Computers and E-documents in a regulated environment
|
|
|
- Francine Powell
- 6 years ago
- Views:
Transcription
1 Computers and E-documents in a regulated environment David R Buckley FQSA info@navigategmp.com enquiries_dba@optusnet.com.au
2 Introduction TGA 2005 name change A&NZ Pharmaceutical Products Therapeutic Goods Act 1989 Licence requirements and conditions Manufacturing Principles Code of GMP same as PICS but! Should and shall mean must ie mandatory Annex 5 (EU 11) computers, Annex 15 validation (includes laboratories) TGA MAS Auditors Powers of entry Training Specialists
3 Regulators USA - FDA increasing scrutiny Expectations Freedom of Information Consent decree Europe - changing UK MCA, keeping pace with and influenced by the FDA Tends to set bench mark for Europe Freedom of Information in preparation Japan - imported drugs are under scrutiny Australia - TGA using European GMP but tending to FDA interpretations FOI but never really tested for inspection reports Independent product review
4 FDA consent decrees Wyeth October 4, 2000 $15,000 per day if fail to adhere to corrective action schedule (up to a $5,000,000) and Pay $30,000,000 to U.S. Treasury Abbott November 2, 1999 $15,000 per manufacturing process, per day, (up to $10 million dollars cap) if fail to adhere to corrective action schedule and Pay $100,000, to U.S. Treasury Schering-Plough May 20, 2002 Pay $500,000, to U.S. Treasury
5 Medical device software associated recalls Number of Recalls USA
6 US Device Software Recalls Radiology InVitro Diagnostic Cardiovascular Anesthesiology General Hospital Other Average Number of Software Recalls Per Year
7 Regulatory Action Once a company is found deficient: Corrective action is difficult, long, very costly Affects all parts of the business Interruption of market supply Independent product review Loss of reputation Loss to shareholder of stock value Financial implications Major company re-organization All parts of TGA get involved
8 Regulatory Inspections Key Regulators are: Becoming more risk averse No risks - no mistakes Introducing Quality Systems approach to inspections PIC/S FDA requiring companies to make global commitments on behalf of all sites Wyeth FDA Issuing Warning Letters to Pharmaceutical, Consumer Healthcare Companies TGA unannounced audits During normal business hours also means 1230pm! Teams of 3 or 4 Auditors with specialist skills
9 The major risk areas USA - FDA increasing surveillance and expectations Europe - rapidly changing UK MCA, keeping pace with and influenced by the FDA, sets standards for Europe Has established a merged company database Moving towards Freedom of Information Japan - imported drugs are under scrutiny Australia - TGA using FDA standards Changes post Pan
10 TGA and Industry need... confidence in the production and management computers and electronic records... Proof of a capable system... Evidence that computer systems are fully validated... Ability to obtain true and accurate copies of e-records, both electronically and on readable hard copy (now and into future)... Secure e-records and traceability. Applies to all systems involved with manufacturing
11 E-doc Applies to Custom/Commercial/Proprietary Software (OTS, COTS) Electronic Batch Records Laboratory Analysis Data Facility Design Documents Records Maintenance Cleaning Calibration Deviations, OOS Procedures Product Label Information Spreadsheets/Databases Validation Records
12 Controls - Policy for operation/responsibilities of system owners - Computerised system requires validation - System to be secure, with appropriate access controls - Authority/device checks for input data - Sequence step control (process control systems) - Training of developers and operators - Controls over system documentation - Change control - Audit trail -Time stamped -Unable to be altered -Computer generated -Not accessible by operator
13 Code comparison 21 CFR part11 EU GMP Annexe 11 GAMP Appendix 4 Electronic Records, Controls Computerised Systems APV Guideline for Closed Systems Validation 11.10a) 11.2, 11.7, , 2.1, 4 Copies of Records 11.10b) 11.12, Record Protection 11.10c) 11.13, 11.14, 11.15, , 2, 2.1, 6 System Access 11.10d) 11.8, , 4 Audit Trails 11.10e) 11.10, Sequencing 11.10f) 11.6, Authority Checks 11.10g) 11.8, 11.9, , 4 Device Checks 11.10h) 11.6, , 4 Training 11.10i) Policies 11.10j) Document Controls 11.10k)1) 11.16, , 2.1, 6 Change Control 11.10k)2) 11.11, , 2.1, 6
14 Pharma E-docs US 21 CFR part 11 applies to: Records in electronic form required under any pre-existing FDA regulation (FDA predicate rules for gmps, glps, gcps) Electronic records submitted to and subject to inspection Hardware, software, documentation associated with both Includes hybrid systems, electronic records that do not include an electronic signature Computer controlled manufacturing processes

15 Pharma E-docs
16 Pharma E-docs - definitions Electronic record any combination of text, graphics, data, audio, pictorial, or other information in digital form that is created, modified, maintained, archived, retrieved or distributed by computer system Electronic signature A computer rendition (biometrics, non-biometrics, digital) of some unique mark that an individual executes, adopts and authorises that is considered to be a legally binding equivalent of a handwritten signature Closed system an environment in which system access is controlled by persons who are responsible for the content of electronic records that are on the system Open system an environment in which system access is not controlled by persons responsible for the content of the electronic records on the system
17 Pharma E-docs - definitions Volatile (transient) vs nonvolatile data Volatile data - Electronically gathered but not archived on electronic media for future retrieval Nonvolatile data - Electronically gathered & stored on durable media, even for short periods of time Validation accuracy reliability consistent intended performance ability to discern invalid or altered records (audit trails)
18 Audit Trails All changes must be recorded Available for inspection and copying Computer generated Date and time stamped Does not obscure previous data Independently recorded Retain for full retention period Attempts of unauthorised access recorded Should not be able to be viewed by operator
19 Pharma E-docs Security System access is limited to authorized individuals; authority checks must be conducted to grant access and/or sign an e-record Retention must be available throughout the records retention period (governed by predicate rule) Copies must be available in both human-readable and electronic form Training Assurance that persons who develop, maintain or use E-dco systems are properly trained utilize vendor audit program and in-house training programs for evidence Documentation Management programs vendor documentation in-house documentation change and revision control on both
20 Pharma E-docs 1. Written Policies defining individual accountability and responsibility for actions initiated under individual s e-signatures and e-identity 2. Operational System Checks forced sequencing of steps, like a workflow or EBR 3. Device Checks to determine validity of source data 4. Open versus closed systems both subject to the e-records requirements open systems must apply additional controls to assure data integrity such as encryption and digital signatures
21 Electronic Signatures Unique Unique to an individual Not reused or reassigned to anyone else Individual s identity verified prior to signature authority Prior to or at time of use, for FDA, the organization must certify to the Agency that e-signatures are the legally binding equivalent of handwritten signatures Must contain printed name of signer date and time when signing executed meaning associated with signature must be readily readable (by a human!) Must be unbreakably linked to associated electronic record Must not be able to be copied, excised or otherwise utilised to falsify a record
22 Electronic Signatures Nonbiometric signatures comprised of at least two distinct components Must be used only by genuine owners Must be managed Use of one person s e-sig by another fraudulent behavior Combination of ID and password constitutes uniqueness must be maintained Continuous signing events allow the use of only one component during the signing period session must be opened with full signature otherwise all signing events considered discrete & all e-sig elements must be used Password aging Loss management of tokens or other identity-bearing devices Include a program for replacements Need a testing program to determine that devices perform properly Intrusion detection Include the use of transaction safeguards (3 strikes) Detecting and reporting in an immediate and urgent manner attempts to sign a record or use a system without having proper access Report attempts to system security and/or organizational management
23 Computer validation requirements 1. Closed System 2. Generation of copies of records - human readable and electronic 3. Protection of records 4. Limited System Access 5. Use of audit trail 6. Operational System Checks 7. Authority checks 8. Key signature requirements - Printed name of signer - Date and time - The meaning 9. Signature controls 10. Signature uniqueness - can not be copied distinct identification components 12. Uniqueness of Passwords and ID s 13. Safeguards to prevent unauthorized use 14. System Validation
24 Validation Validation plan defines strategy, responsibilities, approach, tasks signed by management Validation procedures/protocols how will the testing be carried out and documented? Validation reports - the collated efforts of the testing, specifying quantifiable results rather than qualitative as much as possible Hardware and software IQ OS data base application
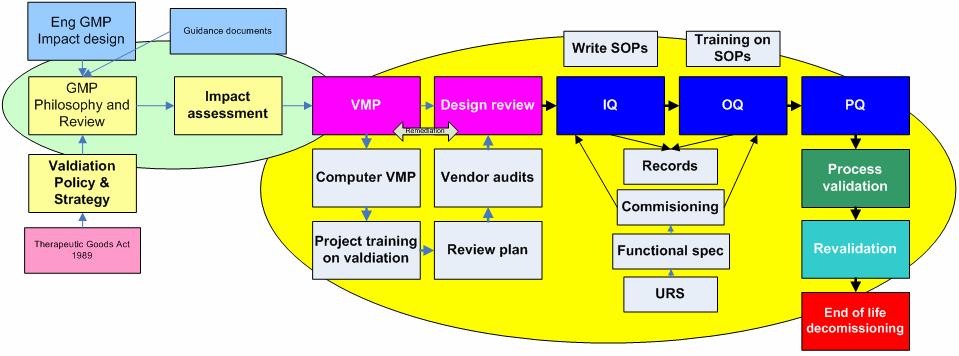
25 Validation expectations

26 Not generally audited

27 Generally subject to audit
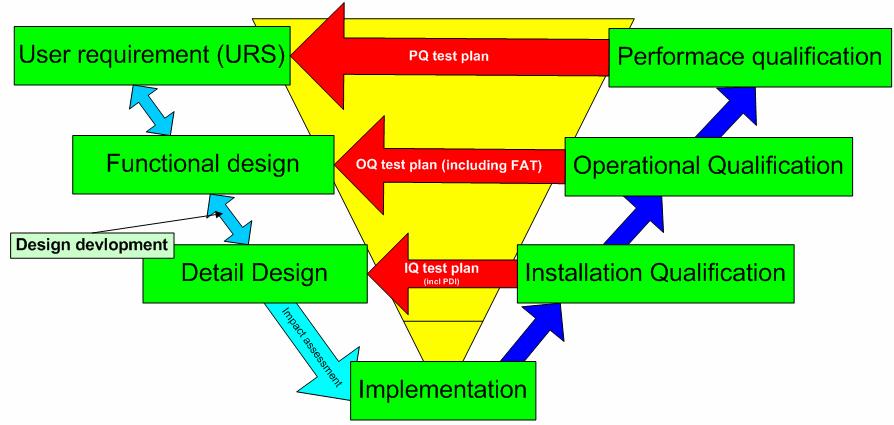
28 V-model
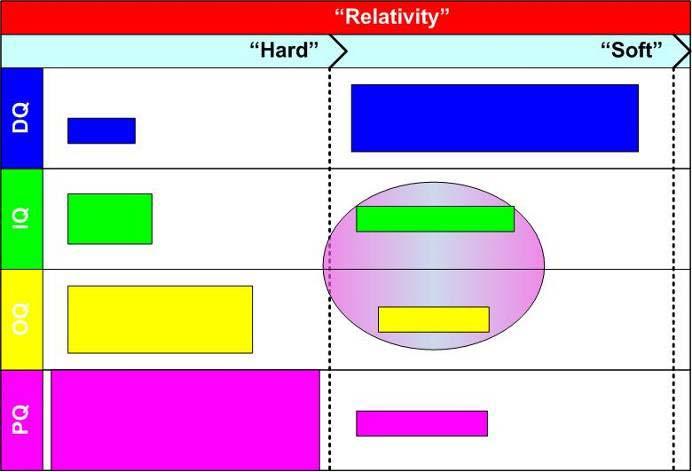
29 Relativity
30 URS requirements Each requirement uniquely referenced Requirement statements not duplicated nor contradicted URS should express requirements not design solutions Each requirement should be testable or verifiable The URS must be understood by both user and supplier: Avoid ambiguity and jargon Requirements should be prioritized URS should distinguish between essential requirements and merely desirable features.
