin
Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland
 Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland AUT-G0095-1 15 AUGUST 2014 This guide does not purport to be an interpretation of law and/or regulations and is for
Guide for Manufacturers and Sponsors on Clinical Investigations Carried Out in Ireland AUT-G0095-1 15 AUGUST 2014 This guide does not purport to be an interpretation of law and/or regulations and is for
KRONOS WORLDWIDE, INC. SAFE HARBOR PRIVACY POLICY Effective December 1, 2009 Amended and Restated as of July 20, 2012
 . SAFE HARBOR PRIVACY POLICY Amended and Restated as of July 20, 2012 I. OBJECTIVES The objective of this policy is to comply with applicable laws and regulations and document the processes and procedures
. SAFE HARBOR PRIVACY POLICY Amended and Restated as of July 20, 2012 I. OBJECTIVES The objective of this policy is to comply with applicable laws and regulations and document the processes and procedures
COUNTRY OVERVIEW: THAILAND. August 2012 SPECIAL REPRINT. By Rarana Phanudulkitti
 August 2012 SPECIAL REPRINT COUNTRY OVERVIEW: THAILAND By Rarana Phanudulkitti Reproduced with the kind permission of Global Regulatory Press from the Journal of Medical Device Regulation, 2012, 9(3),
August 2012 SPECIAL REPRINT COUNTRY OVERVIEW: THAILAND By Rarana Phanudulkitti Reproduced with the kind permission of Global Regulatory Press from the Journal of Medical Device Regulation, 2012, 9(3),
Agenzia Italiana del Farmaco
 Agenzia Italiana del Farmaco European Regulation on Advanced Therapies Cristina Pintus Head of European Relations Unit and Coordinator of the Advanced Therapy Project Italian Medicines Agency Proposal
Agenzia Italiana del Farmaco European Regulation on Advanced Therapies Cristina Pintus Head of European Relations Unit and Coordinator of the Advanced Therapy Project Italian Medicines Agency Proposal