31 Item Function Name Function Description 21 CFR Copy of records Records copy (audit trail, batch report) b 02 PDF files Reports converted to PDF standard c 03 Limited system access Limit system access to authorised users d, g 04 Audit trail Generation of an Audit trail e 05 Record changes Previously recorded info still visible e 06 Storage Storage / archiving of Audit trail data e 07 Signature Signature manifestation a Signature record linking Signature record linking enabled Uniqueness of combo Uniqueness combo UID & password a, a 09 UID in barcode Unique ID + password, UID in barcode a 1 10 Series of signing Series of signing, subsequent signings UID (barcode) should be used a 1 I 11 Password aging Password change after x time b 12 Logout Automatic logout after x minutes a 1 II 13 Unsuccessful logins Unsuccessful logins documented d 14 Blocking of user Block user after x unsuccessful logins d 15 Intrusion, attack Compare system after with before d
32 Validation Dynamic testing normal as well as stress conditions to include boundary testing, errors, etc simulation - conducted offline live, user site - under actual, continuous operating conditions structural - white box to show that the software creator followed SDLC (code reviews) functional - black box to show that inputs yield expected outputs Static testing document and code inspections code walk-throughs technical reviews Extent of validation based on risk system poses to product safety, efficacy and quality, data integrity/authenticity/confidentiality, system s complexity
33 Validation Results review must be independent of execution Configuration management must be in place to include firmware changes, service packs, bug fixes COTS (no access to source code) validate custom changes review of software performance history perform software vendor assessment Internet CAN T BE VALIDATED!! validate data source data destination environments consider additional measures to assure integrity and authenticity eg digital signatures delivery acknowledgements
34 IQ Requirements System description List of applicable specifications Hardware components list System software configuration list Application software configuration list Fixed configurable data list Electrical power requirements Installation and hook-up inspection Documentation requirements Calibration requirements Preventative maintenance requirements System training Deficiency reports
35 OQ requirements Calibration verification Input/output verification Program functional test Screen tests Alarm challenges Program security challenges Power failure recovery Backup and recovery RFI testing EMI testing Report format verification Check against operating procedures
36 PQ requirements System challenges against URS Worst case condition challenges Memory Capacity Multi terminal access Users Boundary and extreme value challenges Abnormal pathway challenges Positive and negative tests
37 Audit trail Time stamps must be based on computer system clocks Accurate Reliable Ideally automatically synchronized to a master clock Clock functions Security to prevent unauthorized changes or/and coupled with employee training where technical restrictions are not available (laptops) Time stamp policing - unannounced checks of computer clocks, spot checking audit trail time stamps for anomalies Time zones - local time dilemma - often server time Assure organization understands how date and time are expressed
38 Maintenance E-records must be available and readable throughout the record retention period Select media carefully Stability? Recovery? Reading hardware? Have a data recovery process for retrieving information from old media Backups of archive media Must be stored under conditions that will not accelerate media deterioration EMF EMR Process should be validated if there is no built in error-checking Approaches to assuring continued e-record availability Save everything hardware software continue to train staff on the system use, even if system no longer used for production only short term needs
39 Maintenance Migration move records from one environment to another Retire or discard old system after reasonable period (?) Assure continued e-record integrity and reliability take into consideration moving from one OS to another one type of storage media to another one file format to another one type of peripheral to another consider use of third party authentications include controls over data, metadata, hardware, software, audit trails, e-signature links as part of migration process After migration should be able to search, sort and process information in the migrated electronic record at least at the same level as what you could attain in the old system
40 Electronic Copies Have to be accurate and complete but not necessarily in the same format at they were created Accurate and complete means including audit trails, metadata and e-signatures Validate the copying process Include considerations of file format, media, built-in error-checking capabilities Any hyperlinked reference is considered part of the copy E-copies of the database queries used to retrieve the copy information should be included with the e-record E-copies should carry some form of authentication to support integrity as the e-records are delivered to the agency Hash value change if file was tampering
41 Citations No revision control on the program Lack of summary validation reports throughout {program s] lifecycle An application program listing was found within the program design binders for the stability application which was not reviewed or approved. Failed to validate the MS Excel spreadsheet file (i.e.; lack of audit trail functions for the spreadsheet file, failure to create and maintain specifications for the spreadsheet file) Lack of high level functional or structural system diagrams throughout the lifecycle of the program [for all applications] Failed to generate or maintain complete software functional or structural design diagrams for all of the applications making up the program throughout the software lifecycle
42 Citations High level specifications for program applications was not a controlled record and it lacked review or approval Change control records were not signed off by QC No approved protocols defining what tests were to be conducted No documented review and approval of test records Continued to use a program after a bug was detected User screens & user menus did allow for access to view the version of the application Failed to document all sites, departments or connections on the network User manuals for the software applications obsolete
43 Citations Sharing a username/password with highest level of access amongst several employees Failed to produce an approved list of personnel currently authorized to share this username/password. Lack of sufficient design control documentation for complete definition of the network (i.e.; high level diagrams identifying all sites / equipment making up the network). Electronic records are not reviewed or approved (i.e.; no electronic signatures of review or approval). Lack of definition documentation and security for a database Lack of review or approval by the QCU of LAN wiring diagram, which is not a controlled record
44 Citations Failed to generate or maintain complete documentation of the myriad (>100) of user interface displays that have been developed and maintained by company personnel for the system Process Control System Security SOP does not adequately describe all the steps performed for security and computer access No written procedure to describe the process used to assign, maintain passwords and access levels to the system Nonviable particle measurements recorded floppy disk and data manually transferred to data base system No formal evaluation to confirm that the measurements printed is an accurate reflection of the data When the diskette is filled, the original data is not retained and is overwritten and/or deleted No established written SOP to describe reuse of the 3.5 floppy disks. SOP was not followed for establishment of validation requirements for GMP computer systems
45 483s North American Science Associates - Nov 1998 failure to maintain adequate controls over computers & systems no current listing of individuals who have access, no audit trail Hydro Med Sciences - Feb 1999 computer software programs have not been validated programs do not secure files from data loss analysts can delete whole data files no security SOPs or back-up Fairbanks Memorial - Apr 1999 failure to control computer or related systems publicly posted employee user name and computer password terminated employees still had access No change control records Linweld - Aug 1999 failure to maintain a computer system with validated program capabilities no testing of system after installation validation protocol incomplete protocol execution pre-dates protocol and requirements approval lack of change control procedure
46 483s Hoffman-LaRoche - Dec 1999 Lab computer system software Intersurgical - May 2000 failure to validate computer software Baxter Healthcare - Aug 2000 electronic cgmp records into conformance Pharmacia - Jan 2001 (2 sites) network program lacked adequate validation Zeus - Mar 2001 firm failed to validate the electronic documentation system Stough Enterprises - April 2001 failure to establish and implement adequate computer security
47 483s Meridien Bioscience - June 2001 database has not been validated nor does it include necessary controls Michigan Instruments - Oct 2001 no documentation to show electronic records meet requirements Luneau SA - Oct 2001 Failure to validate computer software for its intended Earlham College Laboratory using an electronic record system for processing and storage of data from the AA and HPLC instruments Not set up to control the security and data integrity Not password controlled No systematic back-up provision No audit trail of the system capabilities Not appear to be designed and controlled in compliance with requirements
48 Business Stopper! The computer for disposition control of finished, inprocess and purchased materials, controls and maintains stability sample inventories for commercial and development stability programs, controls stability programs [through test schedules], maintains and manages test results, generates stability study reports for presentation in annual reports, annual product reviews, and New Drug Applications.. is not considered validated because.

49 Result v process Result OK?


50 Result v process! Process unacceptable! Patches unacceptable!
51 References GAMP ISPE PDA Good Electronic Record Management FDA groups.yahoo.com/group/21cfrpart11/messages/ Questions?
SIMATIC IT. SIMATIC IT R&D SUITE V7.1 Compliance Response ERES. Introduction 1. The Requirements in Short 2
 Introduction 1 The Requirements in Short 2 SIMATIC IT Meeting the Requirements with SIMATIC IT R&D Suite V7.1 3 SIMATIC IT R&D SUITE V7.1 Evaluation List for SIMATIC IT R&D Suite V7.1 4 Product Information
Introduction 1 The Requirements in Short 2 SIMATIC IT Meeting the Requirements with SIMATIC IT R&D Suite V7.1 3 SIMATIC IT R&D SUITE V7.1 Evaluation List for SIMATIC IT R&D Suite V7.1 4 Product Information
Compliance Response Electronic Records / Electronic Signatures (ERES) for SIMATIC PCS 7 V8.1
 for SIMATIC PCS 7 V8.1") Compliance Response Electronic Records / Electronic Signatures (ERES) for SIMATIC PCS 7 V8.1 SIEMENS AG Industry Sector VSS Pharma D-76187 Karlsruhe, Germany E-Mail: pharma@siemens.com March 2015 A5E35829023-AA
Compliance Response Electronic Records / Electronic Signatures (ERES) for SIMATIC PCS 7 V8.1 SIEMENS AG Industry Sector VSS Pharma D-76187 Karlsruhe, Germany E-Mail: pharma@siemens.com March 2015 A5E35829023-AA
Clarity CDS since version 7.2 supportive tools for compliance with Title 21 CFR Part 11, EudraLex Chapter 4, Annex 11 and other similar legislation
 Clarity CDS since version 7.2 supportive tools for compliance with Title 21 CFR Part 11, EudraLex Chapter 4, Annex 11 and other similar legislation Datasheet Introduction US Part 11 in Title 21 of Code
Clarity CDS since version 7.2 supportive tools for compliance with Title 21 CFR Part 11, EudraLex Chapter 4, Annex 11 and other similar legislation Datasheet Introduction US Part 11 in Title 21 of Code
Compliance Response Electronic Records / Electronic Signatures for COMOS V10.0
 Compliance Response Electronic Records / Electronic Signatures for COMOS V10.0 SIEMENS AG Industry Sector I IA VSS Pharma D-76187 Karlsruhe, Germany E-mail: pharma@siemens.com November 2013 SIEMENS AG
Compliance Response Electronic Records / Electronic Signatures for COMOS V10.0 SIEMENS AG Industry Sector I IA VSS Pharma D-76187 Karlsruhe, Germany E-mail: pharma@siemens.com November 2013 SIEMENS AG
Risk-based Approach to Part 11 and GxP Compliance
 Welcome to our E-Seminar: Risk-based Approach to Part 11 and GxP Compliance 1 Intro Common Discussion Q: Do I really need to do this? Possible Answers A: Of course! (QA) B: Who cares, I have work to do!
Welcome to our E-Seminar: Risk-based Approach to Part 11 and GxP Compliance 1 Intro Common Discussion Q: Do I really need to do this? Possible Answers A: Of course! (QA) B: Who cares, I have work to do!
The Role of the LMS in 21 CFR Part 11 Compliance
 The Role of the LMS in 21 CFR Part 11 Compliance Co-author: Dr. Bob McDowall, Director R.D. McDowall Limited ABSTRACT The purpose of this white paper is to describe how NetDimensions Learning addresses
The Role of the LMS in 21 CFR Part 11 Compliance Co-author: Dr. Bob McDowall, Director R.D. McDowall Limited ABSTRACT The purpose of this white paper is to describe how NetDimensions Learning addresses
ISPE NORDIC COP CLEAN UTILITIES SEPTEMBER TUUSULA FINLAND. Timo Kuosmanen STERIS Finn-Aqua
 ISPE NORDIC COP CLEAN UTILITIES SEPTEMBER 7 2016 TUUSULA FINLAND Timo Kuosmanen STERIS Finn-Aqua Timo_Kuosmanen@steris.com AUDIT TRAIL IN CRITICAL UTILITIES MONITORING CURRENT TRENDS CONTENTS BACKGROUND
ISPE NORDIC COP CLEAN UTILITIES SEPTEMBER 7 2016 TUUSULA FINLAND Timo Kuosmanen STERIS Finn-Aqua Timo_Kuosmanen@steris.com AUDIT TRAIL IN CRITICAL UTILITIES MONITORING CURRENT TRENDS CONTENTS BACKGROUND
CUSTOMER AND SUPPLIER ROLES AND RESPONSIBILITIES FOR 21 CFR 11 COMPLIANCE ASSESSMENT. 21 CFR Part 11 FAQ. (Frequently Asked Questions)
") 21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
21 CFR Part 11 FAQ (Frequently Asked Questions) Customer and Supplier Roles and Responsibilities for Assessment of METTLER TOLEDO STARe Software Version 16.00, including: - 21 CFR 11 Compliance software
Regulatory Expectations, Standards & Guidelines
 Regulatory Expectations, Standards & Guidelines Regulatory Requirements Pharmacopeias Good Automated Manufacturing Practice (GAMP) 21 CFR Part 11 and Annex 11 Consequences of Non-Compliance 22 Regulatory
Regulatory Expectations, Standards & Guidelines Regulatory Requirements Pharmacopeias Good Automated Manufacturing Practice (GAMP) 21 CFR Part 11 and Annex 11 Consequences of Non-Compliance 22 Regulatory
Implement Effective Computer System Validation. Noelia Ortiz, MME, CSSGB, CQA
 Implement Effective Computer System Validation Noelia Ortiz, MME, CSSGB, CQA Session Outline 1 2 3 4 5 Understanding Regulations and Guidelines Pertaining to Computer Systems Integrate SDLC and GAMP 5
Implement Effective Computer System Validation Noelia Ortiz, MME, CSSGB, CQA Session Outline 1 2 3 4 5 Understanding Regulations and Guidelines Pertaining to Computer Systems Integrate SDLC and GAMP 5
PTC s Windchill Product Lifecycle Management (PLM) System Facilitates Part 11 / Annex 11 Compliance
 System Facilitates Part 11 / Annex 11 Compliance") PTC s Product Lifecycle Management (PLM) ystem Facilitates Part 11 / Annex 11 Compliance A White Paper by: Diane Gleinser, r. VP olutions and ervices, UDM Life ciences Michael Prudhomme, olution Director,
PTC s Product Lifecycle Management (PLM) ystem Facilitates Part 11 / Annex 11 Compliance A White Paper by: Diane Gleinser, r. VP olutions and ervices, UDM Life ciences Michael Prudhomme, olution Director,
EU GMP - Annex 11 Computerised systems Versione corrente Nuova versione per commenti (emessa 8 aprile 2008)
") EU GMP - Annex 11 Computerised systems Versione corrente Nuova versione per commenti (emessa 8 aprile 2008) Principle The introduction of computerised systems into systems of manufacturing, including storage,
EU GMP - Annex 11 Computerised systems Versione corrente Nuova versione per commenti (emessa 8 aprile 2008) Principle The introduction of computerised systems into systems of manufacturing, including storage,
GAMP5 Validation for Dynamics 365
 GAMP5 Validation for Dynamics 365 Prepared by: Michael Webster, Business Development Director, RSM US LLP michael.webster@rsmus.com, +1 617 241 1544 Dynamics 365 is an ideal enterprise resource planning
GAMP5 Validation for Dynamics 365 Prepared by: Michael Webster, Business Development Director, RSM US LLP michael.webster@rsmus.com, +1 617 241 1544 Dynamics 365 is an ideal enterprise resource planning
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL. EudraLex The Rules Governing Medicinal Products in the European Union
 EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, 08 April 2008 EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 EU
EUROPEAN COMMISSION ENTERPRISE AND INDUSTRY DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, 08 April 2008 EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 EU
EU Annex 11 US FDA , (g), (i), 11 Orlando López 09-APR
, (i), 11 Orlando López 09-APR") Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Validation of MES and Manufacturing Automation systems
 Validation of MES and Manufacturing Automation systems The FDA Group Presentation APRIL 26, 2017 Chinmoy Roy, B.S. (Hons.) MSCS Industry Consultant 1 Agenda What is a MES Validation concepts Validation
Validation of MES and Manufacturing Automation systems The FDA Group Presentation APRIL 26, 2017 Chinmoy Roy, B.S. (Hons.) MSCS Industry Consultant 1 Agenda What is a MES Validation concepts Validation
GAMP Guideline & Validation Documentation
 GAMP Guideline & Validation Documentation Danilo Maruccia Milano, 21 Marzo 2006 GAMP Guideline & Validation Documentation GAMP Guideline Planning documents Specification Documents Testing Documents Acceptance
GAMP Guideline & Validation Documentation Danilo Maruccia Milano, 21 Marzo 2006 GAMP Guideline & Validation Documentation GAMP Guideline Planning documents Specification Documents Testing Documents Acceptance
Apriso and FDA 21 CFR Part 11
 Apriso and FDA 21 CFR Part 11 JUNE 2010 Table of Contents Executive Summary... 3 Introduction... 4 What is Compliance?... 4 Cost Effective Compliance... 4 Apriso and 21 CFR Part 11... 5 Part 11: Electronic
Apriso and FDA 21 CFR Part 11 JUNE 2010 Table of Contents Executive Summary... 3 Introduction... 4 What is Compliance?... 4 Cost Effective Compliance... 4 Apriso and 21 CFR Part 11... 5 Part 11: Electronic
References Concept. Principle. EU Annex 11 US FDA , (g), (i), 11 Orlando Lopez 2/15/11. Old Annex 11.
, (i), 11 Orlando Lopez 2/15/11. Old Annex 11.") References Concept Principle a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which
References Concept Principle a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which
EU Annex 11 US FDA 211, 820, 11; other guidelines Orlando López 11-MAY-2011
 Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)
![Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)](/thumbs/72/66853561.jpg "Regulatory Overview Annex 11 and Part 11. Sion Wyn Conformity +[44] (0)") Regulatory Overview Annex 11 and Part 11 Sion Wyn Conformity +[44] (0) 1492 642622 sion.wyn@conform-it.com 1 Two Key Regulations Annex 11 21 CFR Part 11 Apply to the regulated company, but often have a
Regulatory Overview Annex 11 and Part 11 Sion Wyn Conformity +[44] (0) 1492 642622 sion.wyn@conform-it.com 1 Two Key Regulations Annex 11 21 CFR Part 11 Apply to the regulated company, but often have a
Understanding GxP Regulations for Healthcare
 Understanding GxP Regulations for Healthcare GxP Guidelines What is GxP? GxP is a collection of quality guidelines and regulations created to ensure that bio/pharmaceutical products are safe, meet their
Understanding GxP Regulations for Healthcare GxP Guidelines What is GxP? GxP is a collection of quality guidelines and regulations created to ensure that bio/pharmaceutical products are safe, meet their
Paperless recorders and 21 CFR part 11 compliance Videographic recorders
 Sales guide SG/RandC/002 EN Paperless recorders and 21 CFR part 11 compliance Videographic recorders Introduction Today's manufacturing environment is becoming more regulated than ever and the most well
Sales guide SG/RandC/002 EN Paperless recorders and 21 CFR part 11 compliance Videographic recorders Introduction Today's manufacturing environment is becoming more regulated than ever and the most well
Introduction to 21 CFR 11 - Good Electronic Records Management
 INTERNATIONAL JOURNAL OF ADVANCES IN PHARMACY, BIOLOGY AND CHEMISTRY Review Article Introduction to 21 CFR 11 - Good Electronic Records Management Pal Tapas Kumar* and Maity Subhasis NSHM Knowledge Campus,
INTERNATIONAL JOURNAL OF ADVANCES IN PHARMACY, BIOLOGY AND CHEMISTRY Review Article Introduction to 21 CFR 11 - Good Electronic Records Management Pal Tapas Kumar* and Maity Subhasis NSHM Knowledge Campus,
COMPUTERISED SYSTEMS
 ANNEX 11 COMPUTERISED SYSTEMS PRINCIPLE This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components
ANNEX 11 COMPUTERISED SYSTEMS PRINCIPLE This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components
Electronic Records and Electronic Signatures (21 CFR Part 11)
") Training Course Computerized System Validation in the Pharmaceutical Industry Istanbul, 16-17 January 2003 Electronic Records and Electronic Signatures (21 CFR Part 11) Hans-Beat Jenny, SwissMedic, Basel
Training Course Computerized System Validation in the Pharmaceutical Industry Istanbul, 16-17 January 2003 Electronic Records and Electronic Signatures (21 CFR Part 11) Hans-Beat Jenny, SwissMedic, Basel
Ensure Data Integrity Compliance Enterprise-Wide
 Ensure Data Integrity Compliance Enterprise-Wide PharmaQual 360 º Conference February 24, 2017 Chris Wubbolt QACV Consulting, LLC www.qacvconsulting.com 1 Objectives What is data integrity and the definition
Ensure Data Integrity Compliance Enterprise-Wide PharmaQual 360 º Conference February 24, 2017 Chris Wubbolt QACV Consulting, LLC www.qacvconsulting.com 1 Objectives What is data integrity and the definition
Regulations in Pharmaceutical Laboratories
 Regulations in Pharmaceutical Laboratories February 2004 Ludwig Huber Compliance Fellow for Life Sciences and Chemical Analysis Chairperson: Rob Sample Regulations and Quality Standards Developed by Industries
Regulations in Pharmaceutical Laboratories February 2004 Ludwig Huber Compliance Fellow for Life Sciences and Chemical Analysis Chairperson: Rob Sample Regulations and Quality Standards Developed by Industries
Computerised Systems. Alfred Hunt Inspector. Wholesale Distribution Information Day, 28 th September Date Insert on Master Slide.
 Computerised Systems Wholesale Distribution Information Day, Alfred Hunt Inspector Date Insert on Master Slide Slide 1 Index What is a computerised system Updates to EU GDPs Expectations Case studies Slide
Computerised Systems Wholesale Distribution Information Day, Alfred Hunt Inspector Date Insert on Master Slide Slide 1 Index What is a computerised system Updates to EU GDPs Expectations Case studies Slide
Could Poor Temperature Data Management be Putting Your GxP Facility at Risk for Data Integrity Violations? we prove it.
 1 How to Meet New MHRA, FDA and WHO Data Integrity Guidelines WHITE PAPER Could Poor Temperature Data Management be Putting Your GxP Facility at Risk for Data Integrity Violations? we prove it. 2 How to
1 How to Meet New MHRA, FDA and WHO Data Integrity Guidelines WHITE PAPER Could Poor Temperature Data Management be Putting Your GxP Facility at Risk for Data Integrity Violations? we prove it. 2 How to
System Requirements (URS)
") System s (URS) System Document Control System Document ID URS000XXX Version 1 DOCUMENT APPROVALS Reason For Signature Name Position Signature Date Prepared by Vic Johnson Project Manager Checked for accuracy
System s (URS) System Document Control System Document ID URS000XXX Version 1 DOCUMENT APPROVALS Reason For Signature Name Position Signature Date Prepared by Vic Johnson Project Manager Checked for accuracy
21CFR11 Compliance and Automated Manufacturing
 Presented at the World Batch Forum North American Conference Woodcliff Lake, NJ April 7-10, 2002 107 S. Southgate Drive Chandler, Arizona 85226-3222 480-893-8803 Fax 480-893-7775 E-mail: info@wbf.org www.wbf.org
Presented at the World Batch Forum North American Conference Woodcliff Lake, NJ April 7-10, 2002 107 S. Southgate Drive Chandler, Arizona 85226-3222 480-893-8803 Fax 480-893-7775 E-mail: info@wbf.org www.wbf.org
Computerised Systems. Inspection Expectations. Paul Moody, Inspector. 18/10/2013 Slide 1. ISPE GAMP COP Ireland Meeting, Dublin, 17 th October 2013
 Computerised Systems Inspection Expectations ISPE GAMP COP Ireland Meeting, Dublin, 17 th October 2013 Paul Moody, Inspector Slide 1 Presentation Contents Brief Introduction to the IMB Regulatory References
Computerised Systems Inspection Expectations ISPE GAMP COP Ireland Meeting, Dublin, 17 th October 2013 Paul Moody, Inspector Slide 1 Presentation Contents Brief Introduction to the IMB Regulatory References
New Data Integrity Regulations and Practical Advice for Life Science Laboratories. we prove it.
 1 How to Meet New MHRA, FDA, EMA and WHO Data Integrity Guidelines white paper New Data Integrity Regulations and Practical Advice for Life Science Laboratories we prove it. 2 How to Avoid Poor Temperature
1 How to Meet New MHRA, FDA, EMA and WHO Data Integrity Guidelines white paper New Data Integrity Regulations and Practical Advice for Life Science Laboratories we prove it. 2 How to Avoid Poor Temperature
Intland s Medical IEC & ISO Template
 Intland s Medical IEC 62304 & ISO 14971 Template Intland s Medical IEC 62304 & ISO 14971 Template codebeamer ALM for Medical Device Development Intland s Medical IEC 62304 & ISO 14971 Template Medical
Intland s Medical IEC 62304 & ISO 14971 Template Intland s Medical IEC 62304 & ISO 14971 Template codebeamer ALM for Medical Device Development Intland s Medical IEC 62304 & ISO 14971 Template Medical
PAI Inspections, Observations and Data Integrity
 PAI Inspections, Observations and Data Integrity Krishna Ghosh, Ph.D. Office of Pharmaceutical Quality Office of Process and Facilities Center for Drug Evaluation and Research November, 2017 20 November
PAI Inspections, Observations and Data Integrity Krishna Ghosh, Ph.D. Office of Pharmaceutical Quality Office of Process and Facilities Center for Drug Evaluation and Research November, 2017 20 November
Computer System Validation Perform a Gap Analysis of your CSV Processes
 Computer System Validation Perform a Gap Analysis of your CSV Processes Chris Wubbolt, QACV Consulting Computer and Software Validation Conference April 27, 2017 www.qacvconsulting.com 1 Objectives Computer
Computer System Validation Perform a Gap Analysis of your CSV Processes Chris Wubbolt, QACV Consulting Computer and Software Validation Conference April 27, 2017 www.qacvconsulting.com 1 Objectives Computer
GxP Compliance for Computerized Systems
 GxP Compliance for Computerized Systems The Second Annual Pharmaceutical Industry Regulatory and Compliance Summit David L. Stone General Manager, Validation Services Glemser Technologies June 13, 2001
GxP Compliance for Computerized Systems The Second Annual Pharmaceutical Industry Regulatory and Compliance Summit David L. Stone General Manager, Validation Services Glemser Technologies June 13, 2001
White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU
 White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU Numerous articles are available comparing the current and previous EU Code of GMP Chapter 4: Documentation, but no comparison exists between
White paper: Code of GMP Chapter 4 Documentation - PIC/S versus EU Numerous articles are available comparing the current and previous EU Code of GMP Chapter 4: Documentation, but no comparison exists between
Computerised System Validation
 Computerised System Validation Considerations for the Validation Lifecycle Paul Moody GMP Conference 12 th November 2014 Regulatory References EU GMP Annex 11 (2011) http://ec.europa.eu/health/files/eudralex/vol-4/annex11_01-2011_en.pdf
Computerised System Validation Considerations for the Validation Lifecycle Paul Moody GMP Conference 12 th November 2014 Regulatory References EU GMP Annex 11 (2011) http://ec.europa.eu/health/files/eudralex/vol-4/annex11_01-2011_en.pdf
Strategies for Risk Based Validation of Laboratory Systems
 Strategies for Risk Based Validation of Laboratory Systems Video Web Seminar September 23, 2004 Ludwig Huber E-mail: ludwig_huber@agilent.com Today s Agenda Background information: why risk assessment,
Strategies for Risk Based Validation of Laboratory Systems Video Web Seminar September 23, 2004 Ludwig Huber E-mail: ludwig_huber@agilent.com Today s Agenda Background information: why risk assessment,
General European OMCL Network (GEON) QUALITY MANAGEMENT DOCUMENT
 QUALITY MANAGEMENT DOCUMENT") General European OMCL Network (GEON) QUALITY MANAGEMENT DOCUMENT PA/PH/OMCL (08) 69 R7 VALIDATION OF COMPUTERISED SYSTEMS CORE DOCUMENT Full document title and reference Document type Validation of Computerised
General European OMCL Network (GEON) QUALITY MANAGEMENT DOCUMENT PA/PH/OMCL (08) 69 R7 VALIDATION OF COMPUTERISED SYSTEMS CORE DOCUMENT Full document title and reference Document type Validation of Computerised
ALCOA AND DATA INTEGRITY: PASTAND FUTURE
 ALCOA AND DATA INTEGRITY: PASTAND FUTURE Marina Figini, PCA S.p.A./Aschimfarma Data Integrity: reliability, quality and competitiveness factors of API manufacturers Pavia, 10 Novembre 2017 Good data management
ALCOA AND DATA INTEGRITY: PASTAND FUTURE Marina Figini, PCA S.p.A./Aschimfarma Data Integrity: reliability, quality and competitiveness factors of API manufacturers Pavia, 10 Novembre 2017 Good data management
Electronic Records Assessment and GAMP Software Categories
 Solutions for Life Science Electronic Records Assessment and GAMP Software Categories Desigo CC V3.0 - White Paper CM110803_en 2017-10-24 Version 1 siemens.com/lifescience Unrestricted Siemens Switzerland
Solutions for Life Science Electronic Records Assessment and GAMP Software Categories Desigo CC V3.0 - White Paper CM110803_en 2017-10-24 Version 1 siemens.com/lifescience Unrestricted Siemens Switzerland
COMPUTERIZED SYSTEM VALIDATION
 www.kevintech.com Current Practices of COMPUTERIZED SYSTEM VALIDATION Kevin ISSUE.1/2017 CONNECT INSIDE - Basics of Computerized System Validation - Life Cycle of Computerized System The less manual the
www.kevintech.com Current Practices of COMPUTERIZED SYSTEM VALIDATION Kevin ISSUE.1/2017 CONNECT INSIDE - Basics of Computerized System Validation - Life Cycle of Computerized System The less manual the
Global Compliance Trends and Warning Letters
 Contact: Charles Lu Director, Quality Carlsbad Tech Phone: (760) 431-8284 Fax: (203) 555-0101 5928 Farnsworth Ct Carlsbad, CA, 92008 www.carlsbadtech.com Global Compliance Trends and Warning Letters Governance
Contact: Charles Lu Director, Quality Carlsbad Tech Phone: (760) 431-8284 Fax: (203) 555-0101 5928 Farnsworth Ct Carlsbad, CA, 92008 www.carlsbadtech.com Global Compliance Trends and Warning Letters Governance
World Journal of Pharmaceutical Research SJIF Impact Factor 5.990
 SJIF Impact Factor 5.990 Volume 4, Issue 9, 444-454. Review Article ISSN 2277 7105 COMPUTER SYSTEM VALIDATION: A REVIEW Patil Yogesh* Mali Kamlesh, Bodhane Mohini, Ram Phad, Shaikh Ismail, Lale Shivam
SJIF Impact Factor 5.990 Volume 4, Issue 9, 444-454. Review Article ISSN 2277 7105 COMPUTER SYSTEM VALIDATION: A REVIEW Patil Yogesh* Mali Kamlesh, Bodhane Mohini, Ram Phad, Shaikh Ismail, Lale Shivam
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL. EudraLex The Rules Governing Medicinal Products in the European Union
 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Public Health and Risk Assessment Pharmaceuticals Brussels, SANCO/C8/AM/sl/ares(2010)1064599 EudraLex The Rules Governing Medicinal Products
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Public Health and Risk Assessment Pharmaceuticals Brussels, SANCO/C8/AM/sl/ares(2010)1064599 EudraLex The Rules Governing Medicinal Products
21 CFR Part 11 Compliance
 ENTERPRISE CONTENT MANAGEMENT ON THE MICROSOFT PLATFORM Table of Contents Page 1 Introduction Page 5-13 Electronic Record Compliance and FirstPoint Applicability Pages 14-18 Electronic Signature Compliance
ENTERPRISE CONTENT MANAGEMENT ON THE MICROSOFT PLATFORM Table of Contents Page 1 Introduction Page 5-13 Electronic Record Compliance and FirstPoint Applicability Pages 14-18 Electronic Signature Compliance
Data Integrity Through the Life Cycle Balancing Quality and Innovation. Sion Wyn Conformity +44 (0)
") Data Integrity Through the Life Cycle Balancing Quality and Innovation Sion Wyn Conformity +44 (0) 1492 642622 sion.wyn@conform-it.com Theme Achieving harmony Balancing quality, safety, and compliance
Data Integrity Through the Life Cycle Balancing Quality and Innovation Sion Wyn Conformity +44 (0) 1492 642622 sion.wyn@conform-it.com Theme Achieving harmony Balancing quality, safety, and compliance
Guidance for Industry - Computerized Systems Used in Clinical Trials
 Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
Page 1 of 14 Regulatory Information Computerized Systems Used in Clinical Trials Guidance for Industry - Computerized Systems Used in Clinical Trials
Oracle Tech Cloud GxP Position Paper December, 2016
 Oracle Tech Cloud GxP Position Paper Page 1 of 29 Oracle Tech Cloud GxP Position Paper December, 2016 Prepared By: Subbu Viswanathan, Head of Solutions Reviewed By: David Blewitt, VP Cloud Compliance Oracle
Oracle Tech Cloud GxP Position Paper Page 1 of 29 Oracle Tech Cloud GxP Position Paper December, 2016 Prepared By: Subbu Viswanathan, Head of Solutions Reviewed By: David Blewitt, VP Cloud Compliance Oracle
Force.com and the FDA CFR 21 Part 11 Requirements
 Force.com and the FDA CFR 21 Part 11 Requirements Overview of Capabilities to Enable Compliance Version 4, Nov 14 Last Updated: November 10, 2014 Disclaimer This statement does not confer legal advice,
Force.com and the FDA CFR 21 Part 11 Requirements Overview of Capabilities to Enable Compliance Version 4, Nov 14 Last Updated: November 10, 2014 Disclaimer This statement does not confer legal advice,
Addressing the Paradigm Shift in Regulatory Inspections
 An Executive Summary Addressing the Paradigm Shift in Regulatory Inspections Understanding the paradigm shift in a regulatory audit and what it means from an electronic system perspective. Humera Khaja
An Executive Summary Addressing the Paradigm Shift in Regulatory Inspections Understanding the paradigm shift in a regulatory audit and what it means from an electronic system perspective. Humera Khaja
Develop a Roadmap for the Implementation of a Global CSV Program. Eileen Cortes April 26, 2017
 Develop a Roadmap for the Implementation of a Global CSV Program Eileen Cortes April 26, 2017 Agenda CSV Regulation Principles CSV Lifecycle Approach CSV and Quality Management Governance Program and CSV
Develop a Roadmap for the Implementation of a Global CSV Program Eileen Cortes April 26, 2017 Agenda CSV Regulation Principles CSV Lifecycle Approach CSV and Quality Management Governance Program and CSV
The Convergence of Software in the Medical Device Industry. Joseph Azary
 The Convergence of Software in the Medical Device Industry Joseph Azary 203-944-9320 Info@azarytech.com FDA Regulations & Quality Requirements Clinical Sterilization FDA Regulations & Quality Requirements
The Convergence of Software in the Medical Device Industry Joseph Azary 203-944-9320 Info@azarytech.com FDA Regulations & Quality Requirements Clinical Sterilization FDA Regulations & Quality Requirements
GCP Basics - refresher
 p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
p. 01 GCP Basics - refresher Agenda: p. 02 Brief History of GCP GCP Regulations Principles of ICH E6 Sponsor Responsibilities Computer Systems Common Compliance Issues Brief History of GCP 3 Brief History
Lessons from Pharmaceutical Laboratory related FDA Warning Letters
 Lessons from Pharmaceutical Laboratory related FDA Warning Letters The Agilent Critical Compliance Seminar 2016 Ludwig Huber Ludwig_huber@labcompliance.com Overview FDA Inspections and reports GMP compliance
Lessons from Pharmaceutical Laboratory related FDA Warning Letters The Agilent Critical Compliance Seminar 2016 Ludwig Huber Ludwig_huber@labcompliance.com Overview FDA Inspections and reports GMP compliance
Current Trends in Data Quality and Integrity Issues in Inspections and Risk Based Approach to Investigations; EU perspective
 Current Trends in Data Quality and Integrity Issues in Inspections and Risk Based Approach to Investigations; EU perspective Mark Birse / Richard Andrews, MHRA Data Integrity has no relationship with product
Current Trends in Data Quality and Integrity Issues in Inspections and Risk Based Approach to Investigations; EU perspective Mark Birse / Richard Andrews, MHRA Data Integrity has no relationship with product
DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS
 GLOBAL PROVIDER, LOCAL SOLUTIONS IN YOUR LANGUAGE DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS Francesco Amorosi PhD Octorber 2017 DATA INTEGRITY & CSV:
GLOBAL PROVIDER, LOCAL SOLUTIONS IN YOUR LANGUAGE DATA INTEGRITY ASSURANCE AS KEY FACTOR FOR SURVIVING CORPORATE AUDITS AND REGULATORY INSECTIONS Francesco Amorosi PhD Octorber 2017 DATA INTEGRITY & CSV:
Managing Validation. Paperless Recorders
 Managing Validation Paperless Recorders Multitrend Plus Validation Background The past five years has seen an increase in the use of computerized systems and products in the pharmaceutical and bio-pharmaceutical
Managing Validation Paperless Recorders Multitrend Plus Validation Background The past five years has seen an increase in the use of computerized systems and products in the pharmaceutical and bio-pharmaceutical
Inspection Trends. American Society for Quality Richmond, VA Section March 8, 2016
 Inspection Trends American Society for Quality Richmond, VA Section March 8, 2016 Brooke K. Higgins, Senior Policy Advisor CDER / Office of Compliance Office of Manufacturing Quality Division of Drug Quality
Inspection Trends American Society for Quality Richmond, VA Section March 8, 2016 Brooke K. Higgins, Senior Policy Advisor CDER / Office of Compliance Office of Manufacturing Quality Division of Drug Quality
A Comprehensive Approach to Find and Remediate Data Integrity Problems
 A Comprehensive Approach to Find and Remediate Data Integrity Problems June 14, 2017 Copyright 2017 QuintilesIMS. All rights reserved. What is data integrity? Whom does it apply to? Definitions matter
A Comprehensive Approach to Find and Remediate Data Integrity Problems June 14, 2017 Copyright 2017 QuintilesIMS. All rights reserved. What is data integrity? Whom does it apply to? Definitions matter
Jean Guichard. Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN
 #: Internet-WP Version: e1.00 /EN") Jean Guichard White Paper Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN Introduction to Good Process Record Management (GxP) 2 / 6 Document History Version Date
Jean Guichard White Paper Introduction to Good Process Record Management (GxP) #: Internet-WP Version: e1.00 /EN Introduction to Good Process Record Management (GxP) 2 / 6 Document History Version Date
SESSION 14. Documentation Requirements and Management for Validation. 30 March Frits Vogt
 Validation Services SESSION 14 Documentation Requirements and Management for Validation 30 March 2017 Frits Vogt fritsvogt@valserv.nl Content Documentation Requirements and Management for Validation I.
Validation Services SESSION 14 Documentation Requirements and Management for Validation 30 March 2017 Frits Vogt fritsvogt@valserv.nl Content Documentation Requirements and Management for Validation I.
Data Integrity Issues & Concerns PDA Meeting Kansas City, MO August 21, 2017
 Data Integrity Issues & Concerns PDA Meeting Kansas City, MO August 21, 2017 CAPT Sharon K. Pederson (Thoma), PharmD National Expert of Pharmaceutical Inspections Food and Drug Administration ORA/OMPTO/OPQO/DPQP/Pharmaceutical
Data Integrity Issues & Concerns PDA Meeting Kansas City, MO August 21, 2017 CAPT Sharon K. Pederson (Thoma), PharmD National Expert of Pharmaceutical Inspections Food and Drug Administration ORA/OMPTO/OPQO/DPQP/Pharmaceutical
Contents. Contents (13) 1 Qualification (21)
 1 Qualification (21)") 1 Qualification (21) 1.A Official requirements (23) 1.A.1 Legal aspects of qualification (23) 1.A.2 Documentation of the qualification (26) 1.A.3 Design Qualification (DQ) (27) 1.A.4 Installation Qualification
1 Qualification (21) 1.A Official requirements (23) 1.A.1 Legal aspects of qualification (23) 1.A.2 Documentation of the qualification (26) 1.A.3 Design Qualification (DQ) (27) 1.A.4 Installation Qualification
GUIDELINES FOR VALIDATING ELECTRONIC CHART RECORDERS
 Institute For Thermal Processing Specialists GUIDELINES FOR VALIDATING ELECTRONIC CHART RECORDERS Various methods may be employed to validate electronic chart and the following recommendations are to be
Institute For Thermal Processing Specialists GUIDELINES FOR VALIDATING ELECTRONIC CHART RECORDERS Various methods may be employed to validate electronic chart and the following recommendations are to be
Summary of Significant Changes. Policy
 This Policy replaces POL251/1 Copy Number Effective 03/04/17 Summary of Significant Changes Addition of requirements of 2016 review of Guidance by MHRA, ALCOA+, and documents related to Policy The purpose
This Policy replaces POL251/1 Copy Number Effective 03/04/17 Summary of Significant Changes Addition of requirements of 2016 review of Guidance by MHRA, ALCOA+, and documents related to Policy The purpose
GMP On Site Series. GMP Essentials
 GMP On Site Series GMP Essentials GMP Basics Objectives 1. State the critical definitions of the pharmaceutical industry. 2. Describe the law as it applies to various critical functions. 3. State the history
GMP On Site Series GMP Essentials GMP Basics Objectives 1. State the critical definitions of the pharmaceutical industry. 2. Describe the law as it applies to various critical functions. 3. State the history
10 "Must-Haves" for the Life Sciences Learning Management System
 10 "Must-Haves" for the Life Sciences Learning Management System Why the Life Sciences LMS Needs to Demonstrate Record Control UL talks to many Life Sciences companies that are exploring learning and development
10 "Must-Haves" for the Life Sciences Learning Management System Why the Life Sciences LMS Needs to Demonstrate Record Control UL talks to many Life Sciences companies that are exploring learning and development
[ WHITE PAPER ] A Basic Overview: Meeting the PIC/S Requirements for a Computerized System INTRODUCTION GOOD MANUFACTURING PRACTICES
![[ WHITE PAPER ] A Basic Overview: Meeting the PIC/S Requirements for a Computerized System INTRODUCTION GOOD MANUFACTURING PRACTICES](/thumbs/93/111368697.jpg "[ WHITE PAPER ] A Basic Overview: Meeting the PIC/S Requirements for a Computerized System INTRODUCTION GOOD MANUFACTURING PRACTICES") A Basic Overview: Meeting the PIC/S Requirements for a Computerized System Lynn Archambault Waters Corporation, Milford, MA, USA INTRODUCTION The Pharmaceutical Inspection Convention and Pharmaceutical
A Basic Overview: Meeting the PIC/S Requirements for a Computerized System Lynn Archambault Waters Corporation, Milford, MA, USA INTRODUCTION The Pharmaceutical Inspection Convention and Pharmaceutical
COMPLIANCE BY DESIGN FOR PHARMACEUTICAL QUALITY CONTROL LABORATORIES INSIGHT FROM FDA WARNING LETTERS
 COMPLIANCE BY DESIGN FOR PHARMACEUTICAL QUALITY CONTROL LABORATORIES INSIGHT FROM FDA WARNING LETTERS Primer CONTENTS INTRODUCTION...3 QUALITY AND COMPLIANCE IN QUALITY CONTROL LABORATORIES...5 Compliance
COMPLIANCE BY DESIGN FOR PHARMACEUTICAL QUALITY CONTROL LABORATORIES INSIGHT FROM FDA WARNING LETTERS Primer CONTENTS INTRODUCTION...3 QUALITY AND COMPLIANCE IN QUALITY CONTROL LABORATORIES...5 Compliance
Water purification in the pharmaceutical industry
 ABB MEASUREMENT & ANALYTICS APPLICATION DESCRIPTION Water purification in the pharmaceutical industry Providing independent verification and validation of the water purification process for compliance
ABB MEASUREMENT & ANALYTICS APPLICATION DESCRIPTION Water purification in the pharmaceutical industry Providing independent verification and validation of the water purification process for compliance
Design Qualification SOP
 1.0 Commercial in Confidence 16-Aug-2006 1 of 13 Design Qualification SOP Document No: SOP_0400 Prepared by: David Brown Date: 16-Aug-2006 Version: 1.0 1.0 Commercial in Confidence 16-Aug-2006 2 of 13
1.0 Commercial in Confidence 16-Aug-2006 1 of 13 Design Qualification SOP Document No: SOP_0400 Prepared by: David Brown Date: 16-Aug-2006 Version: 1.0 1.0 Commercial in Confidence 16-Aug-2006 2 of 13
Good Automated Manufacturing Practices (GAMP)
") Good Automated Manufacturing Practices (GAMP) Klaus Krause, Amgen ISPE/GAMP Americas Steering Committee ISPE San Francisco/Bay Area Chapter Meeting, October 7, 2004 Presentation Overview I. GAMP - Organization
Good Automated Manufacturing Practices (GAMP) Klaus Krause, Amgen ISPE/GAMP Americas Steering Committee ISPE San Francisco/Bay Area Chapter Meeting, October 7, 2004 Presentation Overview I. GAMP - Organization
IT Compliance in the FDA Regulated Industry. IT Responsibilities 10 Years Ago TODAY! 11/17/2008. Business. Security. Compliance. IT & Automation Forum
 IT Compliance in the FDA Regulated Industry IT & Automation Forum November 13, 2007 Caribe Hilton, San Juan Puerto Rico Speaker: Mr. Juan O. Pérez, Principal Consultant joperez@fdcpr.com IT Responsibilities
IT Compliance in the FDA Regulated Industry IT & Automation Forum November 13, 2007 Caribe Hilton, San Juan Puerto Rico Speaker: Mr. Juan O. Pérez, Principal Consultant joperez@fdcpr.com IT Responsibilities
COMPUTERIZED SYSTEM VALIDATION (CSV) IMPLEMENTATION, DEMARCATION AND STRUCTURATION
 IMPLEMENTATION, DEMARCATION AND STRUCTURATION") COMPUTERIZED SYSTEM VALIDATION (CSV) IMPLEMENTATION, DEMARCATION AND STRUCTURATION Audit Security is the high priority for pharmaceutical companies, especially when computerized Systems are used. The everchanging
COMPUTERIZED SYSTEM VALIDATION (CSV) IMPLEMENTATION, DEMARCATION AND STRUCTURATION Audit Security is the high priority for pharmaceutical companies, especially when computerized Systems are used. The everchanging
OECD Working Group on Good Laboratory Practice. Template for submission of comments on draft GLP Guidance Documents. Instructions for Use
 OECD Working Group on Good Laboratory Practice Template for submission of comments on draft GLP Guidance Documents Instructions for Use 1. First, please complete the table below giving the full name of
OECD Working Group on Good Laboratory Practice Template for submission of comments on draft GLP Guidance Documents Instructions for Use 1. First, please complete the table below giving the full name of
GMP Compliance. George Acevedo, CQV Senior Engineer M+W Singapore Pte. Ltd
 GMP Compliance George Acevedo, CQV Senior Engineer M+W Singapore Pte. Ltd June 2017 Overview GMP Definition and Regulation Our Philosophy and Strategy for Compliance Project Lifecycle Deliverables Master
GMP Compliance George Acevedo, CQV Senior Engineer M+W Singapore Pte. Ltd June 2017 Overview GMP Definition and Regulation Our Philosophy and Strategy for Compliance Project Lifecycle Deliverables Master
GMPs: Distribution Centers
 SkillsPlus International Inc. The On-Site Series GMPs: Distribution Centers Learn to apply the GMP regulations as they relate to distribution center operations! FDA Past, Present, and Future and opener
SkillsPlus International Inc. The On-Site Series GMPs: Distribution Centers Learn to apply the GMP regulations as they relate to distribution center operations! FDA Past, Present, and Future and opener
The SaaS LMS and Total Cost of Ownership in FDA-Regulated Companies
 The SaaS LMS and Total Cost of Ownership in FDA-Regulated Companies The SaaS LMS and Total Cost of Ownership in FDA-Regulated Companies By Rob Sims, UL Compliance to Performance When Life Sciences companies
The SaaS LMS and Total Cost of Ownership in FDA-Regulated Companies The SaaS LMS and Total Cost of Ownership in FDA-Regulated Companies By Rob Sims, UL Compliance to Performance When Life Sciences companies
According to USP <1058>
 Analytical l Instrument t Qualification According to USP Scope, Approach, Requirements Dr. Ludwig Huber Chief Advisor, Global FDA Compliance ludwig_huber@labcompliance.com Overview FDA and International
Analytical l Instrument t Qualification According to USP Scope, Approach, Requirements Dr. Ludwig Huber Chief Advisor, Global FDA Compliance ludwig_huber@labcompliance.com Overview FDA and International
ISPE-FDA 3 rd Annual CGMP Conference 2 4 June 2014 Baltimore, MD. Detecting GMP Data Integrity Issues
 Detecting GMP Data Integrity Issues Elaine Eborall Senior Director, GMP Compliance, Americas and Asia Pacific Compliance and External Collaboration ISPE-FDA cgmp Conference Baltimore, Maryland 2-4 June
Detecting GMP Data Integrity Issues Elaine Eborall Senior Director, GMP Compliance, Americas and Asia Pacific Compliance and External Collaboration ISPE-FDA cgmp Conference Baltimore, Maryland 2-4 June
Guidance on 21 CFR Part 11 Validation of Electronic Chart Recorders Used in the Food Processing Industry
 Guidance on 21 CFR Part 11 Validation of Electronic Chart Recorders Used in the Food Processing Industry Draft copy issued for IFTPS membership review only May 5, 2017 Guidance on 21 CFR Part 11 Validation
Guidance on 21 CFR Part 11 Validation of Electronic Chart Recorders Used in the Food Processing Industry Draft copy issued for IFTPS membership review only May 5, 2017 Guidance on 21 CFR Part 11 Validation
Preparing for Successful Data Integrity Audits. Mary Chris Easterly & Richard D. Schlabach 13 October 2017
 Preparing for Successful Data Integrity Audits Mary Chris Easterly & Richard D. Schlabach 13 October 2017 Preparing for Successful Data Integrity Audits U.S. Food & Drug Administration and other regulatory
Preparing for Successful Data Integrity Audits Mary Chris Easterly & Richard D. Schlabach 13 October 2017 Preparing for Successful Data Integrity Audits U.S. Food & Drug Administration and other regulatory
DEC STD ISO Quality Systems - Model for Quality Assurance in Design /Development, Production, Installation and Servicing
 Digital Internal Use Only DEC STD 017-1 - ISO 9001 - Quality Systems - Model for Quality Assurance in Design /Development, Production, Installation and Servicing DOCUMENT IDENTIFIER: A-DS-EL00017-01-0000
Digital Internal Use Only DEC STD 017-1 - ISO 9001 - Quality Systems - Model for Quality Assurance in Design /Development, Production, Installation and Servicing DOCUMENT IDENTIFIER: A-DS-EL00017-01-0000
VALIDATION READY IT PROJECTS OF BEAS INDUSTRY SOLUTIONS BASED ON SAP BUSINESS ONE
 White Paper VALIDATION READY IT PROJECTS OF BEAS INDUSTRY SOLUTIONS BASED ON SAP BUSINESS ONE The comprehensive Solution for small and mid-sized Manufacturing Enterprises in regulated Industries CONTENT
White Paper VALIDATION READY IT PROJECTS OF BEAS INDUSTRY SOLUTIONS BASED ON SAP BUSINESS ONE The comprehensive Solution for small and mid-sized Manufacturing Enterprises in regulated Industries CONTENT
Documenta tion and Records
 Documenta tion and Records Page 1 of 30 Training Outcome of the Module: After completing this module, you will be able to: Recognize the importance of procedures Recognize the importance of record keeping
Documenta tion and Records Page 1 of 30 Training Outcome of the Module: After completing this module, you will be able to: Recognize the importance of procedures Recognize the importance of record keeping
EU Annex 11 update. An ISPE interpretation
 EU Annex 11 update An ISPE interpretation ISPE Interpretation Published by Winnie Cappucci Chris Clark Tim Goossens Sion Wyn Disclaimer ISPE cannot ensure and does not warrant that computerized systems
EU Annex 11 update An ISPE interpretation ISPE Interpretation Published by Winnie Cappucci Chris Clark Tim Goossens Sion Wyn Disclaimer ISPE cannot ensure and does not warrant that computerized systems
Correspondence Between ISO 13485:2016 and 21 CFR Part 820 QMS Requirements
 Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
Correspondence Between and 21 CFR Part 820 QMS Requirements 10411 Corporate Drive, Suite 102, Pleasant Prairie, WI 53158 262.842.1250 262.842.1240 info@rcainc.com rcainc.com 2 4 Quality Management System
Process ERP, an ideal software solution for Life Science industries
 Process ERP, an ideal software solution for Life Science industries Update on FDA's current compliance requirements of GxP regulated computerized systems Ravi Jotwani, Ph.D University of Louisville ProcessManufacturingSoftware
Process ERP, an ideal software solution for Life Science industries Update on FDA's current compliance requirements of GxP regulated computerized systems Ravi Jotwani, Ph.D University of Louisville ProcessManufacturingSoftware
QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES
 Your Company Name QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES Origination Date: XXXX Document Identifier: Date: Document Revision: QMS-00 QMS Policies and Procedures Latest Revision Date Abstract:
Your Company Name QUALITY MANAGEMENT SYSTEM POLICIES AND PROCEDURES Origination Date: XXXX Document Identifier: Date: Document Revision: QMS-00 QMS Policies and Procedures Latest Revision Date Abstract:
CSV Inspection Readiness through Effective Document Control. Eileen Cortes April 27, 2017
 CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
CSV Inspection Readiness through Effective Document Control Eileen Cortes April 27, 2017 Agenda Background CSV Readiness CSV and Change Management Process Inspection Readiness Do s and Don ts Inspection
For analytical laboratories. A primer Good laboratory practice and current good manufacturing practice
 For analytical laboratories A primer Good laboratory practice and current good manufacturing practice For analytical laboratories A primer Good laboratory practice and current good manufacturing practice
For analytical laboratories A primer Good laboratory practice and current good manufacturing practice For analytical laboratories A primer Good laboratory practice and current good manufacturing practice
Framing the Problem. Benefits of Electronic Validation Lifecycle Management (evlm) Systems: A Case Study 2/14/2018
 Systems: A Case Study 2/14/2018") Benefits of Electronic Validation Lifecycle Management (evlm) Systems: A Case Study Charlie Maher Managing Director, BioVoke charlie.maher@cagents.com 808-255-7603 Framing the Problem 1 Last century 3
Benefits of Electronic Validation Lifecycle Management (evlm) Systems: A Case Study Charlie Maher Managing Director, BioVoke charlie.maher@cagents.com 808-255-7603 Framing the Problem 1 Last century 3
Association of American Railroads Quality Assurance System Evaluation (QASE) Checklist Rev. 1/12/2017
 Checklist Rev. 1/12/2017") Company: Prepared By: Date: Changes from previous version highlighted in yellow. Paragraph Element Objective Evidence 2.1 Objective of Quality Assurance Program 2.2 Applicability and Scope 2.3 QA Program
Company: Prepared By: Date: Changes from previous version highlighted in yellow. Paragraph Element Objective Evidence 2.1 Objective of Quality Assurance Program 2.2 Applicability and Scope 2.3 QA Program
Cross-walk between EU Annex 11 and US FDA 211, 820, 11; other guidelines and regulations Orlando López Rev19Dec14
 Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Principle. a. This annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill
Risk Assessment is a vital component in
 This article demonstrates how the risk analysis guidance in GAMP 4 can be applied to GMPs and Good Distribution Practices (GDPs). Reprinted from The Official Journal of ISPE PHARMACEUTICAL ENGINEERING
This article demonstrates how the risk analysis guidance in GAMP 4 can be applied to GMPs and Good Distribution Practices (GDPs). Reprinted from The Official Journal of ISPE PHARMACEUTICAL ENGINEERING
Data Integrity Why is it important? DADM Conference - 22 August 2017
 Data Integrity Why is it important? DADM Conference - 22 August 2017 Maibritt Haugaard Møller, System Validation Expert Mette Ravn, Vice President, Data Management, IT and System Validation Overview of
Data Integrity Why is it important? DADM Conference - 22 August 2017 Maibritt Haugaard Møller, System Validation Expert Mette Ravn, Vice President, Data Management, IT and System Validation Overview of